
Transcriptome-wide analyses of piRNA binding sites suggest distinct mechanisms regulate piRNA binding and silencing in C. elegans
- Wei-Sheng Wu1,3,
- Jordan S. Brown2,3,
- Sheng-Cian Shiue1,
- Chi-Jung Chung1,
- Dong-En Lee1,
- Donglei Zhang2 and
- Heng-Chi Lee2
- 1Department of Electrical Engineering, National Cheng Kung University, Tainan 701, Taiwan
- 2Department of Molecular Genetics and Cell Biology, University of Chicago, Chicago, Illinois 60637, USA
- Corresponding author: hengchilee{at}uchicago.edu
-
↵3 These authors contributed equally to this work.
Abstract
PIWI-interacting RNAs (piRNAs) protect genome integrity by silencing transposon mRNAs and some endogenous mRNAs in various animals. However, C. elegans piRNAs only trigger gene silencing at select predicted targeting sites, suggesting additional cellular mechanisms regulate piRNA silencing. To gain insight into possible mechanisms, we compared the transcriptome-wide predicted piRNA targeting sites to the in vivo piRNA binding sites. Surprisingly, while sequence-based predicted piRNA targeting sites are enriched in 3′ UTRs, we found that C. elegans piRNAs preferentially bind to coding regions (CDS) of target mRNAs, leading to preferential production of secondary silencing small RNAs in the CDS. However, our analyses suggest that this CDS binding preference cannot be explained by the action of antisilencing Argonaute CSR-1. Instead, our analyses imply that CSR-1 protects mRNAs from piRNA silencing through two distinct mechanisms—by inhibiting piRNA binding across the entire CSR-1 targeted transcript, and by inhibiting secondary silencing small RNA production locally at CSR-1 bound sites. Together, our work identifies the CDS as the critical region that is uniquely competent for piRNA binding in C. elegans. We speculate the CDS binding preference may have evolved to allow the piRNA pathway to maintain robust recognition of RNA targets in spite of genetic drift. Together, our analyses revealed that distinct mechanisms are responsible for restricting piRNA binding and silencing to achieve proper transcriptome surveillance.
Keywords
INTRODUCTION
Small noncoding RNAs, such as miRNAs and piRNAs, play important roles in gene regulation by guiding Argonaute proteins to target RNAs with sequence complementarity (Hutvagner and Simard 2008). miRNAs play critical roles in various developmental processes by regulating the expression of endogenous genes (Bartel 2009). PIWI-interacting RNAs (piRNAs) are germline-enriched small RNAs found in diverse animals that silence non-self nucleic acids, such as transposons and viruses (Saito et al. 2006; Brennecke et al. 2007; Carmell et al. 2007; Batista et al. 2008; Das et al. 2008). One unique characteristic of piRNAs is their sequence diversity—tens of thousands of sequence-distinct piRNAs are produced. To identify mRNA targets regulated by these diverse piRNAs, both sequence-based approaches as well as experimental approaches have been reported; using bioinformatic analyses and in vivo reporter assays, previous studies have identified the piRNA targeting rules in C. elegans (Shen et al. 2018; Zhang et al. 2018). In C. elegans, piRNAs identify their targets through base-pairing, requiring nearly perfect complementarity at seed regions and tolerating several mismatches in the remaining sequence. Several predicted piRNA targeting sites have been confirmed to play a regulatory role in vivo, and removal of predicted piRNA targeting sites allows for stable expression of several silencing-prone transgenes (Shen et al. 2018; Zhang et al. 2018). piRNAs are predicted to bind various germline-silenced mRNAs, and surprisingly, many germline-expressed mRNAs as well (Zhang et al. 2018; Wu et al. 2019). CLASH (crosslinking, ligation and sequencing of hybrids) is a powerful biochemical approach to identify in vivo small RNA binding sites (Helwak et al. 2013); protein-bound small RNAs are ligated to their target mRNAs and these hybrid molecules, comprised of small RNAs and mRNA fragments, are sequenced to identify putative direct interactions. A recent study has applied CLASH analyses of C. elegans PIWI (PRG-1) to identify in vivo piRNA binding sites (Shen et al. 2018). Analysis of this data revealed that piRNAs bind both germline-silenced and germline-expressed mRNAs in C. elegans. Therefore, both bioinformatic and experimental approaches suggest that piRNAs alone are not sufficient to distinguish self from non-self nucleic acids. Two types of endogenous small RNAs work with piRNAs to achieve proper transcriptome surveillance. WAGO associated small RNAs are produced at piRNA targeting sites to silence non-self nucleic acids (Lee et al. 2012; Shirayama et al. 2012). In addition, CSR-1 Argonaute and its associated small RNAs have been shown to target germline-expressed RNAs and protect self nucleic acids from piRNA silencing (Seth et al. 2013; Wedeles et al. 2013; Shen et al. 2018). While the mechanism that controls CSR-1 licensing remains largely unknown, these studies suggest an interesting model where CSR-1 and its small RNAs act as a gene-licensing pathway, critical for distinguishing self nucleic acids from non-self in C. elegans.
Our genome-wide analyses of piRNA targeting sites have identified surprisingly few overlapping sites between experimentally identified piRNA binding sites and sequence-based predicted piRNA targeting sites (Wu et al. 2019). These observations suggest that piRNA binding to mRNA targets may be regulated, such as by the CSR-1 pathway. However, CSR-1 protection alone cannot fully explain gene licensing, as CSR-1 knockdown does not result in silencing of many CSR-1 targets (Campbell and Updike 2015; Singh et al. 2021). Currently it is unclear whether additional mechanisms exist to control piRNA recognition.
To gain insight into the mechanisms that regulate piRNA silencing, we compared the in vivo binding sites of piRNAs to the in silico predicted piRNA targeting sites on target mRNAs (Fig. 1A). Specifically, we performed metagene analyses to examine the distribution of piRNA binding sites on mRNAs, and we compared binding density across various genomic features. We also examined the distribution of silencing WAGO small RNAs and licensing CSR-1 small RNAs, and their relationship with piRNA binding sites. Surprisingly, we found that piRNAs preferentially bind to coding regions (CDS) of mRNAs. As a consequence, piRNAs trigger the production of secondary WAGO silencing small RNAs preferentially at the CDS. In contrast, our analyses showed that the sequence-based predicted piRNA targeting sites are slightly enriched in 3′ UTRs, thus the preference for CDS binding by piRNAs cannot be explained by the distribution of predicted piRNA targeting sites. In addition, we found that CSR-1 licenses germline expression by inhibiting both piRNA binding and secondary silencing WAGO small RNA production, but does so through distinct modes of action. Nonetheless, our analyses suggest CSR-1 is not responsible for restricting piRNA binding to the CDS. Together, our analyses identify CDSs as the critical regions that are uniquely susceptible to piRNA silencing in C. elegans and reveal distinct mechanisms that regulate piRNA silencing in vivo.
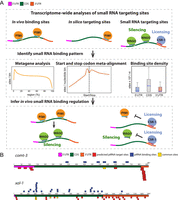
Transcriptome-wide analyses of piRNA binding sites. (A) To investigate in vivo small RNA binding regulation, we analyzed in vivo binding site data, in silico predicted piRNA targeting site data, and small RNA targeting site data. Synthesizing these data sets allowed us to identify small RNA binding patterns across groups of genes and within genomic features. By comparing such patterns in distinct genetic mutants, we infer and propose possible regulatory mechanisms of piRNA silencing. (B) Two examples highlight distinct patterns observed between in vivo piRNA binding sites and in silico predicted piRNA targeting sites. The locations of predicted piRNA targeting sites are shown in red, the experimentally identified piRNA binding sites are shown in blue, and the common sites (defined as sites where the same piRNA was determined by prediction and experimentally to target overlapping mRNA regions) are shown in yellow. The specific regions of the indicated mRNAs, including 5′ UTR, CDS, and 3′ UTR, are also labeled.
RESULTS
piRNAs exhibit a binding preference to the coding regions of target mRNAs in C. elegans
As described above, only a small proportion of predicted piRNA targeting sites were found to be bound by piRNAs in vivo, suggesting that piRNA binding to mRNA targets is regulated by cellular mechanisms (Wu et al. 2019). Intriguingly, while examining two well-characterized piRNA germline target mRNAs (Shen et al. 2018; Tang et al. 2018), we noticed that despite the presence of several predicted piRNA targeting sites in various regions of these mRNAs, including CDS, 5′ UTRs and 3′ UTRs (Fig. 1B and for complete binding site information, see Supplemental Data File 1), the experimentally identified piRNA binding sites are mostly present in the CDSs, with some of these binding sites also predicted as piRNA targeting sites. We therefore wondered whether the observed piRNA CDS-binding preference could be a global trend, applicable to other germline mRNAs.
To test whether our analyses of Argonaute/small RNA binding sites are consistent with previously published results, we first examined the distribution of experimentally identified miRNA binding sites in C. elegans, which are known to be enriched in 3′ UTRs of mRNAs (Broughton et al. 2016). Using previously published miRNA Argonaute ALG-1 iCLIP data, we identified transcriptome-wide in vivo miRNA binding sites. As expected, when we analyzed lin-4 miRNA binding sites on lin-14 mRNA, all in vivo lin-4 binding sites are located in the 3′ UTR of lin-14 (Fig. 2A; Supplemental Data File 1). We then calculated the density of all miRNA binding sites in different regions of the mRNAs, including 5′ UTR, CDS or 3′ UTR (Wu et al. 2022). We observed a higher density for both the number of miRNA binding sites and the miRNA binding events at the 3′ end of mRNAs, especially at the 3′ UTR of mRNAs in C. elegans (Fig. 2B,C; Supplemental Fig. S1A,B). To more closely examine the local change in miRNA binding preference between different regions, we examined the distribution of miRNA binding sites at the borders of CDS and UTRs. As expected, we observed that the number of miRNA binding sites did not change around the start codon but increased sharply immediately downstream from the stop codon (Fig. 2D). Together, these analyses confirmed the previous observations that miRNAs predominately bind to their target mRNAs at their 3′ UTRs (Broughton et al. 2016).
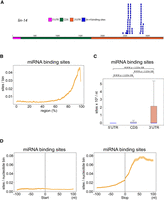
In vivo miRNA binding sites are enriched at the 3′ UTR of mRNAs in C. elegans. (A) lin-4 miRNA in vivo binding sites found in lin-14 mRNA. (B) Metagene trace shows the distribution of miRNA binding sites along an mRNA. The solid line indicates the average number of miRNA binding sites in all mRNAs. The dotted lines indicate the average number plus/minus one standard error. (C) The density of miRNA binding sites in the indicated regions. Blue lines represent median values. The statistical significance (P-value) of the difference in density between two regions was calculated by the Mann–Whitney U-test. (D) The distribution of miRNA binding sites around the start codon (left) or stop codon (right). A 200 nt window centered at start or stop codon is shown. The solid line indicates the average number of miRNA binding sites in all mRNAs. For each position, the average is calculated from all mRNAs that possess that position. For example, not all mRNAs have 5′UTRs, but they all have CDSs. The dotted lines indicate the average number plus/minus one standard error.
We then compared the genome-wide distribution of sequence-based predicted piRNA targeting sites and experimentally identified piRNA binding sites on germline mRNAs, since piRNAs are mostly expressed in the germline (Ortiz et al. 2014; Wu et al. 2018). Using the stringent piRNA targeting rule identified in previous studies (Wu et al. 2018; Zhang et al. 2018), we observed that the predicted piRNA targeting sites are enriched at the 3′ end of germline mRNAs (Fig. 3A), and their densities are highest in the 3′ UTR of germline mRNAs (Fig. 3B). Using published PIWI PRG-1 CLASH data (Shen et al. 2018), we then analyzed the transcriptome-wide in vivo piRNA binding sites on germline mRNAs (Wu et al. 2022). Interestingly, we observed a very distinct distribution of in vivo piRNA binding sites compared to that of the predicted piRNA targeting sites; the overall distribution of piRNA binding sites on germline mRNAs exhibits a peak at the 5′ end, followed by a steady level across the gene body before a decrease at the 3′ end (Fig. 3C). Consistent with the CDS binding preference found in the well-characterized piRNA targeted mRNAs, we found that the density of in vivo piRNA binding sites is highest in the CDS of germline mRNAs (Fig. 3D). Specifically, the piRNA binding sites increased most notably around the start codon and decreased most notably around the stop codon (Fig. 3E)—opposite to the distribution of the predicted piRNA targeting sites (Supplemental Fig. S2A). These observations suggest that piRNAs preferentially bind to CDSs, and this preference cannot be explained by the distribution of predicted piRNA targeting sites.
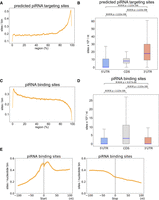
In vivo piRNA binding sites are enriched in the coding region (CDS) of germline mRNAs in C. elegans. (A) Metagene trace shows the distribution of predicted piRNA targeting sites along a germline mRNA. The solid line indicates the average number of piRNA targeting sites in all germline mRNAs. The dotted lines indicate the average number plus/minus one standard error. (B) The density of predicted piRNA targeting sites in the indicated regions of germline mRNAs. Blue lines represent median values. The statistical significance (P-value) of the difference in density between two regions was calculated by the Mann–Whitney U-test. (C) Metagene trace shows the distribution of experimentally identified piRNA binding sites along a germline mRNA. The solid line indicates the average number of piRNA binding sites in all germline mRNAs. The dotted lines indicate the average number plus/minus one standard error. (D) The density of experimentally identified piRNA binding sites in the indicated regions of germline mRNAs. Blue lines represent median values. The statistical significance (P-value) of the difference in density between two regions was calculated by the Mann–Whitney U-test. (E) The distribution of piRNA binding sites around the start codon (left) or stop codon (right). A 200 nt window centered at start or stop codon is shown. The solid line indicates the average number of piRNA binding sites in all germline mRNAs. For each position, the average is calculated from all germline mRNAs that possess that position.
As some miRNAs are also expressed in the germline, we wondered whether the enrichment of piRNA binding sites in CDSs is a feature shared by germline-expressed miRNAs (McEwen et al. 2016). We therefore restricted our miRNA binding site analysis to germline-enriched miRNAs at germline mRNAs. However, we found that miRNA binding sites for these germline-enriched miRNAs remain enriched at the 3′ UTR and 3′ end of germline mRNAs (Supplemental Fig. S2B). Therefore, the piRNA CDS-binding preference is not shared by miRNAs in the germline. We wondered whether binding of miRISC complexes in the 3′ UTR could explain the exclusion of piRNA binding to 3′ UTRs. If so, those mRNAs without miRNA binding sites at their 3′ UTRs should exhibit a reduced CDS-binding preference and enhanced 3′ UTR binding. We analyzed piRNA binding sites in germline mRNA transcripts which contain predicted binding sites of germline miRNAs in their 3′ UTRs and those that do not. We found that piRNA binding is heavily enriched in the CDS for both of these groups (Supplemental Fig. S2C). Additionally, we analyzed piRNA binding site density in germline transcripts with experimentally identified germline miRNA binding sites in their 3′ UTRs and those without (Supplemental Fig. S2D). Again, we found that piRNAs still heavily favor CDS binding even without 3′ UTR germline miRNA binding. Therefore, despite germline miRNAs’ preference for binding 3′ UTRs (Supplemental Fig. S1B), miRNA binding at 3′ UTRs is not sufficient to explain the piRNA binding preference for the CDS. Since previous studies have shown that germline-expressed transcripts are protected from piRNA silencing by CSR-1 and its associated small RNAs (Seth et al. 2013; Wedeles et al. 2013; Shen et al. 2018), we also examined whether there is a difference in piRNA binding distribution on germline-expressed mRNAs (here defined as CSR-1 targets) or germline-silenced mRNAs (here defined as WAGO targets, see more descriptions of WAGO Argonautes below). We found that the CDS enrichment for piRNA binding sites exists for both WAGO targets as well as for CSR-1 targets (Supplemental Fig. S2E; Claycomb et al. 2009; Gu et al. 2009). This observation suggests that the CDS binding preference is likely not caused by CSR-1 (see more about CSR-1 function below). Together, we conclude that the in vivo piRNA binding sites are enriched in the CDSs of both germline-silenced and germline-expressed mRNAs in C. elegans.
piRNAs preferentially trigger the production of secondary small RNAs at mRNA coding regions
piRNAs induce gene silencing by triggering the production of secondary silencing small RNAs that associate with Worm specific ArGOnautes (WAGOs), also known as WAGO 22G-RNAs (Ashe et al. 2012; Bagijn et al. 2012; Lee et al. 2012). These WAGO 22G-RNAs are produced by RNA-dependent RNA polymerase locally at piRNA binding sites. Since piRNAs preferentially bind to CDSs, we wondered whether the production of WAGO 22G-RNAs is also enriched at CDS regions. To test this hypothesis, we compared the density of WAGO 22G-RNAs in distinct regions of WAGO targeted mRNAs. Indeed, for germline-silenced mRNAs (WAGO targets) (Gu et al. 2009), we observed that both WAGO-9 (also known as HRDE-1) and WAGO-1, bound 22G-RNAs are significantly enriched in the CDS (Fig. 4A). In addition, for CSR-1 targeted mRNAs, WAGO 22G-RNAs are also enriched in the CDS (Fig. 4A), despite their overall WAGO 22G-RNA levels being much lower in CSR-1 targets than in WAGO targets. WAGO 22G-RNA production can be induced by piRNAs and other small RNA pathways (Conine et al. 2010; Fischer et al. 2011). Therefore, if piRNA-induced WAGO 22G-RNAs are produced preferentially at the CDS, we expect that in PIWI mutants, which lose all piRNAs, the WAGO 22G-RNAs should be preferentially reduced at coding regions. Indeed, when we compared the levels of WAGO-1 22G-RNAs between wild type and PIWI prg-1 mutants (Barucci et al. 2020), we observed a greater reduction of WAGO-1 22G-RNAs in the CDS than that in 5′ or 3′ UTRs, for both germline-silenced and germline-expressed mRNAs (Fig. 4B). These observations suggest that the preferential piRNA binding at the CDS of mRNAs is functionally relevant, as piRNA binding preference in the CDS leads to a corresponding enrichment of secondary WAGO 22G-RNAs produced at the CDS. Furthermore, this confirms that the piRNA binding pattern we observed was not simply due to a technical bias of the CLASH method, as this independent signature of piRNA binding corroborates our findings. Interestingly, a recent study compared the ability of synthetic piRNAs to trigger gene silencing and showed that those synthetic piRNAs targeting the CDS can trigger robust gene silencing, but for synthetic piRNA targeting 5′ or 3′ UTRs, gene silencing is not as successful (Priyadarshini et al. 2022). These reporter-based experiments support our transcriptome-wide analyses that the CDS is the critical region that is uniquely competent for piRNA silencing in C. elegans.
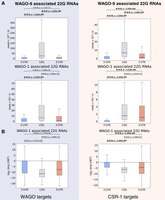
Secondary WAGO 22G-RNAs are preferentially produced in coding regions. (A) The density of WAGO-9 (top) and WAGO-1 (bottom) 22G-RNAs in the indicated regions of WAGO targets (left) or CSR-1 (right) targets. Blue lines represent median values. The statistical significance (P-value) of the difference in density between two regions was calculated by the Mann–Whitney U-test. (B) The ratio of WAGO-1 22G-RNAs in the prg-1 mutant over those in wild type in the indicated regions of WAGO (left) or CSR-1 (right) targets. Blue lines represent median values. The statistical significance (P-value) of the difference in fold change (prg-1 mutant/WT) between two regions was calculated by the Mann–Whitney U-test.
CSR-1 inhibits piRNA binding and WAGO 22G-RNA amplification with distinct modes of action
CSR-1 Argonaute and its associated 22G-RNAs have been reported to protect germline transcripts from piRNA silencing (Seth et al. 2013; Wedeles et al. 2013). However, the mechanism behind CSR-1 Argonaute's anti-piRNA silencing function remains unclear. A previous study suggests that CSR-1 inhibits the piRNA pathway by interfering with piRNA binding (Shen et al. 2018). Nonetheless, it is unclear whether CSR-1 also inhibits downstream WAGO 22G-RNA synthesis. In addition, it is unknown whether CSR-1 binding leads to protection of the whole transcript from piRNA silencing, or CSR-1 only interferes with piRNA silencing locally, proximal to CSR-1 targeting sites. To gain insight into CSR-1's anti-piRNA silencing function, we first examined the distribution of CSR-1 22G-RNAs on germline mRNAs. We found that the levels of CSR-1 22G-RNAs are higher at the 3′ end of CSR-1 targets (Fig. 5A, top) and are produced at both CDSs and 3′ UTRs (Supplemental Fig. S3A, top), consistent with a recent report (Singh et al. 2021). In addition, we found a low level of CSR-1 22G-RNAs produced from WAGO targets. These CSR-1 22G-RNAs are more equally distributed across the gene body, with their levels reduced at both 5′ and 3′ ends (Fig. 5A, bottom), and enriched at the CDS (Supplemental Fig. S3A, bottom).
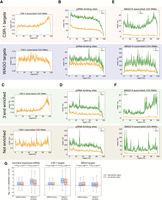
CSR-1 inhibits piRNA binding and WAGO 22G-RNA synthesis with distinct modes of action. (A) Metagene trace shows the distribution of CSR-1 22G-RNAs along CSR-1 (top) or WAGO (bottom) targets. The solid line indicates the average number of CSR-1 22G-RNA read counts in CSR-1 or WAGO targeted mRNAs. The dotted lines indicate the average number plus/minus one standard error. (B) Metagene trace shows the distribution of piRNA binding events in wild type (orange) or in CSR-1 depleted animals (green) mapped to CSR-1 (top) or WAGO (bottom) targets. The piRNA–tRNA hybrid reads, which are expected to be nonspecific ligation products and thus remain unchanged between samples, are used for normalization. The solid line indicates the average number of piRNA binding events (read counts) in CSR-1 or WAGO targets. The dotted lines indicate the average number plus/minus one standard error. (C) Metagene trace shows the distribution of CSR-1 22G-RNAs mapped to the indicated CSR-1 subgroup genes, including 3′ end 22G enriched (top) or not enriched (bottom) CSR-1 targets. The solid line indicates the average number of CSR-1 22G-RNAs read counts in the indicated CSR-1 subgroup genes. The dotted lines indicate the average number plus/minus one standard error. (D) Metagene trace shows the distribution of piRNA binding events in wild type (orange) or in CSR-1 depleted animals (green) mapped to the indicated CSR-1 subgroup genes, including 3′ end 22G enriched (top) or not enriched (bottom) CSR-1 targets. The solid line indicates the average number of piRNA binding events (read counts) in the indicated CSR-1 subgroup genes. The dotted lines indicate the average number plus/minus one standard error. (E) Metagene traces show the distribution of WAGO-9 (HRDE-1) 22G-RNAs in wild type (orange) or in CSR-1 depleted animals (green) mapped to CSR-1 (top) or WAGO (bottom) targets. The miRNA mapped reads, which are expected to be nonspecific RNA cloned from these experiments and thus remain unchanged between samples, are used for normalization. The solid line indicates the average number of WAGO-9 22G-RNA read counts in CSR-1 or WAGO targets. The dotted lines indicate the average number plus/minus one standard error. (F) Metagene traces show the distribution of WAGO-9 (HRDE-1) 22G-RNAs in wild type (orange) or in CSR-1 depleted animals (green) mapped to the indicated CSR-1 subgroup genes, including 3′ end 22G enriched (top) or not enriched (bottom) CSR-1 targets. The miRNA mapped reads are used for normalization. The solid line indicates the average number of WAGO-9 22G-RNAs read counts in the indicated CSR-1 subgroup genes, including 3′ end 22G enriched (top) or not enriched (bottom) CSR-1 targets. The dotted lines indicate the average number plus/minus one standard error. The miRNA mapped reads are used for normalization between samples. (G) The ratio of piRNA binding events or WAGO-9 22G-RNAs in csr-1 depletion over control in high-density CSR-1 targeted windows and low-density CSR-1 targeted windows for germline mRNAs, CSR-1 targets, and WAGO targets. The piRNA–tRNA hybrids and the miRNA reads are used for normalization of piRNA binding and WAGO-9 22G-RNAs, respectively. Blue lines represent median values. The statistical significance (P-value) of the difference in fold change (csr-1 depletion over control) between two windows was calculated by the Mann–Whitney U-test.
To examine the relationship between CSR-1 binding and piRNA binding, we then examined the effect of CSR-1 depletion on piRNA binding (Shen et al. 2018). Our analyses showed that CSR-1 depletion leads to a greater increase of piRNA binding in CSR-1 targets than in WAGO-1 targets, indicating that the greater level of CSR-1 22G-RNAs leads to greater levels of protection from piRNA binding (Fig. 5B; Supplemental Fig. S3B). If CSR-1 inhibits piRNA binding locally, we expect a greater increase of piRNA binding at regions where CSR-1 levels are higher. However, despite the very different distribution of CSR-1 22G-RNA accumulation on WAGO targets and CSR-1 targets, we observed a similar increase of piRNA binding in all regions of the transcripts for both WAGO and CSR-1 targets (Fig. 5B; Supplemental Fig. S3B) upon CSR-1 depletion. Therefore, while the overall level of CSR-1 targeting a given transcript correlates with its protection level against piRNA binding, the location of CSR-1 targeting within the transcript does not seem to matter. To further examine the relationship between CSR-1 localization and piRNA binding, we defined subgroups of CSR-1 targets for CSR-1 targets where CSR-1 22G-RNAs are either enriched at their 3′ UTRs (n = 2082) or are not enriched at any specific regions (n = 763) (Fig. 5C; Supplemental Fig. S3C). Again, upon CSR-1 depletion, we found that piRNA binding sites increased in all regions of both subgroups of CSR-1 targets despite their different CSR-1 distributions (Fig. 5D; Supplemental Fig. S3D). These results suggest that CSR-1's inhibition of piRNA binding does not correlate with local CSR-1 22G-RNA levels.
While our analyses show that CSR-1 inhibits piRNA binding, we observed that piRNAs bind germline-expressed CSR-1 targets with higher density than they bind germline-silenced WAGO targets (Supplemental Fig. S3E). This apparent contradiction could be explained by a significantly higher expression level for CSR-1 targets, providing more opportunities for piRNA binding. To test this hypothesis, we normalized the piRNA binding density to mRNA expression levels. Indeed, we found piRNA density is significantly greater for WAGO targets than for CSR-1 targets on a per transcript basis (Supplemental Fig. S3F). Nonetheless, significantly more WAGO 22G-RNAs (both WAGO-1 and WAGO-9 22G-RNAs) were produced from all regions of WAGO targets compared to CSR-1 targets (Supplemental Fig. S3G), indicating that piRNA binding on CSR-1 targets does not trigger WAGO 22G-RNA production as effectively as piRNA binding on WAGO targets. This observation suggests that CSR-1 not only inhibits piRNA binding, it may also inhibit the downstream production of WAGO 22G-RNAs. To examine the relationship between CSR-1 binding and WAGO 22G-RNA synthesis, we examined the change of WAGO-9 (HRDE-1) 22G-RNAs upon CSR-1 depletion (Singh et al. 2021). Interestingly, for both CSR-1 and WAGO targets, there is a greater increase in WAGO-9 22G-RNAs preferentially at regions where CSR-1 22G RNAs are enriched, including the 3′ end of CSR-1 targets and at the gene body of WAGO targets, respectively (Fig. 5E; Supplemental Fig. S3H). To further examine the relationship between CSR-1 distribution and WAGO 22G-RNA production, we compared those CSR-1 target subgroups mentioned above that have either CSR-1 22G-RNAs enriched at 3′ UTRs or not enriched at a specific region. Upon CSR-1 depletion, we found a greater increase in WAGO 22G-RNA production at 3′ UTRs for those CSR-1 targets with a 3′ CSR-1 targeting enrichment (Fig. 5F; Supplemental Fig. S3I). These observations show that the local CSR-1 22G-RNA levels contribute to the inhibitory strength against WAGO 22G-RNA production, indicating that CSR-1 acts locally to inhibit the production of WAGO 22G-RNAs.
Our analyses suggest that CSR-1 acts to combat piRNA binding in a transcript-wide manner, evidenced by CSR-1 depletion leading to uniformly increased piRNA binding across transcripts (Fig. 5D). However, we also showed that CSR-1 acts locally to prevent WAGO 22G-RNA accumulation, where the most CSR-1 dense regions of a transcript are most protected from WAGO 22G-RNA amplification (Fig. 5F). To directly compare the effects of CSR-1 targeting on piRNA binding and WAGO 22G-RNA synthesis, we identified 100 nt windows within each germline mRNA that are most heavily and most poorly targeted by CSR-1. We then compared the impact of CSR-1 depletion on piRNA binding and WAGO 22G-RNA production within each window. If CSR-1 combats piRNA binding in a transcript-wide manner, we would expect there to be little difference in the increase in piRNA binding density upon CSR-1 depletion between the most heavily and poorly CSR-1 targeted regions. Conversely, if CSR-1 prevents local WAGO 22G-RNA amplification, we expect there would be a significantly greater increase in WAGO 22G-RNA production in heavily CSR-1 targeted regions compared to the production in poorly targeted regions. Indeed, the increase in piRNA binding following CSR-1 depletion is not significantly different between high-density and low-density CSR-1 targeting windows (Fig. 5G). In addition, we found that for WAGO 22G-RNA production, there is strikingly more 22G-RNA accumulation in high-density CSR-1 windows following CSR-1 depletion compared to low-density CSR-1 windows for germline expressed mRNAs (Fig. 5G). A similar trend of piRNA binding and WAGO 22G-RNA production was found (Fig. 5G) for CSR-1 target genes. For WAGO targets, we found that depletion of CSR-1 has little effect on piRNA binding, but leads to a slight insignificant increase in WAGO-22G-RNAs in high density CSR-1 windows. Taken together, our analyses suggest that CSR-1 can counteract the piRNA pathway through the inhibition of both piRNA binding and WAGO 22G-RNA production, but these two functions of CSR-1 exhibit distinct modes of action, where only the inhibition of WAGO 22G-RNA synthesis is sensitive to the levels of local CSR-1 22G-RNAs.
CSR-1 is not responsible for piRNAs’ CDS-binding preference
We then investigated whether CSR-1 contributes to the piRNA CDS binding preference. If CSR-1 is responsible for the piRNA CDS-binding preference, we would expect this preference to diminish upon CSR-1 depletion. However, upon CSR-1 knockdown, piRNAs retain their CDS binding preference for both CSR-1 targets and WAGO targets (Supplemental Fig. S3B). Specifically, the knockdown of CSR-1 did not change the pattern of piRNA binding sites around start or stop codons for either CSR-1 targets or WAGO targets (Supplemental Fig. S4A). As translation has been shown to antagonize CSR-1 22G-RNA synthesis in CDS regions (Singh et al. 2021), we wondered whether translation may also play a role in regulating piRNA binding to the CDS. For both highly translated and lowly translated CSR-1 or WAGO targets (Aeschimann et al. 2017), we found that piRNAs retain their CDS binding preference (Supplemental Fig. S4B). However, since translation of germline mRNAs is a highly regulated process throughout germline development, future studies are needed to further investigate whether translation plays a role in regulating piRNA binding. Together, our analyses suggest that the piRNA CDS binding preference cannot be explained by CSR-1 licensing and indicates that an unknown mechanism promotes or restricts piRNA binding to the CDS.
DISCUSSION
Small RNAs, such as miRNAs and piRNAs, regulate gene expression through base-pairing with their target mRNAs. It has been shown that miRNA targeting sites are enriched at 3′ UTRs and that 3′ UTR sites exhibit more gene regulatory effects than those at the CDS (Baek et al. 2008). While piRNAs are known for their roles in genome defense, it was previously unknown whether piRNA targeting or regulatory potential operate differently among different regions of their mRNA targets. By analyzing transcriptome-wide in vivo piRNA binding sites, we uncovered a new feature regulating the piRNA defense system in C. elegans: C. elegans piRNAs preferentially bind and trigger gene silencing at the CDS of germline mRNAs. Consequently, secondary WAGO 22G-RNAs are preferentially produced at the CDS as well (Fig. 6). Our finding has important implications on using synthetic piRNAs to silence target genes of interest, where piRNAs targeting the CDS are likely going to be more effective. Indeed, a recent publication has confirmed this trend, albeit not on a transcriptome-wide scale (Priyadarshini et al. 2022). Nonetheless, it is worth noting that synthetic piRNAs targeting the 3′ UTRs of transgenes can trigger transgene silencing (Ashe et al. 2012; Lee et al. 2012). In addition, the CDS of oma-1, but not its UTRs has been reported to carry specific licensing properties that counter piRNA silencing (Seth et al. 2018). Future studies will be necessary to further understand the underlying mechanisms that confer such differences in sensitivity to piRNA silencing. Furthermore, our study also provides insight into the applications in the expression of foreign nucleic acids in the germline of C. elegans. piRNAs are known to trigger gene silencing of transgenes that carry foreign nucleic acids, and removal of piRNA sites from the coding regions of these foreign nucleic acids are thus likely to be critical for the successful expression of these foreign nucleic acids in C. elegans. Indeed, previous studies have optimized the coding regions of foreign nucleic acids, including GFP, mCherry or Cas9, which successfully prevent these silencing-prone transgenes from piRNA-induced gene silencing (Zhang et al. 2018).
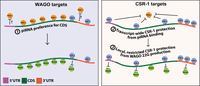
A model depicting three distinct mechanisms that control piRNA binding and gene silencing on WAGO and CSR-1 targets. For WAGO targets (left), a CSR-1-independent mechanism promotes piRNAs binding at CDS (mechanism 1). WAGO 22G-RNA production, which is initiated by piRNA targeting, is therefore following a similar pattern of enrichment. For CSR-1 targets (right), CSR-1 blocks piRNA binding in a transcript-wide mode of action (mechanism 2). For downstream WAGO 22G-RNA production, CSR-1 acts locally to block WAGO 22G-RNA accumulation (mechanism 3).
In addition, our analyses show that while the reported piRNA targeting rules are effective in preventing germline transgene silencing by mutating potential targeting sites, they can be further improved to identify functional piRNA targeting sites by considering other features of targeting events. For example, currently many CDS targeting events may have poor hybridization and are therefore dropped in the in silico prediction models. For these sites, lowering the stringency of hybridization may better recapitulate the in vivo target site landscape. In addition, target sites with high local CSR-1 levels may not be effective to trigger WAGO 22G-RNA production. Furthermore, future analyses are needed to examine and identify noncanonical piRNA binding rules.
Our work also provides insight into the mechanisms by which CSR-1 inhibits the piRNA pathway. Specifically, we found that CSR-1 depletion leads to increased piRNA binding across whole CSR-1 targeting transcripts, while increased secondary WAGO 22G-RNA synthesis preferentially occurs locally at CSR-1 targeting sites. In addition, our analyses showed that CSR-1 poorly targeted regions are just as effective in inhibiting piRNA binding compared to CSR-1 highly targeted regions. While we cannot fully rule out the possibility that low levels of CSR-1 also act locally to inhibit piRNA binding, our analyses showed that two antisilencing functions of CSR-1 exhibit two distinct modes of action, where only the inhibition of WAGO 22G-RNAs but not piRNA binding are correlated with local CSR-1 levels. Taken together, our observations support a model where the mechanisms underlying these two CSR-1 antisilencing processes are distinct. In this model, CSR-1 binding at various locations in a transcript can result in the protection of the entire transcript from piRNA recognition while CSR-1 targeted regions are locally protected against WAGO 22G-RNA production (Fig. 6). Several previous experimental reports have provided evidence that supports our model. First, tethering of CSR-1 to a single locus at the 3′ UTR of an endogenous mRNA can protect that mRNA from piRNA silencing (Cornes et al. 2022). Second, in transgenic worms carrying a silenced GFP::CDK-1 transgene, only foreign GFP sequences produce high levels of WAGO 22G-RNA, while the CDK-1 sequence (which is identical to the endogenous CDK-1 gene and therefore targeted by CSR-1 22G-RNAs) produces low levels of WAGO 22G-RNAs, indicating a local inhibition of WAGO 22G-RNAs at the CSR-1 targeted region (Seth et al. 2013). Nonetheless, understanding the specific mechanisms by which CSR-1 counteracts piRNA binding and WAGO 22G-RNA synthesis requires future investigation. We speculate that CSR-1 binding can somehow remove its bound mRNA from piRNA recognition subcellular hotspots, while CSR-1 inhibits WAGO 22G-RNAs synthesis locally, such as by competing for RNA-dependent RNA polymerases—resources known to be shared by these two Argonautes to produce their associated small RNAs (Gu et al. 2009).
Finally, our analyses also suggest that the CSR-1 pathway is not responsible for piRNAs’ CDS binding preference, suggesting that an unknown mechanism promotes CDS surveillance by piRNAs. As the CDSs of foreign RNAs are expected to be under more selective pressure than UTRs, we speculate that the preferential recognition of CDSs by piRNAs likely offers a mechanism for the piRNA surveillance system to reduce the chance of evolution of foreign RNA variants that evade piRNA recognition.
MATERIALS AND METHODS
Analyses of in vivo miRNA and piRNA binding sites
The iCLIP data of ALG-1 (SRR3882949) and the CLASH data of PRG-1 (SRR6512652/WT and SRR6512654/CSR-1 depletion) were used in these analyses (Broughton et al. 2016; Shen et al. 2018). To identify in vivo miRNA and piRNA binding sites, hybrids of miRNA/piRNA with their target mRNAs were identified by CLASH analyst (Wu et al. 2022) with default settings for data preprocessing and searching algorithms. To compare the amount of piRNA binding in wild type or CSR-1 depleted animals, the levels of piRNA/tRNA hybrid reads were used for normalization. The C. elegans miRNA/piRNA sequences and the mRNA sequences (n = 43040) of Wormbase version WS275 were used. Once hybrid reads are identified, miRNA and piRNA binding sites are defined by the regions of interacting mRNAs with lowest binding energy or highest binding score to corresponding miRNA using RNAup (Lorenz et al. 2011) or to corresponding piRNA using pirScan (Wu et al. 2018), respectively. When the mRNA interacting sequences (CLASH identified regions) are shorter than miRNA or piRNA, they are first extended to the size of miRNA and piRNAs using both the upstream and downstream sequences before they are examined for sites with best pairing energy/score.
Germline mRNAs (n = 27792) are defined as mRNAs that are detected either in the spermatogenic or oogenetic transcriptome (Ortiz et al. 2014). WAGO targets (n = 3644) are defined as genes whose mapped 22G-RNAs exhibit over twofold enrichment from either WAGO-1 IP than that from input 22G-RNAs (Gu et al. 2009) or WAGO-9 IP than that from input 22G-RNAs (Shirayama et al. 2012). CSR-1 targets (n = 15821) are defined as transcripts whose mapped 22G-RNAs exhibit over twofold enrichment from CSR-1 IP than that from input 22G-RNAs (Claycomb et al. 2009). To compare the WAGO-9 IP levels of wild type or CSR-1 depleted animals, the total miRNA mapped reads were used for normalization. The list of germline-expressed miRNAs was obtained from the previous report (Minogue et al. 2018) for prediction and identification of the mRNA targets of germline miRNAs.
Prediction of piRNA targeting sites
Predicted piRNA targeting sites (with stringent targeting rules) on C. elegans germline mRNAs were obtained from piRTarbase (Wu et al. 2019). For stringent rules, the following criteria are used; at seed region, no non-GU mismatches and no more than two GU mismatches are allowed. At the non-seed region, no more than two non-GU mismatches and no more than three GU mismatches are allowed. Furthermore, no more than six total mismatches are allowed from both seed and non-seed regions (Wu et al. 2018).
Measurements of miRNA, piRNA binding sites, and 22G-RNA levels at different regions of mRNAs
The C. elegans transcriptome data (WS275) annotation was used to define the location of 5′ UTRs, CDSs, and 3′ UTRs. The number of sites or read counts from each region were then divided by the nucleotide length of the corresponding regions for each mRNA to obtain the density. Where sites were spanning between two distinct regions, the sites or read counts were split according to the portion that mapped to each region. For measurements of sites/reads around start and stop codons, the 200 nt (±100 nt) window centered at the start or stop codon of each transcript was aligned, and the number of the sites/reads per transcript mapped at each nucleo-tide was calculated. As transcripts have different 5′ and 3′UTR lengths, the mapped sites/read counts at each position were divided by the number of transcripts that contain such position to obtain the sites/read counts per transcript. The following small RNA data were used to analyze the distribution and levels of CSR-1 and WAGO 22G-RNAs: CSR-1 associated small RNAs data (SRR12318132), WAGO-1 associated small RNAs (SRR8482951/WT, SRR8482949/prg-1 mutant) (Barucci et al. 2020), and WAGO-9 (HRDE-1) associated small RNAs (SRR12318140/WT, SRR12318144/CSR-1 depletion) (Singh et al. 2021).
We defined the highly or poorly CSR-1 targeted regions by identifying 100 nt windows in each germline mRNA transcript which contain the most or the least (nonzero) CSR-1 associated small RNAs (SRR12318132). We also required that these highly or poorly CSR-1 targeted regions are not overlapping with each other and exhibit at least a twofold difference in levels of CSR-1 associated small RNAs. The list of CSR-1 regions used in these analyses are provided in Supplemental Data File 2.
Metagene analyses
A custom script was used to divide mRNA transcripts into 100 bins and the normalized reads within each bin was calculated. The average number of sites or read counts at each bin per mRNA transcript was then calculated.
mRNA-seq analyses
The mRNA-seq data from wild-type C. elegans young adult samples (SRR6512642 and SRR6512643) (Shen et al. 2018) were used to define RPKM-standardized piRNA binding densities. Reads were mapped to the WS275 transcriptome with bowtie2. Normalized reads were calculated for each library by summing sense-mapping reads to each transcript using bedtools, standardizing by mRNA length to calculate reads per kilobase million (RPKM), then averaging the two libraries together.
Ribo-seq analyses
The ribo-seq data from the wild-type C. elegans late L4/young adult sample (36 h post hatching, SRR3356499) (Aeschimann et al. 2017) were used to identify the number of ribosome protected fragments (RPF) mapped to each CSR-1 target or WAGO target. The transcripts with the top 10% or bottom 10% amounts of RPF were defined as highly translated and poorly translated transcripts, respectively.
DATA DEPOSITION
All sequencing data analyzed in the manuscript are available at the NCBI GEO or ENA database. The SRR numbers of sequencing data used in specific analyses are provided in the Materials and Methods section above. Scripts to analyze reads within 5′ UTRs, CDSs, and 3′ UTRs, to analyze read accumulation proximal to start and stop codons, and to generate metagene distributions are available at https://github.com/RyanCCJ/RDT.
SUPPLEMENTAL MATERIAL
Supplemental material is available for this article.
ACKNOWLEDGMENTS
This work is supported in part by National Institutes of Health (NIH) predoctoral training grant T32 GM07197 to J.B.; the Ministry of Science of Technology of Taiwan (MOST 108-2628-E-006-004-MY3, MOST 110-2221-E-006-198-MY3, and MOST-111-2221-E-006-151-MY3) grants to W.-S.W., and NIH grant R01-GM132457 to H.-C.L.
Footnotes
-
Article is online at http://www.rnajournal.org/cgi/doi/10.1261/rna.079441.122.
- Received September 13, 2022.
- Accepted December 25, 2022.
This article is distributed exclusively by the RNA Society for the first 12 months after the full-issue publication date (see http://rnajournal.cshlp.org/site/misc/terms.xhtml). After 12 months, it is available under a Creative Commons License (Attribution-NonCommercial 4.0 International), as described at http://creativecommons.org/licenses/by-nc/4.0/.
REFERENCES
MEET THE FIRST AUTHORS


Meet the First Author(s) is an editorial feature within RNA, in which the first author(s) of research-based papers in each issue have the opportunity to introduce themselves and their work to readers of RNA and the RNA research community. Wei-Sheng Wu and Jordan Brown are co-first authors of this paper, “Transcriptome-wide analyses of piRNA binding sites suggest distinct mechanisms regulate piRNA binding and silencing in C. elegans.” Wei-Sheng contributed to this work as a professor in the Department of Electrical Engineering at National Cheng Kung University, Taiwan. Wei-Sheng is an expert in biological data analysis, web tool development, and database construction. Jordan contributed to this work as a PhD student in Heng-Chi Lee's laboratory in the Department of Molecular Genetics and Cell Biology at the University of Chicago.
What are the major results described in your paper and how do they impact this branch of the field?
We found that in C. elegans, piRNAs tend to target the CDS of mRNAs in vivo rather than the 5′ or 3′ UTRs as predicted by in silico targeting rules. These CDS targeting events also tend to more robustly trigger the production of secondary small RNAs. Surprisingly, the Argonaute protein CSR-1 that has been proposed to protect endogenous genes from piRNA silencing does not seem to contribute to this binding pattern. Our results demonstrate the existence of an additional and still uncharacterized level of regulation that enforces piRNAs’ binding preferences in vivo.
What led you to study RNA or this aspect of RNA science?
JSB: The diversity and abundance of noncoding RNAs has always been exciting to me. As the field of noncoding RNAs has matured, whole classes of highly conserved RNAs have been discovered and characterized. The idea that these important but often enigmatic molecules are performing essential functions in our cells, just waiting to be discovered, has fueled my interest in RNA science.
WSW: With advancements in next-generation sequencing (NGS), bioinformaticians play important roles in mining the information embedded in the raw reads. In collaboration with Dr. Heng-Chi Lee (the corresponding author of this paper), I started to analyze the NGS data of small noncoding RNAs in C. elegans. I am looking forward to solving more biological problems related to RNAs using computational approaches.
During the course of these experiments, were there any surprising results or particular difficulties that altered your thinking and subsequent focus?
The Argonaute protein CSR-1 is often thought of as protecting endogenous genes from piRNA targeting, and CSR-1 targeting tends to occur toward the 3′ end of mRNAs. Therefore, we hypothesized that CSR-1 targeting pushes piRNA binding events into the CDS. We were very surprised that CSR-1 protection is actually not sufficient to explain piRNAs’ CDS binding preference.
What are some of the landmark moments that provoked your interest in science or your development as a scientist?
JSB: I grew up in a small town that had a museum called the Discovery Center which was full of exhibits that demonstrated basic principles and natural phenomena like simple machines, atmospheric circulation, electricity, magnetism, and evolution. Most of these exhibits were diagrams or simulations that you could manipulate and then observe how the system changed; for example, how an Archimedes screw moves a ball or how electrical current flows through different materials. The basic hypothesis testing I could do there as a child first got me interested in science.
WSW: I was originally trained as an electrical engineer. During my PhD study, I changed my research field from control theory to computational systems biology. My career goal is to use control theory to not only understand how a cell operates but also to gain the ability to control a cell's behavior. Since RNA biology has emerged as one of the most influential fields in biological/biomedical research, I will continue studying the biological functions of all kinds of small noncoding RNAs using computational approaches.
What were the strongest aspects of your collaboration as co-first authors?
While Jordan is primarily a molecular biologist with bioinformatics expertise, Wei-Sheng is primarily a bioinformatician with molecular biology expertise. Coming from these different but complementary backgrounds, we often formulated questions and strategies using very different approaches. Those differences helped improve our work tremendously here, where we were able to often show that multiple independent analyses arrived at the same result.








