
Functional analysis of 3′-UTR hairpins supports a two-tiered model for posttranscriptional regulation of MAT2A by METTL16
- Corresponding author: Nicholas.conrad{at}utsouthwestern.edu
-
↵1 These authors contributed equally to this work.
Abstract
S-adenosylmethionine (SAM) is the methyl donor for nearly all cellular methylation events, so cells need to carefully control SAM levels. MAT2A encodes the only SAM synthetase expressed in the majority of human cells, and its 3′-UTR has six conserved regulatory hairpins (hp1–6) that can be methylated by the N6-methyladenosine methyltransferase METTL16. Hp1 begins 8 nt from the stop codon, whereas hp2–6 are clustered further downstream (∼800 nt). These hairpins have been proposed to regulate MAT2A mRNA levels in response to intracellular SAM levels by regulating intron detention of the last intron of MAT2A and by modulating the stability of the fully spliced mRNA. However, a dissection of these two posttranscriptional mechanisms has not been previously reported. Using a modular reporter system, we show that hp1 functions primarily when the detained intron is included in the reporter and when that intron has a suboptimal polypyrimidine tract. In contrast, the hp2–6 cluster modulates mRNA stability independent of the detained intron, although hp1 may make a minor contribution to the regulation of decay as well. Taken with previously published reports, these data support a two-tiered model for MAT2A posttranscriptional regulation by METTL16 through its interactions with hp1 and hp2–6. In the upstream tier, hp1 and METTL16 control MAT2A intron detention, whereas the second tier involves METTL16-dependent methylation of hp2–6 to control MAT2A mRNA stability. Thus, cells use a similar set of molecular factors to achieve considerable complexity in the posttranscriptional regulation of SAM homeostasis.
Keywords
INTRODUCTION
Nearly every cellular process is regulated by the methylation of biological macromolecules. S-adenosylmethionine (SAM) is the methyl donor for nearly all of these intracellular methylation reactions (Walsh et al. 2018). Cells therefore carefully control SAM homeostasis by several mechanisms including posttranscriptional regulation of the MAT2A gene, which encodes the only SAM synthetase expressed in most human cells. Previous studies suggest that cells regulate MAT2A mRNA abundance in response to intracellular SAM levels at the level of mRNA stability and by modulating the splicing efficiency of the last intron of MAT2A (Martínez-Chantar et al. 2003; Pendleton et al. 2017; Shima et al. 2017). In both cases, regulation is governed by six cis-acting hairpins (hp1–6) in the MAT2A 3′-UTR that are conserved in vertebrates (Fig. 1A). The N6-methyladenosine (m6A) methyltransferase METTL16 binds the hairpins and methylates them within the conserved sequence and structural motif in the hairpin loop (UACAGARAA; R = A/G; methylated A is underlined) (Pendleton et al. 2017; Shima et al. 2017; Warda et al. 2017; Doxtader et al. 2018). In addition to the structure and sequence conservation, the general organization of the hairpins is also conserved. That is, the UACAGAGA of the 5′-most hairpin (hp1) begins 141–145 nt from the 3′ splice site of the last intron, and a cluster of hairpins (hp2–6) resides further downstream (Fig. 1A). Hairpin-independent regulation of MAT2A by miRNAs has also been reported, but here we focus on the roles of the hairpins (Lo et al. 2013; Tomasi et al. 2017; Simile et al. 2019).
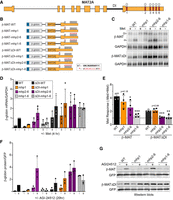
MAT2A hp1, but not hp2–6, requires the presence of the detained intron to regulate mRNA levels. (A) Full-length gene diagram of MAT2A, approximately to scale. Hp1–6 are denoted with red boxes, DI marks the regulated detained intron. (B) Diagrams of the eight β-MAT reporters used throughout this study (not to scale; Fl, flag-tag). The reporters are driven by a CMV immediate-early promoter and have a bovine growth hormone poly(A) signal. Asterisks show mutant hairpin sequences in the inset. (C) Representative northern blot data using RNA from HEK293 cells transfected with the indicated reporters and conditioned in media ±Met for 4 h. The mobilities of the detained intron transcript and mRNA are denoted with DI and m, respectively. (D) Quantification of the β-globin mRNA first normalized to GAPDH then expressed relative to the β-MAT-WT −Met signal, which was set to one for each replicate. (E) Quantification of the Met responsiveness of each reporter. The same data used in panel D were compared in −Met to +Met conditions for each construct. (F) Quantification of protein expression from each of the reporters after overnight incubation with the MAT2A inhibitor AGI-24512. The β-globin signal was first normalized to a cotransfected GFP control, and the β-MAT-WT values were set to one in each replicate (n = 3). The color scheme is the same as panel D above. (G) Representative western blots with anti-Flag antibodies to detect β-globin and anti-GFP which served as a cotransfected loading control. The same exposure is shown for both β-MAT and β-MAT-ΔDI blots. These experiments were performed using quantitative near-infrared fluorescence western blotting (LI-COR Odyssey). For panels D–F data are represented as mean ± standard deviation (SD). For panel E, the data were analyzed by a two-tailed, unpaired Student's t-test comparing each value to the matched wild-type control, β-MAT-WT (left) or β-MATΔDI-WT (right). Significance is annotated as (*) P < 0.05, (**) P < 0.01, or (***) P < 0.001.
Martínez-Chantar et al. (2003) first reported regulation of MAT2A mRNA stability by showing that methionine (Met) depletion increases the half-life of the mRNA. Importantly, they further demonstrated that the stabilization was not due to Met depletion per se, but rather due to the loss of SAM that rapidly occurs upon Met starvation. More recently, the hairpins were shown to control the SAM-responsiveness of a luciferase reporter system (Shima et al. 2017). In addition, the individual hairpins within the hp2–6 cluster displayed redundancy in that mutation of at least four of the five hairpins in the cluster was necessary for the abrogation of SAM-responsiveness. These data suggest that METTL16 methylates hp2–6 to target the mRNA for efficient degradation. This model mirrors the “reader–writer” paradigm used by the METTL3–METTL14 complex that is responsible for producing the vast majority of mRNA m6A methylation marks. In this case, the “writer” methylates mRNAs while the methylation mark targets the transcript for degradation through the activity of m6A-binding “reader” proteins (Shi et al. 2019; Zaccara et al. 2019; Sendinc and Shi 2023).
We previously proposed that splicing of the last intron of MAT2A is regulated by a mechanism involving SAM levels, METTL16, and hp1 (Pendleton et al. 2017, 2018). Our data suggested that when SAM levels are limiting, METTL16 binds hp1 but cannot methylate it due to lack of SAM. When bound, the METTL16 promotes splicing of the otherwise inefficiently spliced “detained” terminal intron. As a result, more mRNA and MAT2A protein are produced in response to low SAM. When SAM is abundant, methylation of hp1 is efficient, so the dwell time of METTL16 is too brief to induce splicing. The MAT2A detained-intron isoform is subsequently degraded in the nucleus and no new MAT2A mRNA is generated. Interestingly, the MAT2A homolog in Caenorhabditis elegans is regulated by its METTL16 homolog, METT-10, in response to SAM by a different mechanism (Mendel et al. 2021; Watabe et al. 2021). In worms, the 3′ splice site itself is methylated in high SAM conditions, which in turn inhibits the binding of splicing factors to the 3′ splice site. Thus, functions for METTL16 as a splicing regulator and SAM sensor have evolutionary roots beyond vertebrates.
Models proposing MAT2A regulation by mRNA stabilization or intron detention are not mutually exclusive. In fact, previous studies have hinted at separable functions between hp1 and the hp2–6 cluster, in pre-mRNA splicing and mRNA stability, respectively (Pendleton et al. 2017; Shima et al. 2017). Nonetheless, these functions have not been tested in parallel using the same reporters and assays. Here, we use a set of modular reporters and cell-based assays to genetically separate the regulation of intron detention and mRNA stability via hp1 and hp2–6, respectively. Taken alongside previously published reports, the data can be rationalized by a two-tiered model for MAT2A posttranscriptional regulation by METTL16. In the upstream tier, hp1 and METTL16 control MAT2A intron detention, and, in the downstream tier, METTL16 methylates hp2–6 to control MAT2A mRNA stability, but hp1 may also contribute to this activity. Thus, METTL16 has interrelated, but distinct, roles in MAT2A regulation that ensure proper control of SAM homeostasis.
RESULTS
Hp1 activity requires the MAT2A detained intron, but Hp2–6 activity does not
To first test the separation of functions of hp1 compared to hp2–6, we examined the effects of mutating either hp1 or hp2–6 in the context of β-globin reporters that fuse MAT2A exons 8 and 9 (β-MAT) and either contain or lack (ΔDI) the detained intron (Fig. 1B). Each mutated hp contains eight changes to the conserved UACAGARAA sequence and one in an invariant C nucleotide 3 bases downstream from the UACAGARAA that forms base pairs in the stem (Fig. 1B). We previously demonstrated that the β-MAT-WT reporter responds to SAM levels similarly to the endogenous RNA, and we validated that the mRNA and detained-intron isoforms are predominantly cytoplasmic and nuclear reflecting the endogenous transcripts (Bresson et al. 2015; Pendleton et al. 2017; Supplemental Fig. S1A). One day after transfection of these constructs into HEK293 cells, the media was changed to media containing or lacking Met for 4 h, cells were harvested, and RNA was analyzed by northern blot (Fig. 1C). While the absolute mRNA levels detected were variable between experiments, two trends were nonetheless apparent. First, the ΔDI constructs generally produced more mRNA than those containing the detained intron, particularly in uninduced (+Met) conditions (Fig. 1D; compare dotted to solid bars). Second, in the induced conditions (−Met), the lowest expressing reporters contained the detained intron and had a mutant hp1 (Fig. 1D; solid orange and gray). These data are consistent with the idea that the hp1 promotes splicing, an upstream step in mRNA biogenesis.
Distinct roles for hp1 and hp2–6 were further confirmed when we determined the Met responsiveness of each reporter by comparing its mRNA levels in ±Met conditions (Fig. 1E). As expected, mutation of all six hairpins negated Met responsiveness regardless of the presence of the detained intron (Fig. 1E, gray bars). When the detained intron was present (solid bars), there was a decrease in Met responsiveness when either hp1 or hp2–6 was mutated, even though the former did not reach statistical significance. In contrast, in the ΔDI reporters, mutation of hp2–6 resulted in a complete loss of Met responsiveness that was indistinguishable from mutation of all six hairpins (Fig. 1E, purple and gray dotted bars). Similarly, the ΔDI construct with all six wild-type hairpins was indistinguishable from the ΔDI with a mutant hp1 (Fig. 1E, black and orange dotted bars), and these were both approximately twofold more Met responsive than ΔDI with either mhp2–6 or mhp1–6 (Fig. 1E, black and orange dotted bars compared to the purple and gray dotted bars). These data demonstrate separable activities for hp1 and hp2–6. They further support the idea that hp1 activity is linked to the presence of the detained intron, while hp2–6 functions even in the absence of the detained intron.
Next, we tested the effects of hairpin mutations on reporter protein production (Fig. 1F,G). Because Met is essential for protein production, we used a MAT2A inhibitor, AGI-24512, to reduce intracellular SAM levels and thereby induce MAT2A expression (Kalev et al. 2021). Most importantly, the trends mirrored those observed with RNA (compare Fig. 1F with D). Protein accumulation in the hp1 mutants containing the detained intron was severely diminished (Fig. 1G, solid orange). Importantly, this loss of expression was also observed in the hp1–6 mutant (Fig. 1G, solid gray), even though hp2–6 were robustly induced (Fig. 1G, solid purple). Finally, we tested the AGI-24512 responsiveness among the reporters (Supplemental Fig. S1B). Although the comparisons were not statistically significant, β-MAT-WT had the largest dynamic range, supporting the idea that both components of the posttranscriptional regulation combine to yield a wide range of control of gene expression. Overall, these data show separable roles of hp1 and hp2–6 and further demonstrate that hp1 activity lies upstream of hp2–6 activity.
Improving MAT2A detained intron splicing negates the effects of Hp1 on mRNA accumulation
If hp1 primarily promotes MAT2A detained intron splicing, then increasing the efficiency of splicing should mimic the effects observed in the ΔDI reporters. To test this prediction, we first needed to determine which aspects of splicing are compromised in the MAT2A detained intron. Consensus splice sites contain a polypyrimidine tract (PPT) immediately upstream of the 3′ splice site that recruits the heterodimeric U2AF complex early in the splicing cycle (Wilkinson et al. 2019). The PPT in the MAT2A detained intron is interrupted by three purines (Fig. 2A, asterisks). Two of these purines are contained in a UGUA element that was previously implicated in regulation, so we focused on these first (Scarborough et al. 2021). As predicted, changing either of these purines to pyrimidines decreased intron detention (Fig. 2B), while mutating the pyrimidines to purines increased intron detention over time after Met depletion (Fig. 2C). Similar to the purine mutations in the UGUA, an A → U mutation at the −7 position relative to the downstream exon enhances splicing efficiency (Fig. 2A,D). Together, these data support the hypothesis that interruption of the PPT in MAT2A detained intron decreases splicing efficiency.
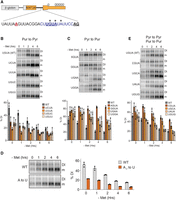
A weak PPT establishes inefficient splicing of MAT2A intron 8. (A) Diagram showing the 3′ splice site of the MAT2A detained intron. The PPT is in purple, the UGUA element is bold underlined, purines interrupting the PPT are marked with asterisks, the 3′ splice site AG is underlined in bold black, and the branch point A is underlined in bold red (Mercer et al. 2015). (B) Representative northern blots (top) and quantification (bottom) of the purine-to-pyrimidine mutations in the UGUA motif in the DI. The cells were transfected and the next day were cultured in Met-depleted media for the 0–6 h prior to RNA harvesting. The quantification is presented as percent detained intron. (C) Same as panel B except pyrimidine-to-purine mutations in the UGUA were tested. (D) Same as B, except the A at −7 position relative to the 3′ splice site was mutated to U. (E) Same as B except pyrimidine-to-pyrimidine and purine-to-purine mutations were tested. Samples in B and C are referenced to the same WT control data.
Consistent with our previous study, mutation of either U in the detained intron UGUA to C decreased splicing efficiency upon Met depletion (Fig. 2E, CGUA, UGCA). Because the CFIm complex component NUDT21 specifically binds UGUA sequences, and it promotes MAT2A splicing, we previously interpreted this result as evidence that this site contributes to CFIm promotion of MAT2A splicing (Scarborough et al. 2021). However, neither the G → A nor the A → G mutation affected splicing even though they also will abrogate CFIm binding (Fig. 2E, UAUA, UGUG; Supplemental Fig. S2B; Yang et al. 2010, 2011). Moreover, the sequence immediately upstream of the UGUA element in the detained intron is sufficient to reduce crosslinking of NUDT21 to an RNA substrate in vitro presumably by structurally blocking the consensus site (Supplemental Fig. S2). These observations are inconsistent with our previous suggestion that binding of CFIm to the detained intron promotes splicing. Instead, the interaction between CFIm and the 3′-UTR may be solely responsible for CFIm’s role in MAT2A splicing (Scarborough et al. 2021). The observed decreased splicing in both U → C mutations reflects a preference of U2AF to bind U over C-containing substrates (Singh et al. 1995; Sickmier et al. 2006; Jenkins et al. 2013). In any case, the data presented here strongly support the conclusion that the purine disruption of the PPT in the detained intron causes suboptimal splicing.
Next, we generated two A → U mutations in the PPT of the detained intron to improve splicing efficiency in the mutant hp1 and hp2–6 β-MAT reporters. We transfected these reporters and monitored β-globin mRNA and DI isoform levels ±Met by northern blot (Fig. 3A). As expected for the wild-type PPT controls, hp1 mutation caused robust accumulation of the DI isoform concomitant with strong decreases in mRNA production, and the hp2–6 mutation increased uninduced levels of mRNA (Fig. 3A–C, solid bars). More importantly, strengthening the PPT increased mRNA from the mutant hp1 reporter (approximately four- to ninefold; Fig. 3C, compare orange solid to striped bars), consistent with its suggested role in promoting the splicing of an otherwise weak intron. An approximately twofold increase in mRNA was observed from these reporters upon Met depletion due to the activity of the wild-type copies of hp2–6. Similarly, inclusion of the improved PPT in the hp2–6 mutants led to high levels of mRNA and no further induction in −Met media (Fig. 3C, purple striped bars), mirroring the observations in the ΔDI reporters (Fig. 1E). While splicing was considerably improved in the mhp1/MutPPT reporter compared to the mhp1/WtPPT reporter, low levels of intron detention were still observed that were not clearly visible in the mhp2–6/MutPPT reporters (Fig. 3A,B). This suggests that splicing of the intron remains somewhat compromised in the double PPT mutants due to the presence of a single G within the PPT, but that hp1 activity overcomes this splicing inefficiency. Overall, these results show that improving the basal splicing efficiency of the detained intron negates the effects of hp1 on mRNA accumulation. Taken with similar results from the ΔDI reporters (Fig. 1), these data strongly support the conclusion that hp1 promotes splicing of the detained intron, and that hp2–6 have a distinct function.
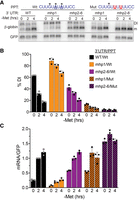
Strengthening the PPT bypasses the activity of hp1, but not hp2–6. (A) Representative northern blot of expression of β-MAT reporters with wild-type or mutant PPT constructs. The two A → U mutations are underlined; one G remains in the PPT, perhaps explaining the low levels of intron detention that persists in the mhp1 constructs. (B) Northern blot data were quantified as %DI. (C) Northern blot data were quantified to show relative mRNA levels. Each value was first normalized to signal from a cotransfected GFP loading control and expressed relative to the wild-type PPT sample after 2 h in −Met media. Bar graphs in B and C show mean values ±SD (n = 3).
Hairpins also regulate mRNA stability
The results above show that hp1 has separable functions from hp2–6. Previous reports demonstrated that hp2–6 regulate protein production from a luciferase reporter lacking the detained intron (Shima et al. 2017). To further validate this observation, we examined the decay rates of the mRNAs produced from our ΔDI reporters. Approximately 18 h after transfection, we added media that contained or lacked Met, and cells were grown for an additional 4 h prior to the addition of Actinomycin D (ActD) to inhibit transcription. Northern analysis of RNA collected at various time points after ActD addition shows that both the wild-type and hp1 mutant reporters have significantly shorter half-lives in Met-replete media compared to Met-lacking media (t1/2 ∼ 2 h versus ∼4 h; Fig. 4A,B black and orange). However, mutation of hp2–6 stabilizes the transcripts regardless of Met levels (t1/2 ≥ 4 h; Fig. 4A,B, purple and gray). Taken with previous studies, these results strongly suggest that the stability of the MAT2A spliced mRNA is regulated by hp2–6.
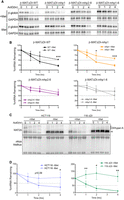
Hp2–6 control stability of the spliced mRNA. (A) Representative northern blots using RNA from cells transfected with the indicated β-MAT reporters after ActD treatment for the indicated times. Cells were conditioned in the presence (top) or absence (bottom) of Met for 4 h then kept in the same media for an additional 1, 2, or 4 h after ActD. GAPDH half-life is significantly greater than 4 h, so it was used as a loading control. (B) Quantification of decay experiments. β-globin signal was first normalized to GAPDH, and samples were quantified relative to the t = 0 sample which was set to 100. Each data point is mean value ± SD (n = 3). (C) Representative northern blots of a decay assay with RNA from HCT116 cells or 116-ΔDI. The latter are HCT116-derived cells that lack the MAT2A detained intron at the endogenous locus. Cells were conditioned in the presence (top) or absence of Met (bottom) for 4 h then kept in the same media for an additional 0, 1, 2, or 4 h with ActD. Methylene blue staining of the membrane serves as a loading control (bottom). The bands with decreased mobility over time in ActD are nuclear RNAs undergoing nuclear hyperadenylation as previously reported (Bresson et al. 2015). (D) Quantification of the decay experiments as in panel C (n = 3). For panels B and D, the data were analyzed by a two-tailed, unpaired Student's t-test comparing each time point ±Met. Significance is annotated as (*) P < 0.05, (**) P < 0.01, or (***) P < 0.001.
To test the stabilization in a more endogenous context, we used a previously described cell line in which the MAT2A detained intron is deleted at the genomic locus of HCT116 cells (116-ΔDI) (Scarborough et al. 2021). In both HCT116 and 116-ΔDI cells, MAT2A mRNA was relatively unstable in Met-replete conditions (t1/2 ∼ 1.5 h) and significantly stabilized upon Met depletion (Fig. 4C,D). Thus, the presence of the detained intron is not necessary to stabilize MAT2A mRNA. Admittedly, these experiments do not directly test hp2–6 function for endogenous MAT2A. However, they lend further support to the existence of separable functions of MAT2A intron detention and mRNA stabilization that combine to regulate MAT2A mRNA levels in response to intracellular SAM concentration.
METTL16 is required for Hp2–6-mediated mRNA stability
Previous reports and the data above suggest that METTL16 methylates the MAT2A hairpins in high SAM conditions to destabilize MAT2A (Shima et al. 2017; Zaccara and Jaffrey 2020). To test this directly, we knocked down METTL16 in cells transfected with the reporters lacking the detained intron and examined their stability in Met-replete media. While the nontargeting siRNAs (siNT) had no effect on reporter stability (t1/2 ∼ 2 h), METTL16 depletion (siM16) significantly increased the stability of both the WT and mutant hp1 mRNAs (t1/2 ∼ 3.5–4 h) (Fig. 5A–C). The increase was not as robust as that observed in hp2–6 mutants or upon Met depletion (Figs. 4B, 5B), but this may be due to residual activity of METTL16 after knockdown (Fig. 5A). Importantly, METTL16 knockdown had no effect on the stability of reporter mRNAs with mutant hairpins (Fig. 5C, bottom right, gray). Interestingly, we observed a slight, but statistically significant increase in the stability of the mhp2–6 mRNA. In fact, a similar trend was observed upon Met depletion (Fig. 4B), but those data do not rise to the level of statistical significance. Thus, in addition to its role in splicing, hp1 may make a minor contribution to the regulation of the stability MAT2A mRNA. These data further support the idea that METTL16 methylates MAT2A hairpins to decrease the stability of the mRNA.
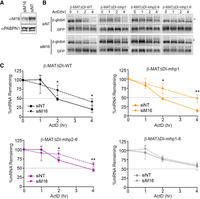
METTL16 destabilizes MAT2A mRNA. (A) Western blot showing knockdown of METTL16 upon treatment with METTL16 siRNAs (siM16) compared to nontargeting control siRNA (siNT). PABPN1 serves as a loading control. (B) Representative northern blots with RNA from cells transfected with the indicated siRNA and the indicated reporters. GFP signal serves as a control for transfection and loading. The asterisk denotes hyperadenylated transcripts as described in Figure 4C. (C) Quantification of the decay experiments as represented in panel B (n = 4). The data were analyzed by a two-tailed, unpaired Student's t-test comparing siNT to siM16 for each time point ([*] P < 0.05, [**] P < 0.01).
DISCUSSION
Taken with previously published work, these findings support a two-tiered model of MAT2A regulation by METTL16 in response to intracellular SAM levels (Fig. 6). In the upstream tier of the regulation (Fig. 6, light gray), the presence of METTL16 on hp1 modulates splicing of the last intron of MAT2A. When SAM levels are low (Fig. 6, left), METTL16 binding to hp1 enhances splicing by a mechanism that remains incompletely understood. However, the mechanism likely involves CFIm-mediated recruitment of U2AF to the weak PPT in the detained intron (see below). We previously proposed that the SAM dependence of the splicing mechanism stems from the increased dwell time of METTL16 on hp1 in low SAM conditions, but this remains to be formally demonstrated. When SAM levels are high, METTL16 efficiently methylates hp1 and the faster turnover of METTL16 on hp1 is insufficient to drive splicing (Fig. 6, right). In the second tier of regulation, high SAM leads to efficient methylation of hp2–6 to destabilize the MAT2A mRNA, mirroring a typical reader–writer mechanism of m6A regulation (Fig. 6, dark gray). Conversely, low SAM leads to lack of hp2–6 methylation and a stable transcript. We refer to this as a two-tiered mechanism because splicing and decay occur at different stages of the mRNA life cycle. However, METTL16 acts cotranscriptionally in splicing (Pendleton et al. 2018), so the regulatory steps are established by METTL16 binding or methylation early in mRNA biogenesis.

Two-tiered model for MAT2A regulation by METTL16 and hp1–6. The model is described in the Discussion. Tier 1 (splicing) and Tier 2 (mRNA stability) are shaded in light and dark gray, respectively. The decay step is likely cytoplasmic, but this has not been fully verified, nor has a reader been fully characterized. For simplicity, the diagram depicts posttranscriptional regulation of the splicing step, but the splicing “choice” and presumably METTL16 methylation of the hairpins occur cotranscriptionally (Pendleton et al. 2018).
The two-tiered model is consistent with nearly all published data on the regulation of MAT2A by METTL16 and hp1–6. For example, the stability data presented here closely mirror those of Shima et al. (2017) in which they demonstrated that mutation of hp1 alone had little effect on a luciferase reporter lacking the detained intron. They additionally showed that mutation of individual hairpins within hp2–6 had an additive effect on expression of that reporter supporting the idea that each individual hairpin within the hp2–6 cluster has redundant activity. Indeed, their model is essentially identical to the second tier of regulation shown in Figure 6. Importantly, if the destabilization of MAT2A mRNA by METTL16 methylation was the only source of regulation, knockdown of METTL16 would be predicted to phenocopy SAM depletion and up-regulate MAT2A. However, knockdown or knockout of METTL16 leads to reductions in basal MAT2A levels and/or loss of its induction in low SAM conditions (Pendleton et al. 2017; Shima et al. 2017; Mendel et al. 2018). The two-tiered model nicely rationalizes these observations because METTL16 knockdown will reduce MAT2A mRNA production by decreasing the efficiency of pre-mRNA splicing. To our knowledge, the only published data that cannot be rationalized with this model are those from Zhang et al. (2022) who reported that MAT2A mRNA was stabilized upon methylation by METTL16. Whether that is due to a unique feature of the hippocampus as suggested by the authors remains undetermined. Of course, the two-tiered model strictly focuses on METTL16's function regulating MAT2A and does not address any of METTL16's other functions in MALAT1 binding, translation, or U6 snRNA methylation (Brown et al. 2016; Warda et al. 2017; Nance et al. 2020; Su et al. 2022; Zeng et al. 2022; Wang et al. 2023).
Both METTL16 and SAM levels have been linked to specific cancer types (Kryukov et al. 2016; Marjon et al. 2016; Mavrakis et al. 2016; Maldonado et al. 2018; Liu et al. 2019; Hou et al. 2020; Li et al. 2020; Wang et al. 2020, 2021; Kalev et al. 2021; Su et al. 2022; Zeng et al. 2022; Han et al. 2023). Since METTL16 is an enzyme, it is reasonable to suggest that METTL16 could be targeted to therapeutically modulate intracellular SAM levels. The two-tiered regulation of MAT2A by METTL16 requires that the specific mechanism of drug action be taken into consideration. For example, any drug that inhibits METTL16 catalysis, but does not alter hairpin binding, will elevate SAM levels by simultaneously enhancing splicing by hp1 binding and by stabilizing the mRNA due to lack of hp2–6 methylation. In contrast, any drug that inhibits both catalysis and hairpin binding could lower MAT2A and SAM levels since it would lead to less efficient splicing and reduced mRNA levels. In this case, it is not clear whether the increase in mRNA stability due to the lack of hp2–6 methylation would be able to compensate for the lower MAT2A mRNA levels produced by inefficient splicing. In addition, catalytic METTL16 inhibitors will inhibit U6 snRNA methylation. While the role of U6 snRNA methylation in mammals is not well characterized, it is reasonable that inhibition of U6 snRNA methylation will be too toxic for therapeutics (Ishigami et al. 2021; Satterwhite and Mansfield 2022). One can also imagine that small molecules could increase METTL16 catalytic activity by inhibiting the autoregulatory K-loop domain that lowers METTL16's SAM affinity (Doxtader et al. 2018). Such compounds would be predicted to simultaneously decrease splicing and MAT2A stability thereby lowering the SAM setpoint in cells. In any case, additional mechanistic studies need to be performed to ascertain whether manipulating METTL16 has therapeutic benefits.
The mechanisms by which METTL16 regulates MAT2A splicing through hp1 and mRNA stability through hp2–6 remain incompletely understood. The work here sheds some light onto the mechanism of hp1-induced MAT2A splicing. First, we show that the inefficient splicing is due to a weak PPT, strongly suggesting that induction of splicing requires enhanced binding of the PPT-binding complex U2AF (Fig. 6, green). Indeed, U2AF can be recruited by CFIm, which we previously showed to be essential for MAT2A splicing induction (Millevoi et al. 2006; Scarborough et al. 2021). Thus, METTL16 binding to hp1 may directly enhance CFIm splicing activity, either by promoting more efficient CFIm binding or CFIm recruitment of U2AF. However, our previous data suggested two CFIm binding sites to be essential, one in the detained intron and one in the 3′-UTR immediately upstream of hp1. The work here excludes the detained intron UGUA as a CFIm binding site (Fig. 2; Supplemental Fig. S2). Nonetheless, the 3′ splice site analysis here lends experimental support to our previous suggestion that CFIm binding promotes splicing by assisting recruitment of U2AF to a weak PPT. To fully understand the coordination of METTL16 with SAM and MAT2A splicing, the precise molecular connections between METTL16, CFIm, and U2AF need to be elucidated.
Even though it was first described 20 yr ago, the mechanism by which SAM levels regulate MAT2A mRNA stability remains incompletely understood (Martínez-Chantar et al. 2003). The most straightforward model proposes a reader–writer paradigm in which METTL16 adds methylation marks in high SAM conditions that are read by cytoplasmic readers to promote decay. While the reader has not been identified, the cytoplasmic YTHDF family of proteins (YTHDF1, YTHDF2, and YTHDF3) bind MAT2A hairpins (Patil et al. 2018). Moreover, YTHDF proteins redundantly function to destabilize m6A-containing RNAs, so they are strong candidates to be the MAT2A m6A reader (Zaccara and Jaffrey 2020). YTHDC1 was proposed to be the reader for MAT2A methylation marks based on reporter assays, but it had no effect on endogenous MAT2A (Shima et al. 2017), and it does not crosslink to the MAT2A 3′-UTR (Patil et al. 2018). While the localization of the decay pathway regulated by hp2–6 has not been determined, it seems likely that it occurs in the cytoplasm since it targets the fully spliced mRNA. Because YTHDC1 is nuclear, it seems unlikely that YTHDC1 is the primary factor stabilizing the RNA (Widagdo et al. 2021). Analogous to its role in splicing, METTL16 could bind the hairpins in low SAM conditions and promote RNA stability, but our data showing that METTL16 is not required for stabilization do not support this idea (Fig. 5). Thus, while it's clear that cells control MAT2A mRNA at the level of stability and splicing, significant gaps remain in understanding the mechanisms driving its regulation.
MATERIALS AND METHODS
Cell culture
HCT116 and HEK293 cells were obtained from ATCC, and the HCT116 derivative 116-ΔDI was previously described (Scarborough et al. 2021). For siRNA knockdown experiments (Fig. 5), we used 293A-TOA cells (Sahin et al. 2010) because we routinely observe more robust knockdown in this line than in standard HEK293 cells. HEK293, HCT116, and 116-ΔDI cells were grown in Dulbecco's modified Eagle medium (DMEM; Sigma D5796) with 10% fetal bovine serum (FBS; Sigma F0926) supplemented with 1× penicillin–streptomycin (Sigma P0781) and 2 mM l-glutamine (Fisher). 293A-TOA were grown in the same with Tet-free FBS (R&D Systems S10350). Cells were grown at 37°C with 5% CO2. For Met-depletion studies, we used Met-free DMEM (Sigma D0422) supplemented with 0.4 mM cysteine and 1 mM sodium pyruvate in addition to 10% FBS, 1× penicillin–streptomycin and 2 mM l-glutamine. For most experiments involving Met depletion, the cells were grown in media supplemented with an additional 200 µM Met.
Plasmid transfections and RNA harvesting
DNA transfections were performed with TransIT-293 (Mirus). Transfections were performed in a 12-well plate with ∼70% confluent cells and 2 µL TransIT-293 reagent per well. A typical reporter assay was performed with 30–100 ng of DNA reporter (higher levels can saturate cellular responses). Where noted, 50 ng of a GFP expressing plasmid was cotransfected as a loading control. The empty vector, pcDNA3, was added to yield a total of 0.7 µg of DNA per 12-well plate. The day after transfection, cells were treated as indicated and RNA was extracted by TRI Reagent (Molecular Research Center, TR118) using the manufacturer's protocol. We note that throughout these studies, biological replicates are defined as experiments using cells that were split, and therefore transfected, on independent days.
Northern blots
Northern blots were performed as previously described (Ruiz et al. 2019). Briefly, 1.0%–1.4% agarose formaldehyde gels were run with ∼3 µg of total RNA. The RNA was transferred overnight to Hybond N+ Nylon membranes (GE Healthcare) by capillary transfer in 20× saline-sodium citrate buffer (SSC; 3 M NaCl, 0.3 M sodium citrate, pH 7.0). After crosslinking with a SpectroLinker XL-1500 (120 mJ/cm2), the membrane was prehybridized in Church's buffer (200 mM sodium phosphate [pH 7.2], 15% deionized formamide, 1 mM EDTA, 1% filtered bovine serum albumin, 7% sodium dodecyl sulfate) for 1 h at 65°C. Probes were generated by in vitro transcription using SP6 or T7 polymerase and templates of either digested plasmid [β-globin; βΔ1,2(B-A); (Conrad et al. 2006)] or a PCR product (GAPDH, GFP), respectively (Supplemental Table S1). Transcription reactions contained ∼200 ng of template, 25 µCi of α-32P-UTP (800 Ci/mmol), 0.5 mM ATP, 0.5 mM CTP, 0.5 mM GTP, and 0.1 mM UTP in 1× transcription buffer (40 mM Tris pH 7.5; 6 mM MgCl2; 10 mM DTT), and 20 U of RNAsin Plus (Promega) and were carried out for 1 h at 37°C. Unincorporated nucleotides were removed using a G-50 spin column (Cytiva 27-5330-01); the probe was heated to 95°C for 5 min and then added to the prehybridization mix. After overnight incubation at 65°C, the blots were washed twice in 2× SSC/0.1% SDS for 15°C at room temperature, then twice more in 0.1× SSC/0.1% SDS at 65°C. Bands were detected and quantified using a Typhoon FLA9500 phosphorimager (Cytiva Amersham) and quantified with ImageQuant (v 8.1).
Nucleocytoplasmic fractionation
Fractionation was performed as described in Bresson et al. (2015) with a few modifications. After harvesting HEK293 cells from an ∼70% to 80% confluent six-well plate, they were resuspended in 100 µL Buffer I (0.32 M sucrose, 3 mM CaCl2, 2 mM MgCl2, 0.1 mM EDTA, 10 mM Tris-HCl [pH 8.0], 1 mM DTT, 0.04 U/µL RNase Inhibitor) supplemented with 150 µg/mL digitonin and incubated on ice for 5 min. After centrifugation at 500g for 5 min at 4°C, the supernatant was added to 1 mL TRI Reagent; this is the cytoplasm fraction. The pellets were twice resuspended in Buffer I supplemented with 150 mM NaCl and 0.5% Triton X-100 and then centrifuged at 500g. While some RNA is lost through these wash steps, they were necessary to generate fractions free of cross contamination. The resulting pellet was resuspended and 1 mL of TRI Reagent was added; this is the nuclear fraction.
siRNA depletion and RNA decay assays
293A-TOA cells were grown in six-well plates to ∼70% confluency and were transfected with 30 nM METTL16 siRNA (Thermo Fisher s35507) using RNAiMax (Thermo Fisher). The next day, the confluent cells were split into 12-well plates. After an additional 2 d, the cells were transfected with 100 ng of reporter plasmids and 100 ng of pcEGFP-stop control plasmid. The following day (4 d after siRNA transfection), fresh media was added containing 1 µg/mL actinomycin D (ActD) (Sigma A9415). RNA was harvested after 0, 1, 2, and 4 h of ActD treatment. For RNA decay assays without siRNA knockdowns, ActD was added the day following reporter DNA transfection.
Plasmids
The β-MAT-WT reporter and the derivatives β-MAT-mhp1, β-MAT-mhp2–6, β-MAT-mhp1–6 were previously described (Pendleton et al. 2017). To make the ΔDI versions, cDNAs were made from cells transfected with each of these plasmids followed by PCR with NC1145 and SP6+ (Supplemental Table S1 lists all primer sequences). The resulting amplicons were digested with EcoRI and XhoI and inserted into gel-purified β-MAT-WT vector cut with the same enzymes. Plasmid pcEGFP-stop (pNC1254) was made by annealing the oligos NC3315 and NC3316 followed by ligation into pcEGFP (pNC1286) digested with HindIII and BamHI (Scarborough et al. 2021); the insert added a stop codon to GFP in the parental plasmid.
Mutagenesis of the PPT in the detained intron was performed by three-fragment Gibson assembly as previously described for β-MAT-DI-UGCA and β-MAT-DI-CGUA (Scarborough et al. 2021). The three fragments consisted of gel-purified XhoI and EcoRI digested β-MAT-WT plasmid, an upstream fragment made by PCR with forward primer NC3339 and a mutation-specific reverse primer, and a downstream fragment made with NC3346 and a mutation-specific forward primer listed in Supplemental Table S1.
In vitro UV crosslinking (label transfer assays)
Crosslinking experiments were performed exactly as described previously except the SUMO-NUDT21 was not treated with SUMO protease Ulp1 in any experiments (Scarborough et al. 2021). RNA substrates were purchased from Sigma (Supplemental Table S1).
SUPPLEMENTAL MATERIAL
Supplemental material is available for this article.
ACKNOWLEDGMENTS
The authors thank Drs. Ashwin Govindan and Minseon Kim for critical review of this manuscript. This work was supported by the National Institutes of Health (NIH) grants R01GM127311 and R01AI123165.
Footnotes
-
Article is online at http://www.rnajournal.org/cgi/doi/10.1261/rna.079695.123.
- Received April 24, 2023.
- Accepted July 29, 2023.
This article is distributed exclusively by the RNA Society for the first 12 months after the full-issue publication date (see http://rnajournal.cshlp.org/site/misc/terms.xhtml). After 12 months, it is available under a Creative Commons License (Attribution-NonCommercial 4.0 International), as described at http://creativecommons.org/licenses/by-nc/4.0/.
REFERENCES
MEET THE FIRST AUTHOR

Meet the First Author(s) is an editorial feature within RNA, in which the first author(s) of research-based papers in each issue have the opportunity to introduce themselves and their work to readers of RNA and the RNA research community. Juliana N. Flaherty is the joint first author of this paper, “Functional analysis of 3′-UTR hairpins supports a two-tiered model for posttranscriptional regulation of MAT2A by METTL16.” She is a PhD candidate in the Biological Chemistry program at the University of Texas Southwestern Medical Center, where she studies the function of the m6A methyltransferase METTL16 in Nicholas Conrad's lab in the Department of Microbiology.
What are the major results described in your paper and how do they impact this branch of the field?
This paper delineates two distinct mechanisms of MAT2A regulation. We provide a thorough investigation of the roles of the six regulatory hairpins in the MAT2A 3′-UTR on both splicing and mRNA decay. This is impactful for those studying MAT2A and SAM homeostasis because prior to this publication, one could interpret splicing and decay to be competing models of MAT2A regulation. However, this paper directly tests and presents plenty of evidence to support the two-tiered model of MAT2A regulation. Not only will this clarify the mechanisms involved in this complex SAM-sensing feedback loop, but it will also guide future mechanistic studies by outlining a way to genetically separate these distinct mechanisms. Furthermore, it underlines the biological importance of MAT2A, as we have evolved multiple mechanisms to regulate it.
What led you to study RNA or this aspect of RNA science?
As a child, I'm sure I nearly drove my parents crazy asking them so many questions about the way the world works. My inquisitive nature followed me into my science classes as I got older, and I began asking questions about the way humans worked too. We are such dynamic and complex systems, how do we stay balanced, and why do our systems fail sometimes? It was in my high school classrooms where I was first introduced to the idea of gene regulation. I remember learning about the term “junk DNA” and my teachers explaining that scientists are beginning to understand that perhaps these noncoding regions are not junk at all. During my undergraduate career, I got some exposure to research in epigenetics and ribosome specialization. This allowed me to dip my toes into gene regulation at different stages of the central dogma, but I was missing one part—RNA! Upon entering graduate school, I was curious how noncoding RNA, alternative splicing, and RNA modifications could also fit into these layers of gene regulation. After joining the Conrad Lab, which focuses on posttranscriptional gene regulation, I was hooked.
During the course of these experiments, were there any surprising results or particular difficulties that altered your thinking and subsequent focus?
When thinking about the role of the hairpins in the two tiers of splicing and decay, it was easy for us to have a binary way of thinking. Hairpin 1 became synonymous with splicing and hairpins 2–6 became synonymous with decay. Although it is true that hairpins 2–6 have no apparent effect on splicing, the inverse is not as simple. We were surprised that when hp2–6 are mutated and the detained intron is deleted, methylation of hp1 alone still has a small but significant effect on decay. While splicing and decay are still distinct mechanisms, hairpin 1 may contribute to both. This makes our model a bit less simple, but science is rarely neat and tidy.
Are there specific individuals or groups who have influenced your philosophy or approach to science?
My high school science teachers Elizabeth Lopez, Frankie Tate, Sheila Jurik, and Tom Shefler were fundamental in developing my love of science. Furthermore, my graduate school mentor Nicholas Conrad has had an immense influence on my approach to science. He has taught me how to conduct robust, hypothesis-driven research and how to consider the nuances of science.
How did you decide to work together as co-first authors?
The decision to work together as co-first authors happened very organically. While students and postdoctoral fellows in the Conrad Lab each have their own project, this project was unique in that there was no one person with obvious “ownership.” We all wanted to pitch in to get this story out there. Olga Hunter performed the fundamental experiments using the β-globin reporters that formed the foundation for this project, Julio Ruiz worked to uncover details of the mechanism of MAT2A intron detention regulation, and I assessed the effect of METTL16 on the stability of MAT2A mRNA and aided Nicholas Conrad in editing the manuscript. This project was truly a group effort.








