
Small-molecule Ro-08-2750 interacts with many RNA-binding proteins and elicits MUSASHI2-independent phenotypes
- Kathryn Walters1,2,
- Marcin Piotr Sajek1,2,3,
- Elisabeth Murphy1,2,
- Aaron Issaian1,
- Amber Baldwin1,2,
- Evan Harrison1,2,
- Miles Daniels1,2,4,
- Julie A. Reisz1,
- Kirk Hansen1,
- Angelo D'Alessandro1 and
- Neelanjan Mukherjee1,2
- 1Department of Biochemistry and Molecular Genetics, University of Colorado School of Medicine, Aurora, Colorado 80045, USA
- 2RNA Bioscience Initiative, University of Colorado School of Medicine, Aurora, Colorado 80045, USA
- 3Institute of Human Genetics, Polish Academy of Sciences, 60-479 Poznan, Poland
- 4Howard University Karsh STEM Scholars Program, Washington DC 20059, USA
- Corresponding author: neelanjan.mukherjee{at}cuanschutz.edu
Abstract
RNA-binding proteins (RBPs) are key regulators of gene expression. Small molecules targeting these RBP–RNA interactions are a rapidly emerging class of therapeutics for treating a variety of diseases. Ro-08-2750 (Ro) is a small molecule identified as a competitive inhibitor of Musashi (MSI)–RNA interactions. Here, we show that multiple Ro-dependent cellular phenotypes, specifically adrenocortical steroid production and cell viability, are Musashi-2 (MSI2)-independent. Using an unbiased proteome-wide approach, we discovered Ro broadly interacts with RBPs, many containing RRM domains. To confirm this finding, we leveraged the large-scale ENCODE data to identify a subset of RBPs whose depletion phenocopies Ro inhibition, indicating Ro is a promiscuous inhibitor of multiple RBPs. Consistent with broad disruption of ribonucleoprotein complexes, Ro treatment leads to stress granule formation. This strategy represents a generalizable framework for validating the specificity and identifying targets of RBP inhibitors in a cellular context.
Keywords
INTRODUCTION
RNA-binding proteins (RBPs) contain RNA-binding domains (RBDs) that bind to specific regulatory elements in mRNAs to control splicing, export, decay, translation, and localization (Keene 2007). These regulatory interactions are crucial for normal physiology and are dysregulated in many human diseases ranging from neurological disorders to cancer (Gebauer et al. 2021). Antisense oligonucleotides (ASOs), such as Nusinersen, which is used to treat spinal muscular atrophy (Bennett et al. 2019), block RBD–RNA regulatory interactions by hybridizing with the RNA in a sequence-specific manner (Singh et al. 2006; Hua et al. 2008). While RBP–RNA interfaces were not historically targeted for small-molecule inhibition, they have recently emerged as a novel space for small-molecule development (Julio and Backus 2021). These small molecules must recognize RBP–RNA interfaces to compete with or prevent RNA binding. Because RBP surfaces are relatively flat and polar, finding a specific ligand can be challenging (Gowthaman et al. 2013). Many small-molecule inhibitors targeting kinases have been shown to exert their cellular effect via off-targets (Klaeger et al. 2017; Lin et al. 2019). ASO off-targets are largely sequence-specific and can be assessed in RNA-seq experiments (Scharner et al. 2020). However, determining functional off-target interactions of RBP small-molecule inhibitors remains challenging.
Substantial effort has been made to identify inhibitors of the Musashi (MSI) family of RBPs (Kudinov et al. 2017; Wu 2020). In mammals, the MSI family of RBPs contains two homologous members: Musashi-1 (MSI1) and Musashi-2 (MSI2). MSI2 is critical for the development and maintenance of embryonic stem cells, hematopoietic stem cells, and neural stem and progenitor cells (Fox et al. 2015). It is also important for fertility in steroid-producing mouse gonadal tissue, which is consistent with our previous findings that MSI2 promotes aldosterone production in human adrenocortical cells (Sutherland et al. 2014, 2015; Fu et al. 2021). MSI2 is a prognostic marker associated with clinical outcomes in several types of cancer including myeloid leukemia, breast cancer, and ovarian cancer among others (Ito et al. 2010; Kharas et al. 2010; Li et al. 2015; Lee et al. 2016; Kang et al. 2017). There are several small molecules that have been reported to be competitive inhibitors of MSI2–RNA binding (Lan et al. 2018; Minuesa et al. 2019; Wang et al. 2020; Zhang et al. 2021). In addition, some ω-9 monounsaturated fatty acids are allosteric inhibitors of MSI2–RNA binding (Clingman et al. 2014). A large chemical screen identified the small-molecule Ro-08-2750 (Ro) as a competitive inhibitor of MSI–RNA interactions (Minuesa et al. 2014). Further structural and biochemical characterization concluded that it binds to the first RNA recognition motif (RRM) domain of MSI2 to displace target mRNAs (Minuesa et al. 2019). Ro treatment reduced disease burden in a murine AML leukemia model, inhibited replication of SARS-CoV-2, and improved muscle function in myotonic dystrophy type 1 (Minuesa et al. 2019; Lim et al. 2021; Sabater-Arcis et al. 2021). While it is clear that Ro binds to MSI2 in vitro, the specificity of the Ro–MSI2 interaction has not been investigated in cells. Furthermore, it is unclear that MSI2 is required for these physiological effects exerted by Ro.
In this work, we investigate the possibility that Ro does not specifically target MSI2 and exerts functional effects independent of MSI2. We show that Ro is a potent inhibitor of human adrenocortical steroidogenesis. However, inhibition of steroidogenesis and other Ro-dependent phenotypes were not rescued by a previously characterized Ro-resistant MSI2 (Minuesa et al. 2019). We used a global approach to experimentally identify proteins that interact with Ro. Specifically, we found a subset of RBPs, many with RRM domains, that both interact with and phenocopy the transcriptomic effects of Ro. Our findings indicate that Ro is not an inhibitor of MSI2, but rather it promiscuously binds and may inhibit multiple RBPs. Finally, this work represents a generalizable framework to assess the protein interactions and functional specificity of small-molecule inhibitors targeting RBPs.
RESULTS
Ro inhibits steroidogenesis in a dose-dependent manner
Angiotensin II (AngII) stimulation of immortalized adrenocortical H295R cells is a robust model for human steroidogenesis (Parmar et al. 2008; Wellman et al. 2021). We previously found that siRNA depletion of MSI2 in AngII-stimulated H295R cells led to decreased aldosterone production (Fu et al. 2021), indicating that MSI2 promotes steroidogenesis. Therefore, we hypothesized that Ro-mediated inhibition of MSI2 would also reduce aldosterone production. Ro inhibited aldosterone production in a dose-dependent manner with an IC50 of 1.50 µM ± 0.154 µM (Fig. 1A). At higher concentrations, Ro caused a decrease in cell viability and proliferation (Fig. 1B,C, respectively). Furthermore, Ro also inhibited cortisol production with an IC50 of 0.682 µM ± 0.056 µM (Supplemental Fig. 1A). These data demonstrated that Ro broadly inhibits steroidogenesis and cell viability albeit at higher concentrations.
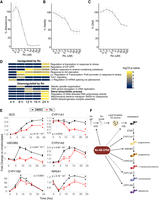
Ro inhibits steroidogenesis. Dose-dependent effects of Ro on the percent of aldosterone (A), cell viability (PrestoBlue) (B), and DNA content (FluoReporter) (C) relative to DMSO in H295R cells stimulated with 10 nM Ang II. Error bars indicate SD from up to three independent rounds of experiments, each containing six independent replicates. (D) Heatmap representing selected enriched BP ontology terms in genes differentially expressed after Ro treatment during AngII stimulation time course (see Supplemental Fig. 1C,D for full results). (E) Fold change and P-values of mRNA levels compared with unstimulated controls for DMSO (black) or 5 µM Ro (red) treated H295R cells at multiple times post-AngII stimulation (10 nM). The likelihood ratio test was used to calculate P-values. (F) Model depicting the regulation of genes involved in key steps steroidogenesis by Ro. Solid lines show genes significantly down-regulated upon Ro treatment.
Ro coordinately down-regulates multiple steps of steroidogenic gene expression
To investigate Ro-mediated inhibition of steroidogenesis, we performed an AngII stimulation RNA-seq time course (six time points in duplicate, 5 µM Ro) in H295R cells treated with either Ro or vehicle (DMSO) and detected 1206 genes with significant expression differences (Supplemental Table 1). In addition to time course analysis, we analyzed gene expression changes at each time point by pairwise comparison between Ro-treated and untreated samples. The number of differentially expressed genes increased over time poststimulation, with 178 at 4 h to 1926 at 24 h (Supplemental Fig. 1B; Supplemental Table 2). We leveraged the temporal resolution to identify genes and pathways with the earliest Ro-dependent expression changes that were sustained throughout the stimulation response representing the most robust and direct Ro-dependent effects. Ro treatment up-regulated multiple stress-dependent pathways as early as 8 h (Fig. 1D; Supplemental Fig. 1C; Supplemental Table 3). The pathway with the earliest and sustained down-regulation was for transcripts encoding proteins involved in steroid and glucocorticoid production (Fig. 1D; Supplemental Fig. 1D; Supplemental Table 3). Ro coordinately down-regulated transcripts encoding enzymes involved in all steps of steroid hormone production from fatty acid and cholesterol metabolism to aldosterone synthesis (Fig. 1E). Taken together, we conclude that Ro coordinately represses multiple steps of the steroidogenic gene regulatory program, including fatty acid and cholesterol metabolism, to potently inhibit aldosterone and cortisol production (Fig. 1F).
Ro-dependent cellular phenotypes are MSI2-independent
We next tested whether the coordinated down-regulation of steroidogenesis upon Ro treatment was dependent on MSI2. We reasoned that expression of a Ro-resistant MSI2 would rescue the loss of aldosterone production. In vitro biochemical analysis identified a mutation in the first RRM domain of MSI2 (R100A) that prevented Ro-binding but retained the ability to bind RNA targets (Minuesa et al. 2019). We created a stable, doxycycline-inducible FLAG-MSI2-R100A H295R cell line (Supplemental Fig. 2A) and found that FLAG-MSI2-R100A induction did not rescue Ro-mediated decreases in aldosterone or cell viability (Fig. 2A; Supplemental Fig. 2B,C). Although the R100A mutation was shown to have similar RNA binding affinity in vitro, we would also observe a lack of rescue if the R100A MSI2 was unable to bind and regulate mRNA targets as well as endogenous MSI2 in vivo. Before testing MSI2-R100A mRNA target binding, we performed RNA immunoprecipitation sequencing (RIP-seq) to identify mRNA targets of endogenous MSI2 in H295R cells to select a set of transcripts to examine. As expected, the MSI2 target transcripts were enriched for UAG motifs in their 3′ UTRs (Supplemental Fig. 2D; Supplemental Table 4). We selected two targets (enriched in MSI2 RIP-seq), CCSAP and SCD, and two nontargets (depleted in MSI2 RIP-seq), ACTB and GAPDH, for characterization. Target and nontarget mRNAs were enriched (or depleted) to the same extent in both FLAG-MSI2-R100A, FLAG-MSI2-WT, and endogenous MSI2 RIP experiments (Fig. 2B; Supplemental Fig. 2E). Next, to test whether R100A regulates targets similar to wild-type MSI2, we designed a NanoLuc luciferase reporter containing seven MSI2 binding sites (based on Numb8) (Lan et al. 2020) in its 3′-UTR (Supplemental Fig. 2F). Dox-induced expression of FLAG-MSI2-R100A increased reporter expression to an extent similar to, if not greater than, FLAG-MSI2-WT in HEK293 cells; this same trend was observed in H295R cells (Fig. 2C; Supplemental Fig. 2G). These experiments demonstrated that FLAG-MSI2-R100A was not distinguishable from its wild-type counterpart with respect to mRNA binding and regulation. We then asked whether Ro altered either R100A or endogenous MSI2 mRNA targeting performing RIP-qPCR on our panel of mRNAs. As expected, Ro did not prevent FLAG-MSI2-R100A targeting (Fig. 2D); however, it also did not alter endogenous MSI2 targeting (Fig. 2E). MSI2 has condition-specific targeting and function (MacNicol et al. 2017; Li et al. 2020; Karmakar et al. 2022), so cell type-specific differences may explain the inability of FLAG-MSI2-R100A to rescue the Ro-mediated phenotypes in H295R cells. Therefore, we turned to K562 cells in which earlier studies demonstrated a Ro-dependent decrease in cell viability (Minuesa et al. 2019) that we also observed (Supplemental Fig. 2H). However, neither FLAG-MSI2-R100A nor FLAG-MSI2-WT rescued Ro-mediated cell death (Fig. 2F; Supplemental Fig. 2H,I). These data demonstrated that Ro did not disrupt MSI2 mRNA targeting or regulation and that multiple Ro-mediated phenotypes across cell lines are independent of MSI2.
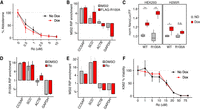
Ro phenotypes are MSI2-independent. (A) Dose-dependent effects of Ro on the percent aldosterone relative to DMSO in H295R FLAG-MSI2-R100A cells with or without 5 µg/mL doxycycline. Error bars indicate the SD of two independent rounds of experiments, each with six independent replicates. (B) RIP-qPCR enrichment of MSI2 targets (CCSAP and SCD) and nontargets (ACTB and GAPDH) normalized to YARS in H295R cells. FLAG-MSI2-R100A RIPs (red, FLAG antibody) were from doxycycline-treated cells and endogenous MSI2 RIPs (gray, endogenous MSI2 antibody) were from untreated cells. Error bars indicate SE from three to six replicates. (C) Ratio of NanoLuc to Firefly luciferase normalized to uninduced cells (no dox, gray) upon expression of either FLAG-MSI2-R100A or FLAG-MSI2-WT protein (dox, red). P-values were calculated using Student's t-test (**) P < 0.001, (*) P < 0.05, (n.s.) ≥ 0.05. Error bars indicate standard error from two (HEK293) or one (H295R) independent rounds of experiments, each containing six (HEK293) or four (H295R) independent replicates, respectively. (D,E) RIP-qPCR enrichment of H295R cells with induced FLAG-MSI2-R100A (D) or endogenous MSI2 (E) treated with 5 µM Ro (red) or DMSO (gray). Error bars indicate SE from three to six replicates. (F) Ro dose-dependent effects on the percent cell viability (Cell-Titer-Glo) relative to DMSO in K562 cells transfected with FLAG-MSI2-R100A plasmid with or without 5 µg/mL doxycycline. Error bars indicate SD from six independent replicates.
Ro binds to many RBPs including the RRM domain containing RBPs
Since Ro treatment caused MSI2-independent cellular phenotypes, we sought to identify Ro-interacting proteins in an unbiased manner. We wanted a method accounting for the compositional complexity of cells rather than the minimal in vitro mixtures; e.g., a solution only containing MSI2, an RNA substrate, and Ro. Therefore, we used thermal proteome profiling, which is used to identify proteins interacting with a small molecule by monitoring protein solubility shifts across a temperature gradient (Martinez Molina et al. 2013). To identify Ro-dependent interacting proteins, we performed a proteome integral solubility alteration (PISA) assay (Fig. 3A; Gaetani et al. 2019). Specifically, Ro was added to the cell lysate to identify proteins that exhibited a shift in thermal stability. We identified 768 proteins that exhibited a statistically significant thermal stability shift upon the addition of Ro to lysates (Fig. 3B). MSI2 exhibited a statistically significant, yet very modest thermostability shift (Fig. 3B). RBPs were the most highly enriched class of Ro-interacting proteins (Fig. 3C; Supplemental Fig. 3A; Supplemental Table 5), consistent with previous in vitro data of Ro-binding to RRM domains of multiple proteins (Minuesa et al. 2019). Nearly 1 in 5 Ro-interacting proteins were RBPs (145 of 768) (Gerstberger et al. 2014). Using annotation of RNA-binding domains in human RBPs (Gerstberger et al. 2014), we found that the RRM domain occurs most frequently in Ro-affected RBPs (Fig. 3D). Ro interacted minimally with MSI2 relative to many other RRM domains containing RBPs in cell lysates containing a wide variety of ribonucleoprotein (RNP) complexes.
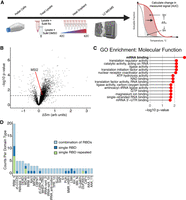
Ro interacts with multiple RRM containing RBPs. (A) Depiction of PISA assay. (B) Volcano plot of the change in relative temperature-dependent solubility of measured proteins with respect to Ro treatment compared with significance (−log10 P-value). (C) Top 15 gene ontology molecular functions enriched in proteins identified as Ro interactors from PISA assay. (D) Bar plot showing the count of each RBD domain type for RBPs enriched as PISA interactors.
Potential inhibition of RBPs by Ro and stress granule formation
PISA identified 145 Ro-interacting RBPs but did not address whether any of these interactions had functional consequences. To identify putative RBP targets of Ro, we screened for cases in which depletion of a Ro-interacting RBP resulted in gene expression changes similar to those from Ro treatment. To do so, we focused our analysis on the 29 RBPs that were both PISA interactors and had shRNA knockdown RNA-seq data in the ENCODE compendium (Fig. 4A, Venn diagram; Van Nostrand et al. 2020). We reanalyzed our RNA-seq data using the ENCODE pipeline and found 1181 genes that were differentially expressed upon Ro treatment. Then we calculated the correlation between Ro-induced and shRNA knockdown-mediated expression changes for these 1181 genes. Several Ro-interacting RBPs had a statistically significant positive correlation in gene expression changes between shRNA depletion and Ro treatment (Fig. 4A). MSI2 depletion exhibited no correlation with Ro treatment, consistent with our earlier data indicating MSI2-independent function of Ro (Fig. 4A). These data supported an alternative model where Ro promiscuously inhibits multiple RBPs to exert its cellular phenotypes. Broad inhibition of RBP-binding could lead to cellular stresses. In our RNA-seq time course experiment, we observed up-regulation of many processes connected to stress response and were particularly interested in arsenite stress, which causes stress granule formation (Supplemental Fig. 1C; Kedersha and Anderson 2002). Moreover, stress granule localized mRNAs (Khong et al. 2017) increased upon Ro treatment (Fig. 4B) and had a statistically significant overlap with mRNAs up-regulated by Ro (Fig. 4C). We then performed immunofluorescence using a stress granule marker (ELAVL1) and found that Ro-induced stress granule formation in a dose-dependent manner (Fig. 4D). These data supported a model in which Ro disrupted multiple RNPs leading to promote stress granule formation.
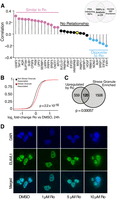
Ro causes dose-dependent stress granule formation. (A) Correlation between gene expression changes in Ro-treated H295R cells and gene expression changes from RBP knockdown in K562 cells (ENCODE) for the subset of genes with Ro-dependent expression changes (see Materials and Methods). RBPs colored by the significance of the correlation coefficient. (Pink) Positive, (gray) not significan, (blue) negative. (B) CDF plot of the log2 fold change in expression between Ro- and DMSO-treated cells for stress granule-localized transcripts (red) and all other transcripts (black). The Kolmogorov–Smirnov test was used to calculate statistical significance. (C) Overlap of Ro up-regulated transcripts and stress granule enriched transcripts. A hypergeometric test was used to calculate statistical significance. (D) Immunofluorescence of a stress granule marker, HuR/ELAVL1 (green), for DMSO- or Ro-treated H295R cells. Nuclei are shown in blue.
DISCUSSION
Ro has profound regulatory effects on the process of steroidogenesis and cell survival that are not dependent on MSI2. In contrast to previous studies, we found that these phenotypes are not rescued by expressing a modified MSI2 (R100A) that binds RNA but not Ro. The biochemical properties of the R100A and other mutations altering RNA and Ro binding were determined via in vitro binding assays (Minuesa et al. 2019). The same study showed MSI2-R100A could rescue Ro-mediated deficiencies in colony formation of mouse MLL-AF9 bone marrow cells. An important difference between this study and our analyses is assay timing: Colony formation assays take weeks, while our cell viability and steroid production assays take days. Therefore, the discrepancy could, in part, be explained by primary versus secondary effects. Another reason for the lack of rescue would be if MSI2-R100A does not phenocopy endogenous MSI2 in cells, which was not formally established in earlier studies. Here, we demonstrate that MSI2-R100A is indistinguishable from endogenous MSI2 with respect to mRNA targeting and regulation. Moreover, Ro did not disrupt endogenous MSI2–mRNA targeting further supporting our contention that Ro does not specifically target MSI2. Indeed, Ro promoted cell death in both wild-type and MSI2 KO HCT116 cells (Zhang et al. 2021). Another recently developed MSI inhibitor, R-12-8-44-3, did not promote cell death like Ro in K562 cells (Bai et al. 2023). Based on our results and other studies, we conclude that Ro-induced phenotypes are independent of MSI2.
Although Ro does not inhibit steroidogenesis (or other phenotypes) via MSI2, one or more of the RBPs we identified as putative Ro targets may regulate steroid hormone production. While our earlier work identified a subset of human RBPs that regulate steroidogenesis (Fu et al. 2021), the putative RBPs inhibited by Ro were not previously associated with steroidogenesis and may represent novel post-transcriptional mechanisms controlling human steroidogenesis. Performing the PISA assays at lower Ro concentrations may help us pinpoint the RBP(s) that regulate steroidogenesis. Regardless, the low steroid phenotype could be explained by the down-regulation of transcripts encoding steroidogenic enzymes (Fig. 1E). Stearoyl-CoA desaturase (SCD) is upstream of both cortisol and aldosterone production and is a plausible target for the observed broad inhibition of steroidogenesis. SCD converts saturated fatty acids to unsaturated fatty acids, promoting the production of lipid droplets that are important sources of cholesterol, an essential precursor to all steroidogenic hormones (Paton and Ntambi 2009). Since Ro caused inhibition of steroidogenesis at lower concentrations than cell death, Ro may have therapeutic utility for treating primary aldosteronism or Cushing's syndrome, which involve excessive production of aldosterone or cortisol, respectively. Additionally, Ro may have therapeutic potential for adrenocortical carcinomas since those with high steroidogenic capacity had the worst survival outcomes (Zheng et al. 2016). Finally, it would also be interesting if Ro repressed cholesterol biosynthesis, which is important for TP53-mediated tumor suppression (Moon et al. 2019). Altogether, Ro may have therapeutic benefits in disease driven or exacerbated by excess steroid hormone production.
Ro also elicits phenotypic changes in other disease models. For example, Ro reduces disease progression in an aggressive murine leukemia model (Minuesa et al. 2019). It prevents excessive autophagy and muscle wasting in models of myotonic dystrophy type 1 (Sabater-Arcis et al. 2021). SARS-CoV-2 nucleocapsid production, a surrogate for viral growth, decreases upon Ro treatment (Kamel et al. 2021; Lim et al. 2021). However, many of these studies do not perform experiments directly testing MSI2-dependence of the Ro phenotype (Kamel et al. 2021; Lim et al. 2021; Sabater-Arcis et al. 2021). Rescue experiments with the drug-resistant MSI2 mutants or small-molecule inhibitors in MSI2 knockout cells are directed approaches to establish target dependence. These approaches are limited by their low throughput. Creation of knockout cell lines is valuable, but time-consuming, not feasible for essential genes, and may additionally require multiple knockouts of functionally redundant paralogs. Additionally, these experiments do not provide alternative targets. To address these limitations, we used an unbiased thermal proteome profiling method, PISA, and discovered that Ro preferentially interacted with RBPs. Using ENCODE data we were able to identify a further refined subset of Ro-bound RBPs for which loss of function created expression changes similar to those from Ro treatment. We propose that Ro alters the balance between RNPs complexes, RBPs, and mRNAs to promote stress granule assembly RBPs (Molliex et al. 2015; Van Treeck et al. 2018; Wang et al. 2018a; Riggs et al. 2020). Indeed, Ro may be a unique tool to study stress granule formation/resolution through the disruption of RNPs in a cellular context. Our overall strategy integrating thermal proteome profiling with small-molecule and RBP-dependent expression changes is a generalizable framework for interrogating RBP inhibitors.
There have been many recently developed small-molecular inhibitors of specific regulatory RBPs (Meisner et al. 2007; Lan et al. 2015; Wu et al. 2015; Wang et al. 2018b; Minuesa et al. 2019; Zhang et al. 2021), spliceosome components (Palacino et al. 2015; Sivaramakrishnan et al. 2017), and translation factors (Bordeleau et al. 2006; Moerke et al. 2007). While they hold promise, the specificity and selectivity of most inhibitors have not been characterized in cells. It is well known that small molecules targeting kinases have off-target effects (Klaeger et al. 2017). The strong electrostatic affinity of the RNA-binding pocket and the sequence/structural conservation of these RBDs among RBPs makes it challenging to identify highly specific small molecules that are competitive inhibitors of RBPs (Wu 2020). Furthermore, many small-molecule inhibitors have micromolar affinity for their RBP target and are likely to bind to other off-target proteins in cells. Our findings, and those of others, demonstrate that the off-target activity is responsible for Ro-mediated cellular phenotypes. Indeed, off-target activity may be more common than expected; a recent analysis of a panel of cancer drugs undergoing clinical trials found that the efficacy of every single small-molecule tested was unaffected by the absence of the target (Lin et al. 2019). Since RBP–RNA interfaces have emerged as promising targets for modulation by small-molecule inhibitors, studies like ours are crucial to establishing target specificity or identifying targets of novel inhibitors.
MATERIALS AND METHODS
Cell culture
H295R cells were cultured in complete media (DMEM/F12 media, 10% cosmic calf serum, 1% insulin–transferrin–selenium) at 37°C and 5% CO2. For all ELISA assays, cells were cultured in low-sera media (DMEM/F12 media, 0.1% cosmic calf serum, 1% insulin–transferrin–selenium). K562 cells were cultured in RPMI10 media (RPMI 1640 +l-glutamate, 10% fetal bovine serum). HEK293 cells were cultured in FBS10 media (DMEM—high glucose, 10% fetal bovine serum).
Steroid hormone and cell viability/proliferation assays in H295R cells
H295R cells were plated in complete media at a density of 20,000 cells/well or 10,000 cells/well for either aldosterone or cortisol experiments, respectively. For cell lines with inducible constructs, 5 µg/mL doxycycline was added. Twenty-four hours after plating, complete media was removed and replaced with 110 µL of low-sera media containing Ro-08-2750 (Tocris) suspended in DMSO or only DMSO as vehicle control, 5 µg/mL dox for inducible experiments, and 10 nM Angiotensin II (Sigma-Aldrich) for stimulation or water for unstimulated cells. For the cortisol experiments, cells were stimulated with 10 µM forskolin (TCI Chemicals). After 24 h, either 100 or 20 µL of supernatant was collected for aldosterone or cortisol competitive ELISA (Invitrogen), respectively. When applicable, supernatants were diluted to a final volume of 100 µL using low-sera media, and the ELISA was performed according to the manufacturer's instructions. The remaining media was removed, and cells received 100 µL of complete media with 1:10 dilution of PrestoBlue cell viability reagent (Invitrogen). This was incubated for 2 h at 37°C and then read on a Synergy HT plate reader (BioTek). All media was removed from the cells, and the plate was frozen at −80°C. Upon thawing, the FluoReporter Blue Fluorometric dsDNA Quantification kit (Invitrogen) assay was run according to the manufacturer's instructions and read on a Synergy HT plate reader. A four parameter logistic (4PL) curve was fitted to a standard curve and used to calculate aldosterone/cortisol concentrations. IC50 values were calculated using the drc R package (Ritz et al. 2015). ELISA data are available at https://github.com/mukherjeelab/2022_RoInhibitor.
K562 viability assay
K562 cells (2 million cells in 100 µL) were transfected with 1 µg of plasmid (MSI2-WT or MSI2-R100A) using the neon transfection system (Invitrogen, 1.450 V, 10 ms, and three pulses). The transfection was split evenly into untreated or 5 µg/mL doxycycline-treated samples. From each condition, 10,000 cells/well were plated in a 96-well plate. Cells were treated with or without 5 µg/mL doxycycline and 0, 1, 5, 10, 15, 20, 50, and 75 µM Ro for 24 h. The remaining cells were plated for western collection. We used the Cell-Titer-Glo assay (Promega) according to the manufacturer's instructions to measure viability, since it was also used in Minuesa et al. (2019). After a 15-min room temperature incubation, luminescence was measured on a Synergy HT plate reader (BioTek).
RNA sequencing
H295R cells (500,000 cells per well) were plated in complete media. After 24 h, complete media was removed and replaced with low-sera media for an additional 24 h. Cells were then stimulated with 10 nM AngII and treated with DMSO control or 5 µM Ro. Cells were harvested at 0, 4, 8, 12, 16, 24 h poststimulation. Cells were collected in TRIzol, and RNA was isolated using Direct-zol Miniprep Plus kit (Zymo) with on-column DNase I digestion. Isolated total RNA was quantified using the Qubit RNA Broad Range Assay kit (Invitrogen) with the Qubit 3.0 fluorometer. Poly(A) RNA was enriched from 1 µg of total RNA using the NEBNext poly(A) mRNA magnetic isolation module (New England Biolab). At the final elution step of poly(A) selection, the RNA-bound beads were resuspended into 1× fragment, primer, and elute (FPE) buffer from the KAPA RNA HyperPrep kit (Roche) and heated for 6 min at 85°C for fragmentation. The resulting RNA was used as input into library preparation at the first strand synthesis step using the KAPA RNA HyperPrep kit (Roche). Libraries were sequenced to a depth of 20 million paired-end 2 × 150-bp reads each on the NovaSeq 6000 (Illumina) at the University of Colorado Genomics and Microarray Core.
RNA-seq analysis
Salmon (Patro et al. 2017) was used for quantifying transcript levels from all libraries using Gencode v26 transcriptome assembly (parameters -l A ‐‐allowDovetail ‐‐validateMappings). All downstream analysis was performed in R. Briefly, Salmon data were imported using the tximport library (Soneson et al. 2015). Differential gene expression was performed using the DESeq2 library (Love et al. 2014) with a likelihood ratio test for whole time course analysis or a Wald test for pairwise comparisons (Padj < 0.05).
Western blot analysis
Cells were induced for 24 h with 5 µg/mL dox and then collected in 1× Laemmli buffer containing 5% β-mercaptoethanol. Samples were heated for 5 min at 95°C to denature and then loaded onto a 15-well, 4%–12% gradient Bis-Tris, 1.0- to 1.5-mm Novex miniprotein gel (Invitrogen). Samples were transferred onto an iBlot mini NC stack nitrocellulose membrane (Invitrogen) using the iBlot2 with P0 transfer program (Invitrogen). Membranes were blocked with 5% nonfat milk in 1× Tris-buffered saline with 0.1% tween (TBST) for 30 min to 1 h at room temperature. Membranes were sequentially incubated with primary antibodies (all 1:2000 in 5% milk TBST) and HRP-labeled secondary antibodies (1:10,000 in 5% milk TBST) (Supplemental Table 6). The signals were detected by chemiluminescence using the Azure Biosystems Sapphire biomolecular imager.
Plasmid creation
pRD-RIPE plasmid was digested with AgeI and BstXI. FLAG-MSI2-R100A and FLAG-MSI2-WT DNA fragments were obtained from Twist Biosciences and amplified to contain homology ends for the digested pRD-RIPE (Supplemental Table 7). Gibson assembly was used to insert the MSI2 fragments into the pRD-RIPE plasmid and transformed into DH5α-competent Escherichia coli generated from the Mix and Go Transformation Buffer kit (Zymo). Plasmids were sequenced for validation.
A fragment containing a bidirectional CMV promoter driving expression of firefly luciferase and NanoLuc luciferase, each containing a PEST domain, was synthesized and inserted into a high-copy ampicillin-resistant expression plasmid by Twist Biosciences (Supplemental Table 7). This plasmid was digested with HindIII to insert a fragment, obtained from Twist Biosciences, containing MSI2 binding sites into the 3′ UTR of the NanoLuc luciferase (Supplemental Table 7). Gibson assembly was used to insert the fragment into the dual-luciferase plasmid and transformed into DH5α-competent E. coli generated from the Mix and Go Transformation Buffer kit (Zymo). The plasmid was sequenced for validation.
Stable cell line creation
A loxP-flanked blasticidin resistance cassette was inserted into the AAVS1 safe harbor locus of H295R cells via CRISPR/Cas9 as has been done previously (Supplemental Fig. 2A; Goering et al. 2022). Once the H295R LoxP cell line was established, MSI2 pRD-RIPE plasmids were electroporated into the cell line using the neon electroporation system (Invitrogen; 1100 V, 30 msec, and two pulses) and allowed to grow for 1 wk at 37°C. The cells were treated with 5 µg/mL puromycin for 1 wk, allowed to recover, and then treated again for another week. Cells were validated using PCR and western blot and screened for steroidogenic potential using an Aldosterone ELISA kit (Invitrogen) as described above.
RNA immunoprecipitation
RNA immunoprecipitation was performed as described previously (Wessels et al. 2016). For MSI2 RIP-seq samples, 70%–80% confluent H295R loxP cells were collected from three 15-cm2 plates per sample. For doxycycline-induced FLAG construct cells, cells were collected from one 70%–80% confluent 10‐cm2 plate. Pellets were weighed and resuspended in equal (1:1 w/v) PLB buffer supplemented with fresh 1 mM DTT, 100 U/µL RNaseOUT, and 1× protease inhibitor cocktail. Pellets were lysed for 5 min on ice followed by clarification by centrifugation. An input aliquot from each sample was set aside for RNA and western blot analysis. For the MSI2 RIP-seq, 850 µg of clarified lysate was immunoprecipitated with 5 µg of rabbit monoclonal MSI2 antibody conjugated to Dynabeads Protein A. For FLAG-MSI2-WT and FLAG-MSI2-R100A IP, ∼150 µg of lysate was immunoprecipitated with 2 µg of mouse FLAG M2 antibody conjugated to Dynabeads Protein G. All IPs were performed for 20 min tumbling end over end at room temperature. Following the IP, the supernatant was removed and the beads were washed five times with NT2 buffer. A 4% sample was removed for IP-western, and the remaining sample was incubated with TRIzol LS to release RNA from RNPs. RNA was isolated from the input, and RIP samples using the Direct-zol Microprep kit (Zymo) with on-column DNase I treatment following the manufacturer's instructions, followed by quantification using the Qubit RNA HS assay. Input, supernatant, and IP samples collected for IP-western were processed for western blot analysis as described above.
RIP RT-qPCR
RNA was reverse-transcribed with random hexamers using the iScript Universal cDNA Synthesis kit (Bio-Rad). The cDNA was diluted within the dynamic range and subjected to qPCR using the iTaq Universal SYBR Supermix (Bio-Rad) and 10 µM primers with three technical replicates for each reaction on the Bio-Rad Opus 384 (primers listed in Supplemental Table 7). The qPCR data were analyzed using the ddCq method.
RIP-seq library preparation and sequencing
rRNA was depleted from matched input and RIP RNA using RNaseH-mediated rRNA depletion (Baldwin et al. 2021). The rRNA-depleted RNA samples were used as input into the KAPA RNA HyperPrep RNA-seq library preparation kit (Roche) following the manufacturer's instructions with dual index adapters and 11 cycles of PCR. Resulting libraries were quantified with Qubit dsDNA HS assay and analyzed with TapeStation 4200 HS D1000. Input and RIP libraries were pooled separately and sequenced on the NovaSeq 6000 (Illumina) for 2 × 150-bp paired-end reads to a depth of 20 million paired-end reads per sample.
RIP-seq analysis
Salmon (Patro et al. 2017) was used for quantifying transcript levels from RIP-seq and input libraries using Gencode v26 transcriptome assembly (parameters -l A ‐‐allowDovetail ‐‐validateMappings). Salmon data were imported to R using the tximport library (Soneson et al. 2015). Differential gene expression was performed using the DESeq2 library (Love et al. 2014) with the Wald test. Genes with Padj ≤ 0.05 and log2 fold change ≥0.322 (1.25 enrichment in RIP-seq vs. input) were considered MSI2 targets.
Nano-Glo dual luciferase assay
H295R cells containing dox inducible FLAG-MSI2-R100A or FLAG-MSI2-WT were plated at a density of 200,000 cells/well in a 24-well plate in complete media. After 24 h, cells were transfected with 500 ng of dual luciferase plasmid containing MSI2 binding sites in the 3′-UTR using Lipofectamine 2000 (Thermo Scientific), according to manufacturer's instructions, and fresh dox was added. After 24 h, cells were harvested into 160 µL of passive lysis buffer and frozen at −20°C. Luciferase activity was measured using the Nano-Glo Dual-Luciferase kit (Promega). Twenty microliters of lysate was used per sample and the assay was run according to the manufacturer's instructions using a GloMax Navigator (Promega) with dual injection.
HEK293 cells at a density of 25,000 cells/well (96-well plate) were transfected with 25 ng of FLAG-MSI2-R100A or FLAG-MSI2-WT plasmid and 25 ng of dual-luciferase plasmid containing MSI2 binding sites in the 3′ UTR using Lipofectamine 2000 (Thermo Scientific), according to the manufacturer's instructions, and fresh dox was added. A total of 125,000 cells was also plated for western blot. After 24 h, cells were harvested and luciferase activity was measured using the Nano-Glo Dual-Luciferase kit (Promega) according to the manufacturer's instructions using a GloMax Navigator (Promega) with dual injection.
Proteome integral solubility alteration assay
The PISA method was performed as previously described (Gaetani et al. 2019). In brief, lysates from cells were adjusted to 1 mg/mL and divided into one-hundred-twenty 40-µL aliquots, 60 treatment, and 60 control. Treated lysate samples were incubated with 100 µM Ro. Samples were heated in a thermocycler (LifeEco, Bioer) at various temperatures (42°C, 42.4°C, 43.3°C, 45°C, 47.2°C, 50°C, 53.4°C, 56.3°C, 58.5°C, 60.4°C, 61.4°C, and 62°C) Samples were isobaric-labeled with TMT reagent (TMT10plex, Thermo Fisher Scientific) as recommended by the manufacturer. Peptide fractionation was performed on a Gemini NX-C18 column (Phenomenex) using a Dionex UltiMate 3000. Peptides were separated using the following gradient: 3% B (0–16.5 min), 3%–45% B (16.5–41.5 min), 45%–65% B (41.5–46.5 min), 65%–100% B (46.5–48.5 min), and 100% B (48.5–53.5 min) with solvent A (10 mM ammonium formate at pH 10.0) and solvent B (10 mM ammonium formate, 75% ACN at pH 10.0) at a flow rate of 0.5 mL/min. TMT-labeled peptides were analyzed by nano-ultrahigh performance (UHP) LC–MS/MS (EASY-nLC 1200, Orbitrap Fusion Lumos Tribrid, Thermo Fisher Scientific). Proteome Discoverer 2.2 (Thermo Fisher Scientific) was used for the database search and TMT quantification. Quantification results were log2-normalized, and the ΔSm value was determined for every protein. Statistical analysis was performed by Perseus (Tyanova et al. 2016).
GO and KEGG enrichment on PISA data
We used the clusterProfiler R library (Yu et al. 2012) to perform GO and KEGG enrichment on PISA interacting proteins. We used a false discovery rate for multiple test correction with a significance cutoff of 0.05. The list of human RBPs and their domains was from Gerstberger et al. (2014).
Immunofluorescence
H295R cells were plated on poly-d-lysine coated coverslips. Cells were treated with DMSO or 1, 5, or 10 µM Ro for 4 h. Cells were then fixed for 10 min in neutral buffered formalin solution. Cells were blocked and permeabilized in CAS-T (CAS-Block [Thermo Scientific] with 0.2% Triton-X) for 30 min followed by incubation for 1 h at room temperature with anti-HuR/ELAVL1 antibody (1:000 dilution in CAS-T) (Supplemental Table 6). Cells were washed with PBS-T (PBS with 0.1% Tween) and incubated for 40 min at room temperature with antimouse Alexa Fluor 488 antibody (1:1000 dilution in PBS-T) (Supplemental Table 6). Coverslips were mounted on slides using VectaShield mounting media containing DAPI and imaged at 60× magnification using a DeltaVision Elite wide-field fluorescence microscope (GE).
Correlation between Ro expression changes and RBP knockdown expression changes
The ENCODE consortium generated RNA-seq data for shRNA depletion of >200 RBPs in K562 cells. We downloaded the kallisto transcript quantification data of shRNA depletion RNA-seq for the subset of RBPs that were in the ENCODE database and were PISA interactors (n = 29) (Fig. 4A), as well as control shRNAs from K562. Data were imported using tximport R library (Soneson et al. 2015). Genes with counts sum ≥10 in all samples were kept for differential gene expression analysis. Differential gene expression was performed using the DESeq2 library (Love et al. 2014) with the Wald test. We applied the same analysis to the H295R RNA-seq from 12 h Ro-treated cells. Namely, transcripts were quantified using kallisto v.0.44 (Bray et al. 2016) (kallisto quant -i /path/to/kallisto.idx -o /path/to/outdir ‐‐plaintext read1.fastq read2.fastq). The kallisto index for gencode GRCh38 v.29 was downloaded from ENCODE (https://www.encodeproject.org/files/ENCFF904UYQ). Data were imported using tximport R library (Soneson et al. 2015). Genes with counts sum ≥20 in all samples were kept for differential gene expression analysis. Differential gene expression was performed using the DESeq2 library (Love et al. 2014) with the Wald test. For the set of 1181 genes differentially expressed after Ro treatment in H295R cells and passed both expression filters, we calculated Pearson correlation coefficients between ENCODE data and Ro-treated cells using Wald statistics (stat) values.
DATA DEPOSITION
RNA-seq data are available from the Gene Expression Omnibus (accession number GSE213218). Processed data and code: https://github.com/mukherjeelab/2022_RoInhibitor.
SUPPLEMENTAL MATERIAL
Supplemental material is available for this article.
ACKNOWLEDGMENTS
We thank David Bentley, Jay Hesselberth, and Sara Johnson for the critical review of the manuscript. This work was supported by the National Science Foundation Graduate Research Fellowship Program under grant number 1000317291-NSFGRFP-Walters (K.W.), the Endocrine Society Research Experiences for Graduate and Medical Students (REGMS) Summer Fellowship Program (K.W.), Polish National Agency for Academic Exchange Bekker Program PPN/BEK/2019/1/00173 (M.P.S.), the University of Colorado Anschutz Medical Campus RNA Bioscience Initiative (N.M.), the Boettcher Foundation Webb-Waring Early Career Investigator Award AWD-103075 (N.M.), and National Institutes of Health 1R35GM147025-01 (N.M.). BioRender was used in Figures 1F, 3A, and Supplemental Figure 2A.
Author contributions: N.M. conceived the project. K.W., A.B., E.H., A.I., M.D., E.M., and J.H. performed experiments and collected data. K.W, M.P.S., A.I., J.H., and N.M. performed formal analysis and conducted the visualization. K.W. wrote the original draft. K.W., M.P.S., A.B., A.A., K.H., and N.M. reviewed and edited the manuscript. K.W., M.P.S, A.A., K.H., and N.M. acquired funding. A.A., K.H., and N.M. provided resources. N.M. supervised the project.
Footnotes
-
Article is online at http://www.rnajournal.org/cgi/doi/10.1261/rna.079605.123.
-
Freely available online through the RNA Open Access option.
- Received January 20, 2023.
- Accepted June 13, 2023.
This article, published in RNA, is available under a Creative Commons License (Attribution-NonCommercial 4.0 International), as described at http://creativecommons.org/licenses/by-nc/4.0/.
REFERENCES
MEET THE FIRST AUTHOR

Meet the First Author(s) is a new editorial feature within RNA, in which the first author(s) of research-based papers in each issue have the opportunity to introduce themselves and their work to readers of RNA and the RNA research community. Kathryn Walters is the first author of this paper, “Small-molecule Ro-08-2750 interacts with many RNA-binding proteins and elicits MUSASHI2-independent phenotypes.” Kathryn is a fourth-year graduate student in the Molecular Biology PhD Program at the University of Colorado School of Medicine in the laboratory of Dr. Neelanjan Mukherjee. Her primary thesis project is to understand how the RNA-binding protein Musashi-2 (MSI2) regulated post-transcriptional regulation during stimulus responses.
What are the major results described in your paper and how do they impact this branch of the field?
In this manuscript, we focus on a small molecule Ro-08-2750 (Ro) that was proposed as a competitive inhibitor of the RNA-binding protein Musashi-2 (MSI2). We discovered that Ro was a potent repressor of human aldosterone and cortisol production. However, and to our surprise, this phenotype and others were not dependent on MSI2. Next, we used an unbiased approach to identify proteins interacting with Ro and leveraged the ENCODE database so that other RNA-binding proteins were the actual putative targets of Ro.
Our paper is important for reiterating the importance of validating the targets of small molecules in a cellular context. Beyond bringing awareness to the research community that Ro acts independently of MSI2, we also provide a framework to identify small molecules with the potential to modulate RBP function. There is a lot of excitement and potential with the development of new inhibitors, especially those targeting RNA-binding proteins, but we as a field must ensure proper validation.
What led you to study RNA or this aspect of RNA science?
I happened to stumble into the field of RNA during my first-year laboratory rotations and was intrigued by the complexity of RNA-binding proteins involved in gene regulation. While exploring options for dynamic loss-of-function models for MSI2, I read about this inhibitor. Despite many unexpected results, I think it provided valuable lessons for us and the greater research community.
What are some of the landmark moments that provoked your interest in science or your development as a scientist?
Growing up in rural North Dakota, the concept of a scientist was an abstract idea, and it definitely was not a career path. I didn't have access to advanced science courses or know anyone beyond medical personnel involved in science, but I was involved in a science competition called Science Olympiad. This organization, and my wonderful friend and coach Mrs. Holecek, helped me foster a love for science. I was encouraged to dig deeper into subjects that interested me, and it gave me the confidence to pursue a university degree in the sciences. After high school, I continued to volunteer as much as possible with this organization and believe it truly changed my life. It is one of few options for students in rural areas to experience how much fun science can be. The opportunities I've had to give back and share my passion for science—through tutoring, volunteering, and mentoring—continue to refresh my spirit and renewed my love for science still today.
If you were able to give one piece of advice to your younger self, what would that be?
Make plans but write in pencil. I've always been a person who likes to plan. I make plans for my laboratory days—packing experiments into every minute, plans for group activities, and plans for years and years ahead. And while this has many benefits, I think I've learned an important lesson in flexibility. Being able to save an assay for tomorrow and grab a drink with coworkers, being open to ideas from others, or changing your research direction when experiments lead you down an unexpected path, as I found in this manuscript, can be important parts of life you don't want to miss as you chase down your perfect plan.
What are your subsequent near- or long-term career plans?
In the future, as I continue my thesis work, I am renewing my focus on MSI2 by using other loss-of-function models besides Ro. I am also developing my skills in the field of computational biology as I seek to move deeper into this field in the future.








