
Type III-A CRISPR systems as a versatile gene knockdown technology
- Walter T. Woodside1,
- Nikita Vantsev2,3,
- Ryan J. Catchpole3,
- Sandra C. Garrett4,
- Sara Olson4,
- Brenton R. Graveley4 and
- Michael P. Terns1,2,3
- 1Department of Microbiology, University of Georgia, Athens, Georgia 30602, USA
- 2Department of Genetics, University of Georgia, Athens, Georgia 30602, USA
- 3Department of Biochemistry and Molecular Biology, University of Georgia, Athens, Georgia 30602, USA
- 4Department of Genetics and Genome Sciences, Institute for Systems Genomics, University of Connecticut Health Center, Farmington, Connecticut 06030, USA
- Corresponding author: mterns{at}uga.edu
Abstract
CRISPR-Cas systems are functionally diverse prokaryotic antiviral defense systems, which encompass six distinct types (I–VI) that each encode different effector Cas nucleases with distinct nucleic acid cleavage specificities. By harnessing the unique attributes of the various CRISPR-Cas systems, a range of innovative CRISPR-based DNA and RNA targeting tools and technologies have been developed. Here, we exploit the ability of type III-A CRISPR-Cas systems to carry out RNA-guided and sequence-specific target RNA cleavage for establishment of research tools for post-transcriptional control of gene expression. Type III-A systems from three bacterial species (L. lactis, S. epidermidis, and S. thermophilus) were each expressed on a single plasmid in E. coli, and the efficiency and specificity of gene knockdown was assessed by northern blot and transcriptomic analysis. We show that engineered type III-A modules can be programmed using tailored CRISPR RNAs to efficiently knock down gene expression of both coding and noncoding RNAs in vivo. Moreover, simultaneous degradation of multiple cellular mRNA transcripts can be directed by utilizing a CRISPR array expressing corresponding gene-targeting crRNAs. Our results demonstrate the utility of distinct type III-A modules to serve as specific and effective gene knockdown platforms in heterologous cells. This transcriptome engineering technology has the potential to be further refined and exploited for key applications including gene discovery and gene pathway analyses in additional prokaryotic and perhaps eukaryotic cells and organisms.
Keywords
INTRODUCTION
Bacteria and Archaea often harbor CRISPR-Cas systems that provide acquired immunity against viruses and other mobile genetic elements (MGEs) (Hille et al. 2018; Makarova et al. 2019). Upon invasion, a portion of cells integrate short (30–40 bp) sequences from the MGE into their CRISPR (clustered regularly interspaced short palindromic repeat) genomic arrays to provide a heritable record of the MGE encounter (Jackson et al. 2017b; McGinn and Marraffini 2019). To carry out an immune response based on these heritable records, the CRISPR arrays are transcribed and the primary CRISPR transcripts processed to generate mature CRISPR (cr)RNAs (Brouns et al. 2008; Carte et al. 2008, 2014). Subsequently, each mature crRNA associates with specific CRISPR-associated (Cas) proteins to form effector crRNPs (crRNA-Cas protein ribonucleoprotein complexes) that mediate crRNA-guided recognition and Cas nuclease-mediated destruction of invasive MGE nucleic acids to prevent further MGE infection (Jackson et al. 2017a; Hille et al. 2018).
CRISPR-Cas systems are diverse and have been categorized into six distinct types (I–VI) that use different effector Cas nucleases that can recognize and destroy either foreign DNA (types I, II, V, and possibly IV) (Brouns et al. 2008; Garneau et al. 2010; Zetsche et al. 2015; Pinilla-Redondo et al. 2020), RNA (type VI) (Abudayyeh et al. 2016), or both DNA and RNA (type III) (Hale et al. 2009, 2014; Staals et al. 2013; Goldberg et al. 2014; Tamulaitis et al. 2014, 2017; Samai et al. 2015; Elmore et al. 2016; Estrella et al. 2016; Kazlauskiene et al. 2016; Han et al. 2017). By harnessing the unique attributes of the various CRISPR-Cas systems (such as nucleic acid binding specificity, nuclease activity, etc.), a range of innovative CRISPR-based DNA and RNA targeting tools and technologies have been developed (e.g., for genome editing, control of gene expression, sequence-specific antibiotics, nucleic acid-based viral, and pathogen diagnostics) (Bikard et al. 2014; Terns and Terns 2014; Gootenberg et al. 2018; Terns 2018; Pickar-Oliver and Gersbach 2019; Smargon et al. 2020). Tremendous potential remains among other CRISPR systems and exploiting this diversity can result in new research tools with associated transformative biotechnological and biomedical applications.
In this study, we sought to investigate the potential of type III-A CRISPR systems (also referred to as Csm systems, Haft et al. 2005) to be harnessed as a gene knockdown platform in prokaryotes, akin to how RNA interference (RNAi) machinery has been used in eukaryotes for gene expression down-regulation and gene discovery applications (Kim and Rossi 2008; Wilson and Doudna 2013; Setten et al. 2019). Type III-A effector crRNPs are composed of a single crRNA stably associated with five (Csm 1–5) Cas protein subunits (Fig. 1A; Staals et al. 2014; Tamulaitis et al. 2014; Kazlauskiene et al. 2016; Ichikawa et al. 2017; Liu et al. 2017; Foster et al. 2018; Dorsey et al. 2019; You et al. 2019). The mature crRNAs within these complexes are generated through site-specific cleavage of type III-A CRISPR array primary transcripts (within the repeat regions) by the Cas6 endoribonuclease (Carte et al. 2014; Ichikawa et al. 2017). The processed crRNAs within the effector crRNPs contain eight nucleotides of repeat sequence at the 5′ end called the 5′ tag (Hale et al. 2009), followed by a ∼30–40 nt guide sequence that base-pairs with the target RNA protospacer. The 3′ ends of the crRNAs within the complexes can have variable lengths of repeat-derived sequence (Hatoum-Aslan et al. 2013; Tamulaitis et al. 2014; Kazlauskiene et al. 2016). While the repeat-derived, 5′ tag element is critical for function, some heterogeneity at the 3′ ends of crRNA species is normally observed and does not appear to impact function (Hale et al. 2012; Tamulaitis et al. 2014; Foster et al. 2018).
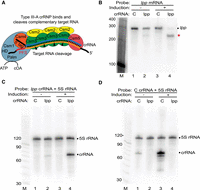
Programmed mRNA cleavage by L. lactis type III-A crRNPs expressed in E. coli. (A) Diagram of a representative type III-A effector crRNP containing Csm1-5 subunits and a crRNA, in the process of cleaving a bound target RNA. Each Csm3 RNase subunit cuts the target RNA once (cleavages indicated by arrows) within the region of the target RNA (orange) that base-pairs with the crRNA guide element (blue). The position of the target RNA protospacer flanking sequence (PFS) is indicated as are the HD (DNase) and Palm (cyclic oligoadenylate [cOA] producing) motifs of the Csm1 subunit. (B–D) Expression of L. lactis type III-A crRNPs containing either a crRNA against the lpp mRNA (lpp) or negative control crRNA (C) was induced (+), and northern analysis was performed using probes against the lpp mRNA (B), lpp crRNA (C), control crRNA (D), or constitutively expressed 5S rRNA (C,D). The positions of the RNAs are indicated, including those of the full-length lpp mRNA (dot) and lpp mRNA cleavage products (red star). The dotted line in B indicates that intervening lanes were omitted from the blot. The sizes of the molecular weight markers (M) are indicated in each panel.
Type III-A systems can function by both DNA and RNA cleavage mechanisms that are each activated via crRNA-guided recognition of target RNA (Staals et al. 2013, 2014; Goldberg et al. 2014; Tamulaitis et al. 2014; Samai et al. 2015; Jiang et al. 2016; Kazlauskiene et al. 2016, 2017; Ichikawa et al. 2017; Liu et al. 2017; Mogila et al. 2019; Sridhara et al. 2022). Target RNA recognition induces conformational changes within the crRNP (Guo et al. 2019; Jia et al. 2019; You et al. 2019; Sridhara et al. 2022) leading to activation of intrinsic DNase and RNase activities as well as triggering production of cyclic oligoadenylate (cOA) from ATP precursors (Kazlauskiene et al. 2017; Niewoehner et al. 2017; Rouillon et al. 2018). In turn, cOA serves as a second messenger that binds to and allosterically activates the RNase activity of a trans-acting enzyme called Csm6 (Kazlauskiene et al. 2017; Niewoehner et al. 2017; Foster et al. 2018; Rouillon et al. 2018; Jia et al. 2019), which has been shown to be capable of acting on both target (crRNA-matching) and non-target (cellular) RNAs (Jiang et al. 2016; Rostol and Marraffini 2019). Collectively, these multiple activities lead to a dual ability of type III-A systems to cleave DNA and RNA targets for viral or plasmid clearance or growth arrest or death of infected cells.
The structural organization of type III-A crRNPs and functional roles of the individual Csm1–6 protein subunits have been investigated and three of the subunits have been shown to be catalytic (Rouillon et al. 2013; Hatoum-Aslan et al. 2014; Staals et al. 2014; Tamulaitis et al. 2014; Liu et al. 2017; Jia et al. 2018; Dorsey et al. 2019; Mogila et al. 2019; You et al. 2019). Csm1 (a Cas10 superfamily member, Makarova et al. 2019) is a large, multiple domain protein that typically contains two highly conserved functional motifs: the HD motif capable of destroying single-stranded DNA (Elmore et al. 2016; Estrella et al. 2016; Kazlauskiene et al. 2016) and the GGDD motif of one of two Palm domains that can convert ATP into cOA second messenger molecules (Fig. 1A; Kazlauskiene et al. 2017; Niewoehner et al. 2017; Rouillon et al. 2018; Foster et al. 2020). Csm3 is an intrinsic RNA endoribonuclease that cleaves the target RNA at regular 6 nt intervals within a region defined by crRNA base-pairing (typically 4–5 cuts are made, depending upon the copy number of Cmr3 RNase subunits within the III-A crRNP) (Fig. 1A; Tamulaitis et al. 2014). As noted above, Csm6 is a cOA-activated, trans-acting ribonuclease (Jiang et al. 2016; Niewoehner et al. 2017; Rostol and Marraffini 2019). The DNase (Csm1) and RNase (Csm3 and Csm6) activities of type III-A systems can each contribute to robust immunity against diverse viral and plasmid invaders with notable differences in the dependency on DNase or Csm6-mediated RNase for the various systems investigated (Hatoum-Aslan et al. 2014; Samai et al. 2015; Cao et al. 2016; Foster et al. 2018; Millen et al. 2019; Lin et al. 2020).
Two distinct regions of the target RNA control type III crRNP activities. Extensive complementary binding between the target RNA protospacer region and the crRNA guide region is required to trigger the activities. However, several studies have revealed a key role for the short, 8 nt sequence that flanks the 3′ end of the RNA protospacer, termed the protospacer flanking sequence (PFS), in controlling type III activities (Fig. 1A). If the target RNA PFS exhibits perfect or significant complementarity to the 5′ tag element of the crRNA (Fig. 1A; Pyenson et al. 2017; You et al. 2019), then that target RNA-bound crRNP becomes incapable of triggering DNase activities or cOA production (Marraffini and Sontheimer 2010; Samai et al. 2015; Elmore et al. 2016; Kazlauskiene et al. 2016), but target RNA cleavage is unaffected (Hale et al. 2012; Tamulaitis et al. 2014).
We have transplanted functional III-A systems from three bacterial species (L. lactis, S. epidermidis, and S. thermophilus) by coexpressing the six Csm proteins, Cas6 (for processing of pre-crRNA transcripts into functional crRNAs), and a CRISPR array on a single, arabinose inducible plasmid in E. coli. The expressed III-A modules were previously reported to specifically eliminate invading plasmids, dependent on crRNA homology, transcription of the DNA target sequence, cOA signaling and RNase activity of Csm6, but independently of DNase activity in Csm1 (Ichikawa et al. 2017; Foster et al. 2018). Here, we have specifically exploited the site-specific and Csm3-mediated RNA cleavage activity of the three distinct type III-A crRNPs to efficiently knock down gene expression of both coding and noncoding RNAs in vivo. We demonstrate that the III-A modules can be programmed to recognize one or more E. coli cellular target RNAs by addition of appropriate crRNA coding sequences to the module. Our findings demonstrate the potential of heterologous type III-A systems as tools for RNA interference and pave the way for expanded utility as functional genomic tools in novel cells and organisms.
RESULTS
Programming L. lactis III-A crRNPs for selective cleavage of target mRNAs in vivo
To harness type III-A crRNPs as a gene expression knockdown platform, we introduced the systems into E. coli cells and expressed them via an arabinose-inducible promoter (Ichikawa et al. 2017; Foster et al. 2018). The E. coli host strain (BL21-AI) lacks endogenous CRISPR-Cas systems. We guided interference by designing crRNA sequences to recognize specific E. coli target mRNAs (Fig. 1). Northern blot analyses, using probes against the target mRNAs as well as an internal control (5S rRNA, untargeted), were carried out to assess the efficiency and specificity of directed cleavage.
Since type III-A interference can lead to both target RNA cleavage and nonspecific RNase activity via Csm6 (Fig. 1A), we designed two approaches to ensure our platform only carried out sequence-specific target RNA knockdown. First, this goal was accomplished by screening potential target RNAs for the presence of 3′ PFS sequences with significant homology with the 8 nt 5′ crRNA tag. Pairing between the crRNA tag and PFS of the target RNA permits Csm3-directed cleavage of crRNA-targeted RNA but prevents the complex from producing cOA second messenger required for activating nonspecific Csm6 RNase activity (Hale et al. 2014; Kazlauskiene et al. 2016; Pyenson et al. 2017; You et al. 2019). Second, as a more versatile strategy, we used type III-A crRNPs that have a mutation in the conserved Csm1 Palm motif (GGDD to GGAA); these mutants have been shown to be incapable of producing cOA/Csm6 activation regardless of the target RNA PFS (Kazlauskiene et al. 2017; Niewoehner et al. 2017; Rouillon et al. 2018; Foster et al. 2020). Mutation of the Palm or HD motif of Csm1 does not influence the ability of the type III-A crRNPs to cleave target RNA (Supplemental Fig. S1).
For the initial test, we assessed the RNA targeting capacity of the L. lactis type III-A crRNP with a crRNA against mRNA for the lpp gene, which encodes a major lipoprotein and is nonessential for viability of E. coli (Baba et al. 2006). The lpp mRNA target sequence was selected because its 3′ PFS has extensive base-pairing potential with the 5′ crRNA tag (PFS bases −1 to −5 and −8 were complementary to positions +1-5 and +8 of the 5′ crRNA tag), so Csm6 nonspecific RNase activity should not be triggered. As a specificity control, the same L. lactis crRNPs were programmed with a control crRNA (C) that does not contain significant complementarity to any E. coli RNA. In the presence of the lpp crRNA, but not the control crRNA, we observed a significant reduction in the steady-state levels of the lpp mRNA (Fig. 1B, compare lanes 3 and 4) that was accompanied by the appearance of a breakdown product of the expected size for cleavage at the crRNA target site (Fig. 1B, red star in lane 4). The crRNA-dependent cleavage of the target lpp mRNA was dependent upon arabinose induced expression of the type III crRNP (Fig. 1B, compare lanes 2 and 4). As expected, the steady-state levels of 5S rRNA remained constant in the analyzed cells (Fig. 1C,D). These same samples were also probed to confirm the expression of each engineered crRNA. The size of the main crRNA species matches that expected for the product following Cas6 cleavage of primary transcripts (71 nt) and low levels of presumably 3′ trimmed crRNAs were also observed (lpp crRNA is shown in Fig. 1C, lanes 2 and 4 and noncognate control crRNA is shown in Fig. 1D, lanes 1 and 3). Mutational analysis of Csm3 (D30A) confirmed that the RNase activity observed for the crRNPs is mediated by the Csm3 backbone subunit cutting within the target RNA protospacer region, as expected (Supplemental Fig. S2). The results show that heterologously expressed L. lactis type III-A crRNPs can be programmed to efficiently and selectively cleave a desired target mRNA in vivo.
S. thermophilus and S. epidermidis type III-A systems can also be programmed to target mRNA destruction
To determine if type III-A crRNPs from other bacterial species were also capable of carrying out efficient and specific mRNA knockdown in E. coli, we programmed the S. thermophilus and S. epidermidis type III systems to target the same lpp endogenous mRNA transcript (Fig. 2). Similar to the L. lactis system (Fig. 1), our results revealed that expression of III-A crRNPs from both S. thermophilus (Fig. 2A–C) and S. epidermidis (Fig. 2D) resulted in specific and efficient knockdown of the lpp mRNA and the accumulation of mRNA cleavage products of the expected sizes for crRNA-directed cleavage. While L. lactis and S. epidermidis share the same 5′ crRNA tag sequence (5′-ACGAGAAC-3′), the S. thermophilus 5′ tag differs at positions 4 and 5; 5′-ACGGAAAC-3′. For this reason, we used the Csm1 Palm motif mutation in S. thermophilus (GGDD motif mutated to GGAA) to ensure that Csm6 nonspecific RNase activity was not triggered by the decreased tag-PFS complementarity. No reduction in 5S signal intensity was observed for any of the three systems (Fig. 2B–D), suggesting there is no detectable nonspecific RNase degradation. Thus, three distinct type III-A CRISPR-Cas systems proved to be functional for programmable RNA targeting in vivo. In subsequent experiments, the Csm1 Palm motif mutation was adopted for all three systems.

Targeted mRNA destruction by S. thermophilus and S. epidermidis type III-A crRNPs. (A–C) Expression of S. thermophilus type III-A crRNPs containing either a crRNA against the lpp mRNA (lpp) or negative control crRNA (C) was induced (+), and northern analysis was performed using probes against the lpp mRNA (A), lpp crRNA (B), control crRNA (C), or 5S rRNA (B,C). (D) Expression of the S. epidermidis (Sep) and L. lactis (Lla) type III-A crRNPs containing either a crRNA against the lpp mRNA or control crRNA was induced (+), and northern analysis was performed using probes against the lpp mRNA and 5S rRNA. The positions of the RNAs are indicated, including those of the full-length lpp mRNA (dot) and lpp mRNA cleavage products (red star). (A–C) The dotted lines indicate intervening lanes were omitted from the blot. The sizes of the molecular weight markers (M) are indicated in each panel.
Type III-A crRNPs are capable of acting at different sites along the length of a target mRNA
Next, we investigated whether the position of the target site significantly impacts the efficacy of target mRNA cleavage. The L. lactis III-A crRNP (with Csm1 Palm motif mutation) was programmed to target five different sites along the lpp transcript. The five distinct crRNAs were tested individually and, in each case, northern analysis was performed with probes specific for either the 5′ or 3′ terminal regions of the lpp mRNA (Fig. 3A). Each of the tested crRNAs led to a major reduction in the steady-state levels of full-length lpp transcript relative to the control crRNA, when probing for either the 5′ or 3′ ends of the mRNA following induction of crRNP formation (Fig. 3B,C). Moreover, 5′ and 3′ expected cleavage products were observed with each tested crRNA (Fig. 3B,C). Following crRNA-guided target RNA cleavage, the 3′ mRNA fragments exhibited generally higher steady-state levels than 5′ mRNA fragments, despite near full cleavage of the full-length transcripts for each of the tested crRNAs. The results indicate that type III crRNPs are capable of targeting mRNA cleavage at multiple sites along the length of transcripts.
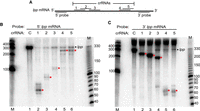
Type III-A crRNPs can cleave at distinct sites along the length of a mRNA. (A) Diagram of the lpp mRNA showing the relative positions of each tested crRNA and northern probes used. (B,C) Expression of the L. lactis type III-A crRNPs containing different crRNAs (1–5) was induced, and northern analysis was performed using probes against the 5′ (B) and 3′ (C) termini of the lpp mRNA. The positions of the full-length lpp mRNA (dot) and cleavage products (red star) are indicated in each panel. The sizes of the molecular weight markers (M) are indicated in each panel.
Type III-A crRNPs can be programmed to cleave multiple distinct RNA targets simultaneously
We next determined if the L. lactis III-A crRNPs could be programmed to degrade multiple mRNAs simultaneously. First, we determined that L. lactis crRNPs efficiently targeted two additional nonessential E. coli mRNAs (Baba et al. 2006) encoding either the cold-shock protein E (cspE) or the outer membrane protein F (ompF), when individually programmed with single crRNAs (Fig. 4B, lane 3 and 4C, lane 4; lpp mRNA cleavage shown in Fig. 4A, lane 2). Next, we attempted simultaneous targeting of lpp, cspE, and ompF mRNAs by expressing a CRISPR array encoding three crRNAs. A similar reduction in full-length transcript levels and appearance of the expected cleavage products was observed in the strain expressing all three crRNAs as compared to individual strains expressing just one crRNA against a single mRNA target (Fig. 4; compare lanes 2 and 5 in A, lanes 3 and 5 in B, and 4 and 5 in C). These results demonstrate the capacity of type III-A crRNPs to carry out multiplexed, simultaneous knockdown of at least three distinct mRNAs.
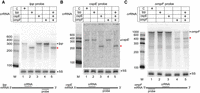
Type III-A crRNPs can effectively target multiple mRNA transcripts simultaneously. (A–C) Expression of the L. lactis type III-A crRNPs with single or multiple crRNAs was induced, and northern analysis was performed with probes against lpp mRNA (A), cspE mRNA (B), ompF mRNA (C), or 5S rRNA (A–C). The positions of full-length mRNAs (dot) and expected cleavage products (red star) are indicated in each panel. The sizes of the molecular weight markers (M) are indicated in each panel. The relative positions of the crRNA and the northern probe for each mRNA are shown below each panel.
Directed cleavage of a noncoding RNA target
We next tested whether L. lactis III-A crRNPs could effectively cleave a noncoding RNA target, specifically one with substantial secondary structure which could potentially interfere with crRNA binding. For this, we chose to target the rnpB RNA/ribozyme, which is the enzymatic component of RNase P and catalyzes 5′ end cleavage of precursor tRNAs (Esakova and Krasilnikov 2010). In E. coli, the rnpB RNA is required for the endonucleolytic separation of the valV-valW pre-tRNA bicistronic transcript (Mohanty and Kushner 2007; Mohanty et al. 2020). We individually tested a panel of seven crRNAs that spanned the length of rnpB RNA (Fig. 5B) for their ability to direct L. lactis III-A crRNPs to destroy the RNase P RNA (Fig. 5C) and lead to decreased processing of the bicistronic pre-tRNA transcript (Fig. 5E).
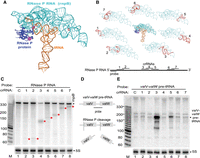
Targeting the noncoding RNA component of RNase P. (A) Structure of the T. maritima RNase P RNA (rnpB RNA in cyan) in complex with the RnpA protein (purple) and substrate tRNA (orange) (PDB 3Q1R). (B) The relative positions of each tested crRNA (1–7) are indicated on the lower diagram and are superimposed (red) onto the rnpB RNA structure (cyan). (C,E) Expression of the L. lactis type III-A crRNP containing a crRNA targeting rnpB (1–7) was induced, and northern analysis was performed with probes against the rnpB RNA (C), valV-valW pre-tRNA (E), or 5S rRNA (C,E). The positions of the rnpB RNA full-length (dot) and cleavage products (red stars) and sizes of molecular weight markers (M) are indicated. (D) Schematic of the valV-valW pre-tRNA processing by RNase P and binding location of probe used to selectively recognize the unprocessed pre-tRNA transcript (E).
Northern analysis revealed that the extent of rnpB RNA degradation varied among the selected crRNAs (Fig. 5C). All seven tested crRNAs could cleave rnpB RNA as evidenced by the accumulation of expected size cleavage products, relative to the control crRNA (Fig. 5C, compare lanes 2–8 with lane 1). However, only one of the seven tested crRNAs resulted in a clear reduction in the steady-state levels of the rnpB target RNA as compared to the control lane (Fig. 5C, lane 4 versus lane 1) and a corresponding accumulation of unprocessed valV-valW bicistronic transcript (Fig. 5E, lane 4). Accumulation of bicistronic pre-tRNA above background levels was also observed for a second tested crRNA (Fig. 5E, lane 6). Given that this RNA exists in a complex secondary structure, we considered the possibility that RNA folding influences crRNA access or binding. To explore this, we compared the E. coli RNase P RNA with that of T. maritima, which has a solved three-dimensional X-ray structure of the folded rnpB RNA in complex with tRNA substrate and associated RNase P protein (Fig. 5A; Reiter et al. 2010). The RNase P RNA is highly conserved between the two organisms, and we note that the two most effective crRNAs (3 and 5) are each predicted to bind to regions of the folded rnpB ribozyme that do not interact with pre-tRNA or RNase P protein (Fig. 5B). From these results we propose that type III-A crRNPs are a feasible tool for knocking down expression of noncoding RNAs, but RNA folding and/or interactions with other RNAs and proteins may reduce targeting efficacy and require careful crRNA design.
Transcriptome-wide effects of type III-A knockdown
Northern blot analyses gave clear evidence that our type III-A knockdown platform efficiently degraded target RNA. Next, we used total RNA-sequencing to examine target and off-target effects in a transcriptome-wide manner (Fig. 6). We focused on L. lactis crRNPs, and used systems programmed with individual targets (lpp, ompF, and cspE) and a multiplexed system with all three individual targets above plus one crRNA against the lacZ mRNA. These systems used the wild-type type III-A crRNP, and in addition, we examined knockdown of the lpp target in a modified L. lactis crRNP (referred to as the Δcsm6, Csm1-HDmut + Palmmut crRNP) which has csm6 deleted, and HD and Palm motif mutations in csm1 (the same mutations used in experiments described above) to eliminate potential nonspecific DNase (Csm1-HD) or RNase activities (Csm1-Palm and Csm6). While some type III-A systems show a nonspecific DNase activity mediated by the Csm1 HD domain (Samai et al. 2015; Jiang et al. 2016; Kazlauskiene et al. 2016; Lin et al. 2020), we previously reported that the systems investigated here do not rely on this activity for immunity against plasmid DNA (Foster et al. 2018). Based on that finding, we did not anticipate nonspecific DNase activity in our knockdown platform. However, to confirm this prediction, we included the Δcsm6, Csm1-HDmut + Palmmut crRNP and compared its lpp knockdown profile with that of the wild-type crRNP. For all systems, total RNA from both arabinose induced and uninduced samples was sequenced. In addition, a mock sample (empty plasmid vector, both induced and uninduced) was included.
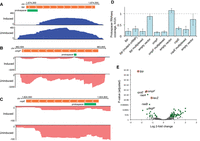
RNA-seq analysis of transcript abundance after expression of type III-A crRNPs. (A–C) Genome browser tracks show stranded total RNA-seq read density over selected regions of the host (E. coli) chromosome. RNA-seq reads aligned to the top strand are colored blue; bottom strand reads are pink. Orange bars show the position of ORFs for targeted genes; green bars show the position of the programmed protospacer for each gene. The x-axis indicates cumulative read depth. Inducing type III-A crRNP expression led to a reduction in read density for the targets lpp (A), ompF (B), and cspE (C). All three replicates were similar; representative replicates are shown. (D) To quantify the reduction in RNA-seq read density at the target site, all reads overlapping the protospacer position were counted. The counts for the induced replicates are shown as a proportion of the average counts for the uninduced replicates. The mutant crRNP lacked the csm6 gene and contained HD and Palm active site mutations in Csm1. (E) Genome wide differences in RNA expression were determined for a type III-A crRNP programmed to target lpp, ompF, cspE, and lacZ as compared to the cspE individual target crRNP. Plots show the log2–fold change (log2fc) in transcript abundance and adjusted P-value (Padj) for each expressed gene; genes for which the Padj was less than 0.1 and log2fc was greater than 0.58 are plotted in green; genes which did not meet this threshold are in gray. The programmed targets lpp, ompF, and lacZ are circled in red. Several genes in the rpoE-rseABC regulon are labeled.
For all three targeted genes, inducing expression of the III-A crRNP resulted in a significant reduction in RNA-seq read coverage over the length of the mRNA and particularly at the site of crRNA binding (Fig. 6A–C). While some RNA upstream and downstream from the target site remained, consistent with the northern blot results, the bisected mRNA would not generate full-length protein so we focused on the lost RNA-seq read coverage over the target site as a proxy for gene knockdown. We took the average number of reads overlapping the crRNA binding site in the uninduced replicates and set that as the baseline expression (equal to one), then determined the proportion of overlapping reads in the induced replicates. With crRNP expression induction, the number of reads overlapping the crRNA binding site dropped to 17%–34% of the baseline, corresponding to knockdown between 64% and 83%, depending on the sample (Fig. 6D). There was little to no difference in the extent of knockdown for an individual target versus that same target in the multiplexed crRNP (Fig. 6D). Likewise, percent knockdown of lpp was the same for the wild-type crRNP as for the Δcsm6, Csm1-HDmut + Palmmut crRNP (Fig. 6D).
Both the northern blot and RNA-seq data indicated that the type III-A platform carried out efficient knockdown of target RNA. Next, we explored off-target effects in our L. lactis system: We generated RNA-seq read count tables for all annotated genes in the E. coli BL21 genome and then used DESeq2 (Love et al. 2014) to look for gene transcripts which were significantly altered upon induction of the crRNPs. Arabinose treatment itself led to large expression changes in genes related to induction and sugar metabolism. In this context, the decreases in coverage for the crRNP-targeted genes were relatively modest, particularly since read counts were determined for the entire length of a gene, while degradation by the programmed crRNP was most pronounced at the crRNA binding site (Fig. 6A–C). Therefore, this comparison was likely not sensitive enough to detect off-target RNA degradation. When comparing gene expression for the induced crRNP systems with the induced empty vector, significant decreases in expression were observed for the target genes; however, there were also large expression changes in both the plasmid-encoded cas genes (as expected) and changes related to a lacI gene that is present on the empty vector but removed from the crRNP-encoding plasmids. To home in on the changes that were exclusive to crRNAs, we compared the cspE individual crRNP with the multiplexed crRNP sample (Fig. 6E; Supplemental Data Table S5). In this comparison, the two sets of samples are identical with respect to induction and plasmid-encoded genes; they differ only in their programmed crRNAs. In this case, we detected significantly reduced expression for the three programmed gene targets, besides cspE (Fig. 6E, target genes are circled in red; all genes with Padj < 0.1 and log2fchange >0.58 are colored green; genes below that threshold are colored gray).
Aside from these three intended mRNA targets, we noted two genes with comparably significant reductions in mRNA abundance, rpoE and rseA, along with several dozen genes with either negative or positive changes in expression that passed the threshold noted above (Fig. 6E, genes plotted in green). Decreased expression could presumably come from either indirect effects related to loss of lpp, lacZ, or ompF or direct effects due to off-target interference. Interestingly, rpoE and rseA are part of the rpoE-rseABC operon, whose protein products regulate the response to cell envelope stressors such as the accumulation of misfolded membrane proteins (De Las Penas et al. 1997). Two additional genes involved in this regulatory pathway, rseB and degP, were also reduced to a lesser extent (Fig. 6E). The transcription factor σE, encoded by rpoE and regulated by RseA and RseB, induces expression of outer membrane stress response proteins, such as DegP (a protease which degrades misfolded outer membrane proteins), chaperones to help fold membrane proteins, and transport machinery to correctly traffic them (Ge et al. 2014; Gottesman 2017). While rpoE, rseA, and rseB are encoded on a single operon and thus could all be diminished by one off-target crRNA binding event, degP lies elsewhere on the genome, and would be a separate transcript. This might indicate that reduced expression of these genes is an indirect effect rather than off-target interference. Furthermore, RNA-seq coverage was reduced across the entire rpoE-rseABC operon (Supplemental Fig. S3), rather than in discrete locations as was observed for the programmed targets (Fig. 6A–C).
In addition to scanning for off-target expression changes in an unbiased, transcriptome-wide manner, we also identified and examined several potential off-target crRNA binding sites based on sequence identity. The programmed crRNAs in our study were designed to match unique regions in target genes and thus there were no close matches elsewhere in the genome. To look for degenerate matches, we aligned the programmed spacers to the genome using bowtie2 with highly permissive parameters (see Materials and Methods). This returned three partial matches: two for lpp with 68% and 59% identity, and one for ompF with 63% identity (Supplemental Fig. S3B). Induction, in the respective strain, did not lead to reduced expression of the potential off-target genes overall nor did we see reduced RNA-seq coverage under the partial crRNA matches (Supplemental Fig. S3C–E). In the mulitiplexed versus cspE comparison, none of the three genes with partial matches to lpp or ompF showed statistically significant changes in expression (Supplemental Data Table S5). Drawing these results together, we conclude that any off-target RNA degradation with our type IIIA platform is minor enough that it cannot be easily detected.
DISCUSSION
Gene knockdown technologies such as RNA interference (RNAi), play an important role in gene function discovery (especially for essential genes that cause lethality when knocked out) and therapeutic applications (Kim and Rossi 2008; Wilson and Doudna 2013; Setten et al. 2019). However, these tools are limited to eukaryotes since the RNAi machinery required for small interfering RNA (siRNA) or short hairpin RNA (shRNA) mediated gene knockdown is present in many eukaryotes but absent from prokaryotes. In this study, we sought to harness the type III-A CRISPR system as a post-transcriptional gene knockdown platform that functions in prokaryotic cells. Our proof-of-principle studies demonstrate that type III-A crRNPs from three distinct sources (L. lactis, S. epidermidis, and S. thermophilus) can be conveniently expressed from an “all-in-one” plasmid and can be readily programmed with crRNAs to selectively cleave both mRNA and noncoding RNA in vivo in a heterologous prokaryotic (E. coli) host cell. Moreover, we show that more than one target mRNA can be efficiently cleaved simultaneously when multiple crRNAs are concurrently expressed, paving the way for important applications such as cellular pathway discovery and manipulation. The type III-A gene knockdown technology established here has future potential as an important functional genomics tool for use in a wide range of prokaryotic and potentially even eukaryotic cells.
Programmable knockdown of diverse RNA transcripts
Our findings showed that type III-A systems provide a versatile platform for controlling the levels of endogenous transcripts in vivo at a post-transcriptional level. We found that each of the crRNAs that we designed and tested were highly effective at selectively reducing the steady-state levels of targeted mRNAs when programmed individually (Figs. 1⇑⇑–4, 6) or in a multiplexed fashion (Figs. 4, 6). Furthermore, crRNAs targeting various locations along the length of a mRNA were comparably effective (Fig. 3) showing that cleavage by III-A crRNPs is not restricted to particular regions of mRNA. Because the guide elements of the type III-A crRNAs that we tested are naturally relatively long (35–37 nt), this likely contributes to the observed effectiveness and specificity of targeting in vivo. When multiple crRNAs were expressed simultaneously, a high degree of sequence-specificity was still observed (Figs 4, 6).
In contrast to the efficient targeted destruction of specific mRNAs when several crRNAs were tested, the noncoding RNA component of RNase P (rnpB) proved to be more recalcitrant to type III-A crRNP-mediated RNA cleavage (Fig. 5). While each of the seven crRNAs did reduce ribozyme levels and block pre-tRNA processing to some degree, they were not all equally effective. Of note, the two crRNAs found to be most effective both mapped to regions of the RNA that are predicted to be solvent exposed, based on the three-dimensional structure. These regions are not thought to be engaged in RNA–protein or RNA–tRNA interactions (Fig. 5), implying that those types of interactions can be a barrier to crRNP-mediated cleavage. It remains to be determined if noncoding RNAs will generally be more refractory to III-A-mediated RNA knockdown or if this RNA target, which is particularly highly folded and highly interactive, is an exceptional case. A prudent general strategy for targeting either a specific mRNA or noncoding RNA of interest would be to pursue the multiplexing route. Simultaneous expression of multiple crRNAs against different regions of the desired target RNA molecule is expected to increase the probability of efficient cleavage and gene knockdown.
Target RNA cleavage products accumulate in vivo
The observation that target RNA cleavage products of the expected sizes relative to the site of crRNA interaction were readily detectable provides strong evidence that the destruction was directed by type III-A crRNPs. However, this phenotype was surprising given a priori expectations that cleavage of the phosphodiester bonds within RNA polynucleotide chains would lead to rapid degradation of the cleavage fragments, particularly since mRNAs in E. coli have short half-lives in general (typically 3–5 min) (Bernstein et al. 2002). It is unclear why RNA fragments located both 5′ and 3′ to the site of cleavage persisted and were readily observable under steady-state conditions. Type III crRNP cleavage results in RNA products with 5′ hydroxyl and 2′,3′-cyclic phosphate moieties rather than more typical 5′ phosphate and 3′ hydroxyl ends created by the action of other RNases (Hale et al. 2009; Zhang et al. 2016). It is possible that these particular RNA chemical end groups impede further RNA turnover by E. coli exoribonucleases. Alternatively, the unexpected stability of the 5′ and 3′ degradation fragments might result from the III-A crRNP sterically protecting these ends from being recognized and destroyed by cellular ribonucleases. However, strong in vitro evidence revealed that type III crRNPs normally rapidly dissociate from target RNAs following cleavage making this possibility less likely (Estrella et al. 2016; Rouillon et al. 2018).
RNA knockdown is highly specific
The ideal RNA knockdown platform would not only efficiently degrade target RNA, it would also be highly specific, with no loss of transcripts other than the one targeted by design. Our 5S integrity data and our transcriptome-wide expression profiles both suggested that there is little to no off-target interference with this platform (Figs. 1, 2, 6E). Several genes in the rpoE-rseABC regulon were decreased to about the same degree as some of the target genes (lpp, ompF, and lacZ) in the multiplexed crRNP (Fig. 6E). However, this reduction is likely due to an indirect effect related to knockdown of lpp, which encodes a lipoprotein, and/or ompF, which encodes an outer membrane protein. When outer membrane proteins are misfolded or mistrafficked, they activate the intermembrane protease DegS. DegS cleaves the anti-σ factor RseA, leading to activation of σE. σE acts as a transcription factor and induces expression of chaperones to help fold OMPs, transport machinery to place beta-barrel proteins in the membrane, and periplasmid proteases that degrade misfolded outer membrane proteins (Ge et al. 2014; Gottesman 2017; Hews et al. 2019). DegP is one such protease induced by σE, and it has been shown to degrade OmpF (Ge et al. 2014). Although DegP protein is functionally linked to rpoE-rseABC, it is not part of the operon and is located elsewhere in the genome. Therefore, the coupled loss of both degP and rpoE-rseABC RNA would have to arise from two separate off-target events in their respective transcripts, a single off-target event that then precipitated a change in expression for the other member(s) of the regulon, or it would have to arise as a functional response to loss of Lpp and/or OmpF. Misfolded OmpF and perturbation of the cell envelope, of which Lpp is a part, both stimulate the rpoE-rseABC mediated membrane stress response (Hews et al. 2019). Given that the rpoE-rseABC regulon is known to interact with OmpF and Lpp, we believe that the last possibility is the most likely. We speculate that loss of OmpF and Lpp induces down-regulation of the rpoE-rseABC regulon in order to limit further loss of these outer membrane proteins through, for example, degradation by DegP.
Although it is possible that decreased expression of rpoE-rseABC and degP was due to off-target interference, we found no matches between these genes and our guide crRNAs that could explain such targeting. We did identify three partial matches between our designed crRNAs and other gene transcripts; however, none of the three genes appeared to be knocked down upon crRNP induction (Supplemental Fig. S3; Supplemental Table S5). It would appear then, that although type III systems may be less sensitive to mismatches compared to other CRISPR-based platforms (Manica et al. 2013; Pyenson et al. 2017), matches with identity of near or <70% do not lead to off-target interference in our system.
Comparison with other RNA targeting CRISPR systems
Most types of CRISPR systems (types I [Cas3], II [Cas9], V [Cas12]) act through crRNA guided Cas nucleases that destroy DNA targets (Hille et al. 2018; Makarova et al. 2019). In contrast, type III (Csm3 and Csm4) and type VI (Cas13) CRISPR systems naturally recognize and target destruction of RNA substrates. Thus, there has been a recent push to develop CRISPR-based systems to fulfill the need for RNA targeting research tools with novel applications (Terns 2018; Smargon et al. 2020).
Previous work showed that endogenous type III-B (also known as Cmr, Haft et al. 2005) systems could be programmed with engineered crRNAs to guide destruction of target RNAs in hyperthermophilic archaea including Pyrococcus and Sulfolobus species (Hale et al. 2012; Zebec et al. 2014; Liu et al. 2018). In these systems, a CRISPR mini-array plasmid supplies the engineered crRNAs and the endogenous III-B crRNPs carry out the knockdown. The optimal growth temperatures of these extremophilic organisms are between 70°C and 100°C, making it unlikely that their type III-B system would work if transferred into mesophilic host cells. The type III-A systems that we have established in this work could potentially function in a broader range of prokaryotes, particularly mesophiles (e.g., those growing around 37°C) which could be of commercial and medical interest. We also expect that the platform would be widely applicable, given that the entire system (crRNP and crRNAs) has been encoded on one plasmid.
Type VI (Cas13-based) systems have also been developed as RNA targeting tools (Abudayyeh et al. 2017; Konermann et al. 2018; Wessels et al. 2020). However they come with a major potential drawback: Cas13 RNase is known to cleave both the target RNA as well as “bystander” RNAs, that is, cellular RNAs can be degraded in a sequence-nonspecific fashion, following activation by the crRNA-target RNA interaction (Gootenberg et al. 2017; Smargon et al. 2017; Konermann et al. 2018; Meeske et al. 2019; Wang et al. 2021). While the type III-affiliated Csm6 (III-A) and Csx1 (type III-B) ribonucleases are also known to induce collateral RNA destruction (Gootenberg et al. 2018; Rostol and Marraffini 2019), these proteins work in trans and are not required for the sequence-specific RNA cleavage mediated by the type III crRNPs (Hale et al. 2012, 2014; Tamulaitis et al. 2014). In this work, we circumvented collateral RNA cleavage in several ways. First, we prevented Csm6 RNase activity through mutation of the Csm1 Palm motif essential for cOA generation and Csm6 RNase activation. We also achieved this same effect by deleting the csm6 gene from the expression plasmids (Supplemental Fig. S2). We propose that the Δcsm6, Csm1-HDmut + Palmmut crRNP would be the best option for future studies and applications as the potential for collateral DNA and RNA damage has been inactivated and target degradation is still comparable to that of the WT crRNP (Fig. 6D).
Future promising applications for type III-A systems
Further studies are required to understand the full potential of the type III-A crRNPs for effective post-transcriptional gene knockdown, as well as to develop additional applications. It will be important to determine if these CRISPR research tools are equally functional outside of E. coli, and if so, which prokaryotic and eukaryotic cells/organisms are amenable to this platform. The ability to assemble functional crRNPs through expression of all required components on a single expression plasmid makes this a facile system for testing these possibilities. Recent success with ectopically expressing similarly complex type I crRNPs, consisting of up to six Cas proteins and a CRISPR RNA, in human cells is encouraging. The type I crRNPs were capable of genome editing/transcriptional control in vivo (Cameron et al. 2019; Pickar-Oliver et al. 2019) which offers hope that type III-A systems will also be useful for in vivo applications in eukaryotic cells. Also of note, a rare Type III-E system that uses a multidomain, single effector Cas protein, was recently established as a promising programmable RNA knockdown/editing tool in human cells (Catchpole and Terns 2021; Ozcan et al. 2021; van Beljouw et al. 2021).
The ease by which both type III-A and III-B crRNPs can be expressed and purified as functional complexes (Hale et al. 2012; Staals et al. 2013, 2014; Tamulaitis et al. 2014; Osawa et al. 2015; Elmore et al. 2016; Estrella et al. 2016; Kazlauskiene et al. 2016; Foster et al. 2018; Dorsey et al. 2019; You et al. 2019; Sridhara et al. 2022; Zhang 2022) offers additional opportunities for delivering programmed, preassembled crRNPs directly into cells. Of note, S. thermophilus type III-A crRNPs expressed and purified from E. coli have recently been shown to efficiently and specifically knockdown maternal mRNAs when microinjected into early zebra fish embryos (Fricke et al. 2020). Moreover, as with type VI (Cas13-based) systems, there is the potential to expand beyond RNA destruction using ribonuclease defective type III-A crRNP variants (through inactivating point mutations to create an RNase-defective or dCsm3 subunit) (Supplemental Fig. S2; Tamulaitis et al. 2014; Samai et al. 2015) as well as effector domain fusions. With these modifications, the type III-A platform could be harnessed to influence target RNA splicing, base editing, translation, degradation, and to track the intracellular localization of the transcripts using fluorescent-based microscopy of GFP-fusion systems (Terns 2018; Smargon et al. 2020). Purified type III-A crRNPs also offer potential to expand proven CRISPR-based powerful molecular diagnostic tools (so far used with Cas13 and Cas12) capable of detecting viral and bacterial pathogens and cancer mutations from patient fluids (Gootenberg et al. 2017, 2018; Santiago-Frangos et al. 2021; Sridhara et al. 2021; Steens et al. 2021). In summary, the work described here provides an important step in the direction of harnessing the potential of type III-A systems as versatile RNA-targeting CRISPR-based research tools with important future applications.
MATERIALS AND METHODS
Plasmid construction
pCsm plasmids for expressing L. lactis, S. epidermidis, or S. thermophilus type III-A crRNPs were described previously (Ichikawa et al. 2017; Foster et al. 2018) with the new crRNAs designed to be complementary to a particular target RNA region. For each crRNA guide sequence (Supplemental Table S1), a pair of complementary oligonucleotides, 35 bp for L. lactis and S. epidermidis, 39 bp for S. thermophilus, and 37 bp for the negative control (Supplemental Table S2), was designed with 4 nt 5′ overhangs that match 5′ overhangs of the pCsm vector left by linearization with BbsI (NEB). An amount of 10 pmoles of each oligonucleotide set were annealed in 1× CutSmart Buffer (NEB) and 0.1 pmole of the annealed products were ligated with 50 ng of the linearized pCsm vector with T4 DNA ligase (NEB). The multispacer array was constructed by amplification of individual spacer arrays with oligonucleotides that were appended with BsaI (NEB) restriction sites such that PCR fragments could be combined in a Golden Gate Assembly (Supplemental Table S2). The ligation and assembly reactions were used to transform chemically competent TOP10 E. coli cells (Thermo Fisher). Plasmids were purified (ZymoPURE Miniprep, Zymo Research) and verified by DNA sequencing before transformation of chemically competent BL21-AI E. coli cells (Invitrogen). The BL21-AI E. coli expression strain has a T7 RNA polymerase under the control of an arabinose inducible promoter.
Splicing overlap extension PCR was used to generate the L. lactis Csm1 Palm (D576A, D577A) and HD (and H13A, D14A) motif mutations as well as the S. thermophilus Csm1 Palm (D575A, D576A) motif. The gel purified DNA fragments were digested with PspXI and NdeI (L. lactis) or BamHI and NdeI (S. thermophilus) before ligation into linearized pCsm vector. L. lactis Csm3 D30A, ΔCsm6, and Csm6 H360A mutations were transferred from previously generated constructs (Foster et al. 2018) by digestion and ligation. The oligonucleotide sequences used to generate the mutant constructs are provided in Supplemental Table S3 and all mutations were verified by DNA sequencing.
Type III-A crRNP expression
Single E. coli colonies were grown at 37°C shaking in Miller's lysogeny broth (Invitrogen) until reaching an OD600 of 0.1 when they were induced with a final concentration of 10 mM arabinose to express Csm1-6, Cas6, and crRNA. After 120 min of induction, 1–1.5 mL of each culture was centrifuged, the supernatant was aspirated, and the cell pellets were flash frozen in a dry ice and ethanol bath and stored at −80°C until RNA extraction.
Northern analysis
Total RNA was prepared using the RNAsnap protocol (Stead et al. 2012). RNA used in Figure 5 was further purified by phenol chloroform isoamyl alcohol extraction (pH 4.5) and ethanol precipitation. The RNA samples were quantified using a Qubit Fluorometer 2.0 (Thermo Fisher) and equal amounts of RNA (5–10 µg) were heat denatured for 5 min at 95°C immediately before electrophoresis on 6%–8% polyacrylamide, 8M urea gels in TBE buffer (89 mM Tris base, 89 mM Boric acid, 2 mM EDTA, pH 8.0) at 400 volts. 5′-radiolabeled, molecular weight markers were RiboRuler Low Range RNA Ladder (Thermo Scientific) or 10 bp DNA Ladder (Invitrogen). The RNA was transferred by electro-blotting with a semidry transfer apparatus (Bio-Rad Trans-Blot SD) to positively charged nylon membrane (Nytran SPC, Whatman). ULTRAhyb buffer (Invitrogen) was used for hybridization with oligonucleotides 5′-end labeled with 6000 Ci/mmol γ-32P ATP (PerkinElmer) using T4 polynucleotide kinase (NEB). One million counts per minute of radiolabeled probe was added for each mL of hybridization buffer. Hybridization was performed at 42°C for 12–16 h. Membranes were washed of unbound probe with a prewarmed (42°C) 2× saline-sodium citrate (SSC) buffer and detected by phosphor imaging (Storm 840, GE Healthcare).
RNA-seq analysis
To evaluate RNA expression patterns, RNA sequencing was done on cultures grown either with or without arabinose induction. Briefly, cultures were pelleted, decanted, and frozen at −80°C. Pellets were thawed and resuspended directly in lysis buffer and RNA was isolated using the PowerBiofilm RNA Isolation kit (Qiagen). Stranded, total RNA libraries (without rRNA or tRNA depletion) were prepared using the Illumina TruSeq kit and were sequenced on an Illumina NextSeq instrument, generating paired 2 by 75 bp reads. Reads were demultiplexed by index, adapter trimmed, and aligned to the reference sequences (E. coli BL21 chromosome and type III-A crRNP encoding expression plasmid) by bowtie2 (Langmead and Salzberg 2012). The alignment outputs were then sorted and processed (Li et al. 2009; Quinlan and Hall 2010) to generate custom genome browser tracks viewable on the University of California Santa Cruz Genome Browser (https://genome.ucsc.edu). To determine the RNA-seq read density over crRNA target sites, we used bedtools (Quinlan and Hall 2010) to count all reads with alignment coordinates overlapping that of the protospacer. To look for on-target and off-target changes in transcript abundances in a genome-wide unbiased manner, we took the aligned RNA-seq data and generated read count matrixes using a custom python script and the available gene annotations for E. coli BL21. The read count matrix was then imported into DESeq2 (Love et al. 2014) and samples were evaluated for changes in gene expression.
To identify partial matches for the programmed crRNAs, each sequence was aligned to the E. coli BL21 reference sequence using bowtie2 (Langmead and Salzberg 2012) with the following options: ‐‐local ‐‐all -D 20 -R 3 -N 1 -L 8 -i S,1,0.50 ‐‐all ‐‐score-min G,8,6. This returned three partial matches in three different genes; the genes and locations were examined on the UCSC Genome Browser tracks to look for differences in RNA-seq coverage.
SUPPLEMENTAL MATERIAL
Supplemental material is available for this article.
ACKNOWLEDGMENTS
We are grateful to Sidney Kushner and Bijoy Mohanty for expert technical advice, helpful discussions, and probe sequence recommendations. We thank members of the Terns laboratory for helpful discussions and Rebecca Terns for her early mentorship role. This work was supported by the National Institutes of Health (R35GM118160) to M.P.T. and (R35GM118140) to B.R.G.
Footnotes
-
Article is online at http://www.rnajournal.org/cgi/doi/10.1261/rna.079206.122.
- Received April 21, 2022.
- Accepted May 9, 2022.
This article is distributed exclusively by the RNA Society for the first 12 months after the full-issue publication date (see http://rnajournal.cshlp.org/site/misc/terms.xhtml). After 12 months, it is available under a Creative Commons License (Attribution-NonCommercial 4.0 International), as described at http://creativecommons.org/licenses/by-nc/4.0/.








