
Comparative study on tertiary contacts and folding of RNase P RNAs from a psychrophilic, a mesophilic/radiation-resistant, and a thermophilic bacterium
- Michal Marszalkowski1,3,
- Andreas Werner2,4,
- Ralph Feltens1,5,
- Dominik Helmecke1,
- Markus Gößringer1,
- Eric Westhof2 and
- Roland K. Hartmann1
- 1Philipps-Universität Marburg, Institut für Pharmazeutische Chemie, D-35037 Marburg, Germany
- 2Université de Strasbourg, Institut de biologie moléculaire et cellulaire du CNRS, Architecture et Réactivité de l'ARN, F-67084 Strasbourg, France
- Corresponding author: roland.hartmann{at}staff.uni-marburg.de
Abstract
In most bacterial type A RNase P RNAs (P RNAs), two major loop-helix tertiary contacts (L8–P4 and L18–P8) help to orient the two independently folding S- and C-domains for concerted recognition of precursor tRNA substrates. Here, we analyze the effects of mutations in these tertiary contacts in P RNAs from three different species: (i) the psychrophilic bacterium Pseudoalteromonas translucida (Ptr), (ii) the mesophilic radiation-resistant bacterium Deinococcus radiodurans (Dra), and (iii) the thermophilic bacterium Thermus thermophilus (Tth). We show by UV melting experiments that simultaneous disruption of these two interdomain contacts has a stabilizing effect on all three P RNAs. This can be inferred from reduced RNA unfolding at lower temperatures and a more concerted unfolding at higher temperatures. Thus, when the two domains tightly interact via the tertiary contacts, one domain facilitates structural transitions in the other. P RNA mutants with disrupted interdomain contacts showed severe kinetic defects that were most pronounced upon simultaneous disruption of the L8–P4 and L18–P8 contacts. At 37°C, the mildest effects were observed for the thermostable Tth RNA. A third interdomain contact, L9–P1, makes only a minor contribution to P RNA tertiary folding. Furthermore, D. radiodurans RNase P RNA forms an additional pseudoknot structure between the P9 and P12 of its S-domain. This interaction was found to be particularly crucial for RNase P holoenzyme activity at near-physiological Mg2+ concentrations (2 mM). We further analyzed an exceptionally stable folding trap of the G,C-rich Tth P RNA.
Keywords
INTRODUCTION
Transfer RNAs (tRNAs) are essential adaptor molecules of cellular protein synthesis machineries. They are synthesized as nonfunctional precursor transcripts (pre-tRNAs) with flanking sequences at their 5′- and 3′-ends. Ribonuclease P (RNase P) is an essential enzyme that catalyzes the 5′-end maturation of pre-tRNAs (Guerrier-Takada et al. 1983; Waugh and Pace 1990; Gößringer et al. 2006; Hartmann et al. 2009; Klemm et al. 2016). In the majority of Bacteria, RNase P consists of a catalytically active RNA subunit (P RNA, ∼340 to 400 nt) and a small protein subunit (P protein, ∼13 kDa) that interacts with nucleotides in the pre-tRNA 5′-leader (Hartmann et al. 2009; Klemm et al. 2016).
Bacterial P RNA is a highly structured molecule, with approximately two-thirds of its nucleotides participating in base-pairing interactions. The basic RNA secondary structure, including structural variations, and major tertiary interactions were inferred from phylogenetic/comparative sequence analyses (Brown et al. 1996; Massire et al. 1997, 1998). Deletion studies revealed that only a subset of helices is strictly essential for catalytic function. These helices form the core structure of all bacterial P RNAs, while several peripheral helices that are attached to the conserved core structure display a variable occurrence and can be individually deleted with only moderate effects on catalytic function (Waugh et al. 1989; Darr et al. 1992; Haas et al. 1994; Schlegl et al. 1994; Siegel et al. 1996). Based on the different peripheral structural elements, bacterial P RNAs can be classified into two major architectures termed type A (ancestral, e.g., Escherichia coli) and type B (Bacillus-like) (Haas et al. 1996; Haas and Brown 1998; Brown 1999).
RNase P RNA is formed from two independently folding units (Loria and Pan 1996), the specificity (S-) domain and the catalytic (C-) domain. In bacterial P RNAs of type A architecture, the two functional domains are oriented toward each other with the help of two to three interdomain loop-helix contacts (L18–P8, L8–P4, L9–P1; Fig. 1A–C). Type A RNAs adopt a rather stretched, slightly concave or open umbrella-like overall shape, formed by two layers of helices lying on top of each other. The bound tRNA occupies the position of the umbrella shaft (Reiter et al. 2010; illustrated in Supplemental Fig. S1). The smaller helical layer consists of the peripheral helices P18 and P13/14 that stabilize the stretched P RNA conformation via their tertiary contacts. The P8/P9 stack is at the interface between the C-domain and the more distal parts of the S-domain (P13/P14 and P7/P10/P11/P12). Structurally, the L8–P4 and L1–P9 contacts support the side-by-side arrangement of the P1/P4/P5 and P8/P9 helical stacks (for review, see Gößringer et al. 2021). Among those interdomain contacts, L8–P4 is common and ubiquitous to bacterial P RNAs of type A and B (Massire et al. 1997, 1998). In Archaea, the RNA subunit forms a holoenzyme with five protein subunits (Jarrous and Gopalan 2010), and folding of the P RNA into its active three-dimensional conformation is more dependent on RNA–protein and protein–protein interactions. This led, for example, to the loss of the L8–P4 interdomain contact in archaeal P RNAs of type M present in Methanococcales. Also, the L9–P1 contact, although basically able to form in type M RNAs (Lai et al. 2017), appears to require structural support from interaction of P1/P9 with protein subunits Rpp21/Rpp29 (Wan et al. 2019).
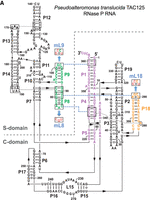
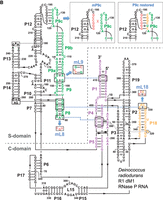
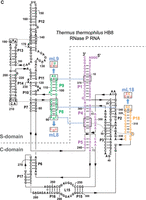
Secondary structure presentation of (A) P. translucida (strain TAC125), (B) D. radiodurans (strain R1 dM1) and (C) T. thermophilus (strain HB8) P RNAs according to Massire et al. (1998), with the three interdomain tetraloop-helix contacts L18–P8, L8–P4, and L9–P1 indicated by dashed blue lines and the involved structural elements in color. Nucleotide exchanges in L8, L9, and L18 (mL8, mL9, and mL18) that disrupt these tertiary interactions are highlighted in red. In the Dra P RNA, the mP9c and “P9c_restored” mutations are illustrated in the boxes. In the P. translucida P RNA (A), the second bp in P1 is shown in small letters to indicate that the bp was mutated from A:U to G:C (for details, see text). Numbering in C is according to Schlegl et al. (1994); numberings in A and B are based on the rnpB genes annotated in the P. translucida strain TAC125 chromosome I (GenBank CR954246.1) and the D. radiodurans strain R1 dM1 chromosome I (GenBank CP031500.1), respectively.
The long-range interactions in P RNAs of type A, originally inferred from phylogenetic covariation analyses, were later confirmed in the P RNA and holoenzyme crystal structures of Thermotoga maritima RNase P (Torres-Larios et al. 2005; Reiter et al. 2010). Yet, our knowledge on the individual contributions of these long-range interactions to P RNA function and how they mutually affect folding of the two domains is still incomplete. To address these questions, we have disrupted the L9–P1, L8–P4, and/or L18–P8 interaction by mutation in three model type A P RNAs: the thermostable, G,C-rich P RNA of the thermophile Thermus thermophilus (Tth), the one from the psychrophilic bacterium Pseudoalteromonas translucida (Ptr) and the P RNA of Deinococcus radiodurans (Dra), a mesophilic, radiation-resistant bacterium phylogenetically related to T. thermophilus. These three type A P RNAs were chosen because of the following outstanding features. Tth P RNA is among the most thermostable P RNAs known. Thermostability of its S-domain was inferred to be achieved by conversion of strategic base pairs to G–C, by decreasing surface accessibility of the native fold and by increasing the amount of structure formation in the native folding transition (Baird et al. 2006). In addition, its two domains form a reinforced L9–P1 contact that contributes to thermostability (Marszalkowski et al. 2008). Dra P RNA deviates substantially form canonical type A structures owing to a P9 stem extension, in which L9 has become an internal loop and where the P9 extension forms a pseudoknot (P9c) with L12, likely to reinforce the S-domain structure (Fig. 1B). The P RNA of the psychrophilic bacterium P. translucida TAC125 (originally named Pseudoalteromonas haloplanktis TAC125; Medigue et al. 2005) may provide hints as to whether adaptation of its host to growth at low temperatures has left traces in the evolution of its P RNA structure. P. translucida grows at temperatures between 4°C and 30°C (Corsaro et al. 2004), D. radiodurans has an optimal growth temperature of ∼ 30°C (Airo et al. 2004; Appukuttan et al. 2006) and T. thermophilus grows between 47°C and 85°C, optimally thriving at 65°C to 72°C (Oshima and Imahori 1974).
To shed light on the role of individual interdomain contacts on P RNA folding and function, we analyzed mutated P RNAs with disrupted interdomain contacts using (i) UV melting to study the effect of the tertiary contacts on RNA folding, (ii) pre-tRNA processing to determine their role for activity, and (iii) complementation of a conditionally lethal E. coli (Eco) P RNA (rnpB) mutant strain to assess their in vivo function in a mesophilic host. We further investigated an exceptionally severe trap in the folding pathway of the thermostable Tth P RNA. The RNA was shown to be unable to function in the mesophilic Eco host and strictly requires a Mg2+-dependent preincubation step at ∼55°C for folding in vitro into its active structure (Marszalkowski et al. 2008). We thus analyzed this folding trap in more depth via folding kinetics and RNA structure probing.
RESULTS
Mutant construction
The L18–P8 tertiary contact was disrupted in the three P RNAs (Ptr, Dra and Tth) by mutating the GNRA tetraloop of L18 to an UNCG loop (Fig. 1). Both loops maintain an interaction between positions 1 and 4 of the tetraloop (Pomeranz Krummel and Altman 1999) to preserve the hairpin-stabilizing ability. The L8–P4 interdomain contact was disrupted by changing the L8 sequence from 5′-AAC to 5′-CGU in the three RNAs (Fig. 1). The UNCG tetraloop mutation was further introduced into L9 of Ptr and Tth (Marszalkowski et al. 2008) P RNAs to disrupt the L9–P1 contact (Fig. 1A,C). In Dra P RNA, the contact between the internal L9 loop and P1 was abolished by changing the bulged A on the 3′-side of L9 to U and by converting the two flanking G•A pairs into G–C pairs (Fig. 1B). For Dra P RNA, we also disrupted the idiosyncratic pseudoknot P9c formed between the apical regions of P12 and the extended P9 element by mutation of the involved L12 nucleotides (Fig. 1B). Disruption of this intra-S-domain interaction enabled us to assess to which extent this interaction contributes to RNA folding and function.
UV melting studies
Folding of P RNA proceeds from an unfolded (U) state, traverses a counterion-dependent intermediate state (I) to reach the final Mg2+-dependent native (N) state. In the I state, the majority of base-pairing and stacking interactions have formed. The transition of the P RNA structure from the I to the U state (and vice versa) can be measured by UV spectroscopy. However, near-UV circular dichroism (CD) is required to study the second transition (I to N) that mainly changes the molecular compactness and helicity parameters (Pan and Sosnick 1997). Mg2+ ions not only contribute to charge shielding, but also occupy specific Mg2+ binding sites. In addition, Mg2+ ions can stabilize kinetic folding traps. As this complicates UV melting profiles, we performed melting experiments in the presence of 100 mM Na+ and absence of Mg2+ to confine the effects of counterions to electrostatic shielding. We also noticed that folding in the presence of Na+ was always more reproducible than with Mg2+, associated with the additional advantage that the RNA could be subjected to repeated heating and annealing cycles while avoiding the risk of metal ion-induced P RNA degradation at high temperatures.
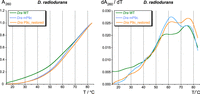
The first derivative dA260/dT curves of the three wild-type (WT) P RNAs (green curves in Fig. 2, profiles on the right) revealed major melting transitions at ∼60°C for Ptr P RNA, at ∼57°C and ∼74°C for Dra P RNA and at ∼75°C for Tth P RNA (Fig. 2A–C). A substantial contribution to the 74°C transition of the Dra P RNA may originate from the long G,C-rich P8/P9/P9a/P9b/P9c stack (Fig. 1B).
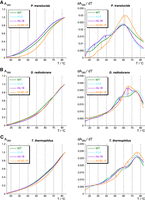
UV absorbance (left) and derivative (right) melting profiles for Ptr (A), Dra (B), Tth (C) P RNAs and mutants thereof, measured in 10 mM sodium cacodylate pH 7.0, 0.5 mM EDTA, and 100 mM NaCl (RNA concentration 25 nM, averages of three to four curves each). Changes in the melting transitions between wild-type (WT) and corresponding mutant P RNAs are indicated by vertical arrows in the first derivative plots on the right.
The effects of mL8 and mL18 mutations (disrupting the L8–P4 and L18–P8 interdomain contacts, respectively) on UV melting profiles depended on the type of P RNA. Both mutations individually had a destabilizing effect on Ptr P RNA, where they caused some partial structures to melt at lower temperatures (approximately between 20°C to 40°C) than in the WT P RNA. The melting profile of Tth P RNA was only marginally affected by either the mL8 or mL18 mutation, while both mutations individually stabilized the Dra P RNA, as inferred from the observed reduced unfolding between 50°C to 57°C (Fig. 2B, profiles on the right). Particularly the mL18 mutation changed the Dra P RNA melting profile to a major peak at ∼64°C in the first derivative curve. However, simultaneous disruption of both interdomain contacts, L18–P8 and L8–P4, stabilized all three P RNAs, in the sense that more structure unfolded concertedly at higher temperatures (Fig. 2, profiles on the right). For Ptr P RNA, this went along with an upshift of the major peak to ∼62°C (mL8/L18) and markedly decreased hyperchromicity at lower temperatures (Fig. 2A). For Dra and Tth P RNA, the double mutation decreased melting of structures in the temperature range of ∼40°C (Dra) or ∼30°C (Tth) to almost 60°C (Fig. 2B,C). Overall, the stabilizing effect upon disrupting the L18–P8 and L8–P4 interdomain contact was most pronounced for Ptr P RNA, the most A, U-rich of the three compared RNAs.
Measurements with the isolated Ptr S-domain revealed that the major unfolding transition of the WT Ptr P RNA (∼59°C) is split into two peaks at 62°C and 72°C, respectively (Fig. 3, right panel). Thus, the Ptr S-domain is more stable than the full-length WT P RNA, and its major transition at 62°C coincides with that of the full-length P RNA carrying the mL8/L18 double mutation (Fig. 3, right panel). The effects of long-range interactions on the UV melting profile are not confined to interdomain contacts. Disruption of the intra-S-domain L12–L9c pseudoknot in the P9c mutant had a similar effect as the mL18 mutation in the context of the full-length Dra P RNA, resulting in a more concerted and upshifted melting transition at 62°C (Figs. 4, 2B, right panels). Restoring pseudoknot formation by introducing compensatory mutations into the P9c mutant again reduced the prevalence of the transition at ∼62°C similar to the WT Dra P RNA (Fig. 4).

UV absorbance (left) and derivative (right) melting profiles of the Ptr P RNA S-domain (brown) compared to the full-length WT (green) and mL8/L18 mutant Ptr P RNA (blue). Assay conditions were the same as in Figure 2.
A folding trap of T. thermophilus RNase P RNA
In vitro transcripts of T. thermophilus P RNA require a preincubation step at temperatures higher than 37°C (e.g., at 55°C) for activation (Hartmann and Erdmann 1991; Marszalkowski et al. 2008). This suggested that the native folding transition has an extraordinarily high activation energy barrier so far unprecedented
among bacterial P RNAs. Here we analyzed the nature of this temperature-dependent folding transition in more detail. The requirement
for a 55°C preincubation step was seen at Mg2+ concentrations between 10 to 100 mM Mg2+ (Supplemental Fig. S2A). In contrast, preincubation of Eco P RNA at 55°C versus 37°C had no or at most a twofold effect, depending on the Mg2+ concentration (Supplemental Fig. S2B). Folding kinetics of Tth P RNA were further investigated at 20 nM P RNA and 200 nM pre-tRNA in the RNA-alone reaction in the presence of 20 mM Mg2+. Preincubation of T. thermophilus P RNA for 15 min at 37°C instead of 55°C resulted in slow substrate turnover and sigmoidal kinetics (Fig. 5, curve a versus b). When the RNA was preincubated for 5 h at 37°C before pre-tRNA addition, the rate of substrate cleavage
increased, but still did not reach that obtained after preincubation for 15 min at 55°C (Fig. 6, curve c versus b). We also analyzed if the presence of the substrate may assist (or inhibit [Pan and Sosnick 1997]) T. thermophilus P RNA folding into an active conformation. After P RNA preincubation for 15 min at 37°C, addition of substrate and incubation
for 285 min (without withdrawal of aliquots), fresh pre-tRNA substrate (200 nM) was added and the reaction monitored for another
285 min by withdrawal of aliquots and analysis by 20% denaturing PAGE. The resulting curve d (Fig. 6) was almost identical to curve c (P RNA preincubated for 5 h min at 37°C), suggesting that the presence of substrate had
little effect on the slow refolding kinetics at 37°C and 20 mM Mg2+. The sigmoidal curve “a” could be fitted to Equation 1 (see below), with kobs describing the pseudofirst order rate constant for cleavage when essentially all P RNAs are folded (as assumed to be accomplished
by preincubation for 15 min at 55°C), k1 the first order rate constant for the slow folding step at 37°C, c the concentration of substrate at any time point, and c0 the concentration of substrate at the start of the reaction.
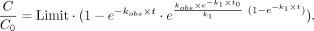 (1)
(1)
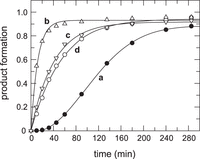
Multiple turnover kinetics of pre-tRNAGly (200 nM) cleavage by T. thermophilus P RNA (20 nM) at 20 mM Mg(OAc)2, 100 mM NH4OAc, 50 mM Hepes, pH 7.0, were conducted at 37°C, but using different preincubation procedures. Curve a: preincubation of P RNA for 15 min at 37°C before substrate addition. Curve b: preincubation of P RNA for 15 min at 55°C before substrate addition. Curve c: preincubation of P RNA for 5 h at 37°C before substrate addition. Curve d: preincubation of P RNA for 15 min at 37°C; after addition of substrate, the reaction was further preincubated for 285 min (4.75 h; without withdrawal of aliquots), whereupon fresh substrate (200 nM) was added (final pre-tRNA plus mature tRNA concentration: 400 nM) and the processing reaction monitored for another period of 285 min.
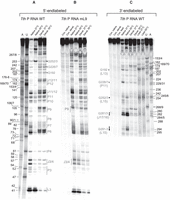
RNase T1 probing of 5′-endlabeled T. thermophilus wild-type (WT) (A) and mL9 mutant P RNA (B), and 3′-endlabeled WT P RNA (C). Lanes A and U: AMPαS- or UMPαS-substituted T. thermophilus WT P RNA used to generate A- and U-specific ladders by iodine hydrolysis; lanes “Native 37°C” and “Native 55°C”: P RNA preincubated for 10 min at 37°C or 55°C in the nondenaturing buffer B before addition of RNase T1 and limited hydrolysis for 10 min at 37°C; lanes “Denat. 37°C” and “Denat. 55°C”: P RNA preincubated for 10 min at 37°C or 55°C in the denaturing buffer A before addition of RNase T1 and limited hydrolysis for 10 min at 37°C; lanes “Con. native” and “Con. denat.”: P RNA preincubated for 10 min at 55°C in buffer B or buffer A, respectively, followed by incubation 10 min at 37°C without RNase T1. Cleavage fragments generated by iodine hydrolysis (lanes A and U) are assigned at the left (A) and right (C) margins according to the T. thermophilus P RNA numbering system shown in Figure 1C; note that shorter fragments of A and U ladders in panel C show double bands, which we attribute to end heterogeneity of the 3′-endlabeled P RNA. In A and C, regions of T. thermophilus P RNA that showed reduced or enhanced RNase T1 accessibility after preincubation at 55°C versus 37°C are marked by gray lettering as well as open and filled circles, respectively, or gray vertical lines; in C, structural elements are given in parentheses on the left, below the indicated G residues; J17/16: junction between P17 and P16. Differences in the RNase T1 protection pattern between T. thermophilus wt and mL9 P RNA are marked by vertical gray-dotted lines in B. The following regions became protected from RNase T1 cleavage upon preincubation of 5′-endlabeled Tth P RNA at 55°C versus 37°C (A): P11 (3′-strand, G226/227), L13 (G192), J11/12 (G127/129/132/134), P10 (114/5, weakly), P9 (G101–104), P6–8 (G74–76; G79–81; G87/88/93), P4 (G60), J3/4 (G55/56), and the 3′-side of P3 (G49/50/51/53). 3′-endlabeled Tth P RNA (C) provided partially overlapping information, such as protection at G226/7 or signal enhancement (filled dots) at G252/3 upon preincubation at 55°C; signal intensities were also increased for cleavages in the 3′-part of L15 (G291–3) and in J17/16 (G281/3).
If the rate of cleavage after preincubation at 55°C (Fig. 5, curve b, kobs = 0.083 min−1) is taken as kobs in Equation 1, a k1 value of 0.0016 min−1 for the folding step at 37°C is calculated. The overall folding rate of Eco P RNA was determined as 0.54 min−1 (Kent et al. 2000), thus being more than 300-fold faster under similar conditions (25 mM Mg2+, 120 mM KCl, pH 7.0, 37°C). We further measured the rate of folding to the native state via processing activity assay as a function of Tth P RNA preincubation time at 55°C. The folding rate was determined as 0.13 min−1 (Supplemental Fig. S3A). When P RNA preincubation was performed at 3 M urea, a folding rate enhancement of up to fourfold was observed (Supplemental Fig. S3B), indicating the presence of one or more kinetic traps along the folding pathway (Pan and Sosnick 1997).
A preincubation step at 55°C for P RNA activation was also required in the holoenzyme reaction and efficient P RNA folding depended on the presence of Mg2+ (4.5 mM) during this step (Supplemental Fig. S4A,B). We also analyzed the effect of the preincubation step performed at 37°C versus 55°C on RNA alone activity for the Tth mL8, mL9, mL18, and mL8/L18 mutant P RNAs (Supplemental Fig. S4C). As for the WT Tth P RNA, substantial activity was only obtained upon P RNA preincubation at 55°C.
Analysis of T. thermophilus P RNA folding by RNase T1 probing
The structure of Tth P RNA was probed by RNase T1 digestion under nondenaturing conditions (Fig. 6). RNase T1 cleaves 3′ of exposed G residues in flexible regions, and it was already shown for the Tth S-domain that accessibility of G residues largely decreases from the I to the N state (Baird et al. 2006). For RNase T1 probing, 5′- or 3′-endlabeled P RNA was preincubated at 37°C or 55°C, followed by probing at 37°C. The following regions became protected from RNase T1 cleavage upon preincubation of 5′-endlabeled Tth P RNA at 55°C versus 37°C (Fig. 6A, gray vertical lines on the right): P11 (3′-strand), L13 (G192), J11/12, P10, P9, P6–8, P4, J3/4, and the 3′-side of P3. 3′-endlabeled Tth P RNA provided partially overlapping information, such as protection at G226/7 or signal enhancement (filled dots) at G252/3 upon preincubation at 55°C; signal intensities were also increased for cleavages in the 3′-part of L15 (G291-3) and in J17/16 (G281/3) (Fig. 6C).
We also analyzed the Tth mL9 mutant RNA (Fig. 1C) by RNase T1 probing as a function of preincubation temperature. The pattern of protection from RNase T1 hydrolysis upon preincubation at 55°C was very similar to that obtained with the WT RNA (Fig. 6B vs. A). The protection of J3/4 upon preincubation at 55°C was less pronounced than for the Tth WT P RNA, suggesting that formation of the L9–P1 contact helps burying these residues more tightly in the catalytic core. This minor difference compared with the WT P RNA is consistent with the moderate activity decrease of the mL9 variant in the RNA-alone reaction at high temperatures (55°C) and at low Mg2+ concentrations in the holoenzyme reaction (Marszalkowski et al. 2008).
In summary, RNase T1 probing revealed substantial structural compaction and surface burial in the S-domain upon preincubation at 55°C, particularly in P6–9, P11 and J11/12. Similar protections from RNase T1 cleavage were observed with the isolated Tth S-domain upon increasing the Mg2+ concentration from 0.05 to 1.6 mM, conditions that were considered to represent the intermediate (I) and native (N) fold of the S-domain, respectively (Baird et al. 2006). The overlap between these previous results and our findings suggests that the folding state of the S-domain within the full-length RNA and before preincubation at 55°C resembles the I state of the isolated S-domain in the study by Baird et al. (2006).
In the C-domain, major compaction occurred in the P6 region (Fig. 6A), suggesting that this structural element forms during the 55°C temperature step. Another area of compaction is the 3′-side of P3, J3/4 and helix P4 near its U bulge in the catalytic core. In addition, RNase T1 accessibility in the P15–17 region changed from the I to the N state (Fig. 6C).
Enzyme kinetic effects upon disruption of L8–P4 and/or L18–P8 interdomain contacts
The functional role of interdomain contacts in Tth, Ptr and Dra P RNAs was analyzed in single turnover kinetic RNA-alone reactions at 37°C (Table 1). Disruption of the L8–P4 contact had little effect on Tth P RNA (1.4-fold decrease in kreact/Km(sto) relative to the WT RNA) but affected Ptr and Dra P RNAs more severely (37- and 92-fold decrease in kreact/Km(sto); Table 1). A similar trend was observed for P RNA variants with a disrupted L18–P8 interaction (approximately six-, 37-, and 28-fold decrease in kreact/Km(sto) for Tth, Ptr and Dra P RNA, respectively; Table 1). For Tth P RNA, the kinetic defect upon disruption of the L18–P8 interaction was more pronounced in cleavage assays performed at 55°C (under otherwise similar conditions; Schlegl et al. 1994) instead of the 37°C uniformly applied in the present study.
Summary of kinetic data of RNA-alone activity assays at 37°C
Simultaneous disruption of the L8–P4 and L18–P8 contacts (variants mL8/L18) impaired activity more severely. Catalytic efficiency [kreact/Km(sto)] for the Tth mL8/L18 variant was now ∼22-fold decreased and ∼430-fold and ∼3000-fold for the Ptr and Dra mL8/L18 variants, respectively (Table 1). Surprisingly, simultaneous disruption of the two interdomain contacts was most detrimental to Dra P RNA function. This may be related to the extension of the P8–P9 stem that is further constrained by the P9c pseudoknot in Dra P RNA. In the absence of the L8–P4 and L18–P8 tertiary contacts, the extended and rigidified P8–P9 stem may be difficult to keep in a productive orientation toward the C-domain or may even lead to steric clashes with the substrate. As a general trend, the thermostable Tth P RNA structure turned out to be more robust than P RNAs from Ptr and Dra, as inferred from the lower sensitivity of the former RNA toward loss of individual interdomain contacts under the conditions tested, in line with previous findings (Marszalkowski et al. 2008). Overall, these findings are consistent with a basic assumption of our model presented in Figure 7, namely that one functional interdomain contact, L8–P4 or L18–P8, can principally maintain domain orientation, while the absence of both contacts severely depletes the fraction of P RNA molecules with a productive domain arrangement.
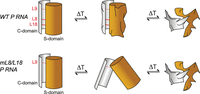
Schematic model to explain the changes in the P RNA melting profiles upon elimination of the L18–P8 and L8–P4 tertiary contacts relative to the wild-type (WT) P RNA.
Analysis of the L9–P1 interaction in Ptr and Dra P RNAs
We extended our studies to the L9–P1 contact in Ptr P RNA. P9 of this RNA harbors a 5′-GCGA-3′ tetraloop that is predicted to dock onto the second and third bp in P1. For cloning and expression purposes we changed the second P1 bp of Ptr P RNA from A:U to G:C (Fig. 1A). We considered it unlikely that this bp exchange would substantially weaken the L9–P1 interaction (see Supplemental Fig. S5 and its discussion in the Supplement). For the Ptr mL9 mutant RNA, we neither saw a defect in the RNA-alone reaction at 37°C (Table 1) nor in the holoenzyme reaction with the heterologous E. coli or B. subtilis P protein (Table 3). This suggested that the P1–L9 interaction does not make a substantial contribution to the overall stabilization of Ptr P RNA at 37°C, in line with the corresponding findings for Eco P RNA (Pomeranz Krummel and Altman 1999; Marszalkowski et al. 2008). A possible explanation is that the L9–P1 contact forms only concomitantly (opportunistically) when the L8–P4 and L18–P8 contacts are in place, but in this native context disruption of the L9–P1 contact remains phenotypically silent. To clarify whether the L9–P1 contact can yet form independently, we compared the kinetic performance of Ptr mL8/L18 double and Ptr mL8/L18/L9 triple mutants. In both mutants, we also restored the native A2:U360 bp in P1 to entirely exclude any effect of the G2:C360 bp exchange on the L9–P1 interaction. Single turnover RNA-alone kinetics under our standard conditions (see Table 1) revealed no significant difference between the Ptr mL8/L18 P RNA used in Table 1 (with bp G2:C360) relative to the Ptr mL8/L18 P RNA with the native A2:U360 bp. However, the Ptr mL8/L18/L9 P RNA triple mutant (with bp A2:U360) showed a twofold increase in Km(sto) and a twofold decrease in kreact (based on ≥11 individual kinetic experiments conducted in parallel for each of the three P RNAs). These findings indicate that the L9–P1 contact can form independently in Ptr P RNA, but its contribution to stabilizing the active fold at 37°C is minor.
Holoenzyme kinetics for P. translucida, T. thermophilus, and D. radiodurans P RNAs and mL9 variants thereof reconstituted with the E. coli or B. subtilis P protein
We further analyzed activity of the Dra mL9 and mP9c mutant P RNAs. The Dra mL9 mutant P RNA showed a minor (1.4-fold) reduction of catalytic efficiency in RNA-alone assays performed at 37°C (Table 1). Likewise, holoenzymes assembled from the Dra mL9 P RNA showed essentially no difference compared with holoenzymes containing the WT P RNA when assayed at 4.5 mM Mg2+, and only moderately reduced cleavage rates at 2 mM Mg2+ (1.5-fold with E. coli RnpA and ∼6.7-fold with B. subtilis RnpA; Table 2). This suggests that the L9–P1 contact forms in this P RNA but makes only a minor contribution to interdomain orientation, as observed for Ptr P RNA. In contrast, much more severe defects were observed for the Dra mP9c mutant RNA particularly in holoenzyme reactions performed at 2 or 4.5 mM Mg2+ (activity losses of three to four orders of magnitude; Table 2).
In vivo complementation analysis
The WT and mutant P RNAs were tested for their capacity to rescue the lethal phenotype of the Eco P RNA (rnpB) mutant strain BW (Wegscheid and Hartmann 2006). Among the heterologous rnpB genes, only that of Ptr was able to rescue growth of the mutant strain when provided on a low copy plasmid (Table 3). The failure of Tth rnpB genes (Marszalkowski et al. 2008) to functionally replace Eco rnpB can be explained by the fact that this RNA of thermophilic origin requires temperatures >>37°C (e.g., 55°C) for efficient folding (see above), suggesting insufficient folding in the E. coli host cultivated at 37°C or 43°C. For Dra P RNA, the failure may be related to its unique architecture in the P9 and P12 regions (Fig. 1), possibly entailing idiosyncratic requirements for the folding process in vivo. This might also be related to expression of a noncanonical P protein in D. radiodurans that is predicted to carry a carboxy-terminal extension according to genome annotations. Hence, we could only test the Ptr mutant P RNAs for complementation in strain BW. Whereas the mL8 and mL8/L18 variants were nonfunctional, a partial rescue of the growth phenotype was seen for the mL18 variant (Table 3). This suggests that, in vivo, the L8–P4 contact is more crucial for P RNA function than the L18–P8 interaction, which is consistent with its presence in both types A and B P RNAs and the observation that the P18 element has been lost in P RNAs of some bacterial clades, such as in the Aquificales and the genus Chlorobium (Haas et al. 1994; Marszalkowski et al. 2006).
In vivo complementation screen in E. coli rnpB mutant strain BW
Full rescue of growth of E. coli strain BW under nonpermissive conditions was observed upon expression of the Ptr mL9 mutant P RNA (Table 3). Remarkably, complementation in E. coli was also successful at 43°C, thus substantially above the maximum growth temperature (30°C) of P. translucida. The finding supports the notion that the L9–P1 interaction is of low relevance for function of Ptr RNase P if the L8–P4 and L18–P8 interactions are in place.
DISCUSSION
The present UV melting analyses have revealed that formation of the L8–P4 and L18–P8 contacts affect structural transitions in one or both P RNA folding domains. Disabling one of the two tertiary contacts has a destabilizing effect in the context of Ptr P RNA, a stabilizing effect on Dra P RNA and a marginal effect on Tth P RNA. Such differential effects on melting profiles of Ptr versus Dra P RNA may be related to some A,U-rich, low stability structural elements in the Ptr P RNA (such as P8 or P18; Fig. 1A) that are stabilized in the native fold. Yet, these partly opposing effects occurred in a context where one of the major interdomain struts (L8–P4 or L18–P8) is assumed to keep a substantial fraction of molecules in the functional interdomain orientation (see below). However, upon simultaneous disruption of both interdomain contacts (mL8/L18 double mutant), interactions between the C- and S-domains are largely lost. The L9–P1 contact is too weak to compensate for the combined disruption of the L18–P8 and L8–P4 contacts. As a result, a uniform overall effect was observed for all three P RNAs: C- and/or S-domains were stabilized such that melting occurred more concertedly with less unfolding at lower temperatures. The net stabilizing effects seen in the melting profiles of the double mutants may have masked minor destabilizing effects, such as those seen for Ptr P RNA upon disruption of the L18–P8 or L8–P4 contact. Since the folding processes are reversible in denaturation and renaturation UV spectroscopy experiments (Supplemental Fig. S6), we conclude that one domain acts as an unfolding chaperone of the other. When the two domains tightly interact, each domain lowers the inherent stability of the other domain (see model in Fig. 7). This may be due to exchange of interactions from intra- to inter-domain contacts, especially, but not solely, at the interaction interface (L18–P8 and L8–P4). More indirect effects are conceivable as well, for example that the tertiary interactions induce deviations in coaxial stackings between helices that are part of helical stacks in P RNA.
In line with the UV melting results, the mL8/L18 double mutation showed the most severe catalytic defect for all three P RNAs, supporting the above assumption that the fraction of P RNA molecules that adopt a productive interdomain orientation is largely decreased in the absence of the L8–P4 and L18–P8 contacts. This depletes the pool of catalytic RNA molecules that are able to position the pre-tRNA substrate for concerted recognition of the tRNA D/T loop by the S-domain and docking of the cleavage site to the active site in the C-domain. The single mL8 and mL18 mutations caused less severe catalytic defects, in line with smaller changes in the UV melting profiles relative to the mL8/L18-double mutant. This supports the notion that maintaining one of the two tertiary contacts still allows a substantial fraction of P RNAs to adopt a productive interdomain orientation. The thermostable Tth P RNA showed the lowest activity decreases upon disruption of the L8–P4, L18–P8 or both contacts. One reason could be its strengthened L9–P1 contact (see below) combined with its reduced conformational flexibility at 37°C, which is well below the growth temperature range of T. thermophilus (∼50°C and 85°C). This may have increased the fraction of mutant P RNAs that adopt an active fold relative to P RNAs of mesophilic or psychrophilic origin.
While we stressed the role of the L18–P8 contact for domain orientation, some previously reported functional and structural consequences of its disruption are noteworthy. For Eco P RNA, deletion of P18 caused a substrate binding (KM) defect in vitro at 25 mM Mg2+ and 1 M NH4+. This defect could be rescued by increasing the NH4+ concentration to 3 M (Haas et al. 1994). Likewise, enzyme activity of Eco P RNA with a mutated L18 loop (5′-UUCG) could be largely rescued by elevating the Mg2+ concentration to 190 mM at 100 mM NH4+ (Pomeranz Krummel and Altman 1999). This suggested electrostatic repulsion effects between enzyme and substrate RNAs upon disruption of the L18–P8 contact. Kirsebom and coworkers probed Eco WT and mL18 (5′-CUUG) P RNA with Pb2+ ions and RNase T1 (Mao et al. 2018). They observed structural changes in P11 and at the junction between P14 and P11 in the S-domain of the mL18 mutant RNA with disrupted L18–P8 interaction. In addition, there was a change in RNase T1 susceptibility at helix P5 in the C-domain, close to the active site. Generally, higher Pb2+ concentrations were required for the mL18 versus WT P RNA to induce backbone hydrolysis at high affinity metal ion binding sites (Mao et al. 2018). This can be explained by a more flexible structure of the mutant RNA, resulting in affinity losses at Mg2+ binding sites. Even charge distribution at the substrate cleavage site was found to be affected by disruption of the L18–P8 interaction. This was analyzed with a substrate carrying a protonable 2′-NH2 group at nucleotide −1 near the canonical cleavage site. The mL18 mutation caused changes in the fraction of substrate that was cleaved at the canonical cleavage (−1/+1) site at different pH values (Mao et al. 2018). The authors proposed that the L18–P8 interaction acts as structural mediator between the substrate (T stem–loop) binding region in the S-domain and the active center of the C-domain. The contact supports the binding of structurally and catalytically relevant Mg2+ ions, supports native P RNA folding particularly in the S-domain and optimizes positioning of chemical groups and catalytic Mg2+ ions during catalysis. Noteworthy, Eco WT and mL18 P RNAs were also analyzed by UV melting in a buffer containing 0.4 M NH4+ and 1 mM Mg2+ (Pomeranz Krummel and Altman 1999). The mL18 mutation had a destabilizing effect on Eco P RNA, as we observed for Ptr P RNA (Fig. 2A), underscoring the resemblance of the two bacterial type A RNAs.
Certain thermophilic bacteria, such as T. thermophilus, have a 5′-GYAA L9 tetraloop and a P1 receptor site consisting of a G–C bp tandem (Massire et al. 1997, Marszalkowski et al. 2006). Such tandem type I/II G–C:A nucleoside triples, where the consecutive A residues interact with the entire (type I) or half (type II) minor groove of the respective G–C bp, represent the most stable combination for this kind of tertiary contact (Doherty et al. 2001). The L9–P1 interdomain contact was shown to contribute to activity of Tth P RNA at elevated temperatures and physiological Mg2+ concentrations. Its disruption by L9 loop mutation prevented formation of a compact conformer observable in native PAA gels (Marszalkowski et al. 2008). In contrast, L9 mutations in Eco P RNA, where two A residues are predicted to interact with a G–U and A–U bp (Massire et al. 1997), had no or little effect on function in vitro and in vivo (Pomeranz Krummel and Altman 1999; Marszalkowski et al. 2008). Likewise, we demonstrated here that a loss of the L9–P1 contact in Ptr P RNA does not abrogate the ability to replace the native Eco P RNA in vivo. Consistent with this finding, the Dra mL9 P RNA showed only a minor reduction in catalytic efficiency in vitro. By testing the mL9 mutation in Ptr P RNA in the context of disrupted L18–P8 and L8–P4 contacts, we were able to carve out that the L9–P1 contact can form independently. However, its functional contribution was found to be minor. This supports the notion that contacts L8–P4 and L18–P8 are most important for maintaining the functional interdomain orientation in bacterial P RNAs of type A. The enhanced functional role of the L9–P1 contact in thermostable P RNAs with a 5′-GYAA L9 and a tandem G–C bp receptor in P1 may also include contributions from the more rigid structural context of the contact. This includes the G–C bp tandem in P1 and the general stabilization of helices, including P1 and P9, by helix extension and/or deletion of nucleotide bulges (Baird et al. 2006; Marszalkowski et al. 2006). This implies that the L9–P1 contact might form more stably in Ptr P RNA at physiological temperatures of its host bacterium (4°C to 30°C) owing to reduced conformational dynamics of the RNA relative to 37°C.
The disruption of the idiosyncratic P9c pseudoknot contact in Dra P RNA moderately affected catalytic performance (approximately sevenfold) in the RNA-alone reaction at high Mg2+ concentration (100 mM; Table 1). However, the mP9c mutation drastically reduced holoenzyme activity at low Mg2+ (∼10−3-fold at 4.5 mM Mg2+, Table 2). Reducing the Mg2+ concentration from 4.5 to 2 mM further reduced holoenzyme activity (≤10−4-fold), indicating that P9c pseudoknot formation is particularly crucial in the holoenzyme context at physiological Mg2+ concentrations.
Although the psychrophilic bacterium P. translucida grows in the range of 4°C to 30°C, its P RNA can functionally replace Eco P RNA in vivo at temperatures above this temperature range (37°C or 43°C, Table 3); likewise, the Ptr P protein was fully functional in B. subtilis at 37°C (Gößringer and Hartmann 2007), showing that Ptr RNase P components are stable at temperatures above the growth temperature limit of the bacterium (30°C). This may suggest that bacterial P RNA ribozymes require a certain minimum overall stability, that is, a minimum number of G–C base pairs, to fold into their functional architecture without the help of protein cofactors. Examples of more A/U-rich P RNAs are only found in eukaryotic organelles, where these RNAs depend on multiple protein cofactors for folding and activity (Schencking et al. 2020). Nevertheless, the Ptr P RNA has a reduced G,C content (∼54%) relative to the mesophilic Eco P RNA (∼62%), suggesting increased structural flexibility of the P RNA as an adaptation to psychrophilic growth, in line with the pronounced changes in UV spectra upon introducing the mL8 and/or mL18 mutation(s) (Fig. 2A). Indeed, several helical elements in Ptr P RNA show hallmarks of reduced stability/increased flexibility (increases in A–U and non-Watson–Crick base pairs, additional bulges; see P1, P3, P8, P18, P11, P12, P17 in Fig. 1A). In this context, we compared the pre-tRNA processing kinetics at assay temperatures of 37°C versus 15°C (Supplemental Tables 1, 2). The cleavage rate constant kreact at 15°C was indeed the highest for Ptr P RNA (compared with Eco, Dra, and Tth P RNAs) in the RNA-alone reaction (Supplemental Table 1), possibly reflecting an adaptation to activity at low temperatures. However, this should be interpreted with caution, because we only measured the processing reaction itself at 15°C, while RNA folding and holoenzyme assembly were conducted at 55°C/37°C. Thus, these experiments did not address effects of low temperature on P RNA and tRNA folding as well as holoenzyme assembly; furthermore, the assembly of the Ptr holoenzyme was performed with the Eco P protein (Supplemental Table 2).
While D. radiodurans R1 optimally grows at ∼30°C, its P RNA has characteristics of a thermostable RNA: its G + C content is ∼69% (71.4% in Tth P RNA) and its processing activity peaks at ∼50°C (that of Tth P RNA at ∼60°C; measured at 25 mM MgCl2 and 1 M NH4Cl). This might contribute to the versatile resistance of the bacterium to stresses including gamma and UV radiation (Setlow and Boling 1965; Battista 1997; Airo et al. 2004), as backbone breaks in thermostable RNAs are less detrimental to RNA function than in mesophilic RNAs. For example, in vitro activity losses of the thermostable Tth P RNA upon Pb2+-induced fragmentation were less pronounced than for Eco P RNA (Ciesiolka et al. 1994).
The group I self-splicing intron is an example of an RNA that first and rapidly folds into a nucleating core structure (the P4/P6 domain), which then serves as a scaffold for folding of its second domain (Doherty and Doudna 1997). In contrast, the two domains of bacterial P RNAs (S- and C-domain) fold in parallel and complete folding of both domains is kinetically indistinguishable (Pan et al. 1999; Kent et al. 2000). RNAs can be classified as slow and fast folding, strongly depending on temperature and ionic conditions. The nature of the rate-limiting step (from the I to the N state) for the slow-folding class can be assigned to disruption of nonnative or prematurely formed native structures (folding traps). In contrast, the fast-folding class is thought to solely involve small-scale conformational changes, for example the rearrangement of prebound metal ions (Fang et al. 2002; Sosnick and Pan 2004, and refs. therein). It was shown for the mesophilic B. subtilis P RNA that the full-length RNA belongs to the class of slow folding RNAs (∼0.2 min−1; measured at 20 mM Tris, pH 8.1, 10 to 20 mM Mg2+ and 37°C; Pan and Sosnick 1997), whereas its isolated C-domain is a member of the fast-folding class (390 min−1; measured at 37.5°C, 20 mM Tris, pH 8.1, 10 mM Mg2+; Fang et al. 1999). The B. subtilis S-domain folds at a rate of 4.5 min−1 (Sosnick and Pan 2004), suggesting that slow folding of the full-length P RNA involves searches for intra- and interdomain contacts during transition to the native structure. Baird et al. (2006) studied the I-to-N transition of the isolated Tth P RNA S-domain by CD spectroscopy relative to the S-domain of Eco P RNA. The Tth S-domain folded at a lower Mg2+ concentration with increased Mg2+ cooperativity and increased denaturant-sensitive surface burial relative to the mesophilic S-domain. Increased surface burial is in line with stronger protection of the Tth S-domain from RNase T1 cleavage when changing from the I to the N state compared with the Eco S-domain (Baird et al. 2006). Folding equilibria for the two S-domains were analyzed at 37°C in the study by Baird et al. (2006) but using an extended time window for the Tth S-domain (up to 70 min) relative to the mesophilic counterpart (up to 9 min). This indicates that already the Tth S-domain folds at a slower rate than the Eco S-domain at 37°C, but its activation energy barrier for the I-to-N transition is clearly lower than for the Tth full-length P RNA that requires a temperature >>37°C for efficient folding (Fig. 5). The high temperature requirement for Tth P RNA folding and the approximately fourfold acceleration of the folding rate by the presence of 3 M urea (Supplemental Fig. S3) indicate that the major activation barrier along the RNA folding pathway is caused by formation of nonnative structures.
These findings are in line with results from previous folding studies of E. coli and B. subtilis P RNAs, showing that formation of P6 and P7 is among the late events in the folding pathway (Zarrinkar et al. 1996; Kent et al. 2000). Backbone discontinuity between P5 and P7 in a circularly permuted B. subtilis P RNA (with the natural 5′- and 3′-ends connected by a loop; termed cp240 RNA) enhanced folding 15-fold at 37°C, supporting the idea of a folding trap involving the P7 region (Zarrinkar et al. 1996; Pan et al. 1999). Tth P RNA in silico folding with RNAfold (default parameters; Lorenz et al. 2011) predicts most secondary structural elements (including P5 at the domain interface) except for the pseudoknots P4 and P6. However, an alternative structure in the S-domain close to the domain interface, involving pairings between 5′-P6/3′-P7 and 5′-P7/3′-P10/P11, is predicted as well (Supplemental Fig. S7). Such a misfold would be consistent with the reported findings cited above.
Our probing experiments showed compactions in several regions of the Tth S-domain upon preincubation at 55°C, prominently in 5′-P6 and 5′-P7 at the junction between the two domains, but also in the C-domain in P3, J3/4 and helix P4 near its U bulge in the catalytic core as inferred from changes in RNase T1 accessibility (Fig. 6). Formation of the L8–P4 contact might be a major factor contributing to this structural adjustment of the catalytic core. We further noticed some changes in RNase T1 accessibility in the P15–17 subdomain (approximately nucleotide 250–295; Fig. 6A,C). This may be interpreted as suggesting that this subdomain is reoriented upon formation of P6. In summary, the Tth P RNA belongs to a small group of natural thermostable RNAs with very high G,C content, resulting in enhanced stability of its secondary and tertiary structure. The RNA is assumed to fold into its active structure without assistance of protein cofactors. The high G,C content and structural stability of the RNA is associated with higher activation barriers that need to be surmounted for proceeding from intermediate folding states to the native fold or to resolve off-pathway folding traps. Here we showed that an intermediate, likely involving nonnative interactions of sequences at the junction of C- and S-domain, can only be resolved at temperatures as high as 55°C. Folding from the intermediate to the native fold resulted in substantial structural compaction and loss of surface accessibilty, in line with the results of the previous study on the isolated Tth S-domain (see above; Baird et al. 2006).
MATERIALS AND METHODS
UV melting
UV melting profiles were recorded on a Uvikon XL UV/Vis spectrophotometer coupled to a Peltier thermosystem (Biotek) and an automated cell changer for 10 cells and two references. The temperature controller was ramped at a rate of 0.5°C/min from 10°C to 90°C, and data points were collected every 0.4°C as determined by a temperature probe inserted into a cuvette containing the final buffer. Samples were measured in 10 mM sodium cacodylate buffer pH 7, 0.5 mM EDTA, and 100 mM NaCl at an RNA concentration of 25 nM. For data analysis, a script was written to convert concatenated Uvikon report text files into separate denaturation and annealing segments for each position. These were subsequently subjected to piece-wise linear interpolation and smoothed over a 5°C window. From this data, the first derivative of absorbance curves (dA260/dT vs. T) were calculated and plotted as a function of temperature, using either Kaleidagraph 4.0 (Synergy Software), or Xmgrace. Xmgrace was also used to fit Gaussian normal distributions to derivative melting profiles. Initial constraints were the number of distributions and the starting Tm, which were iteratively adapted until convergence was obtained.
Construction of plasmids encoding mutant P RNAs used as transcription templates
Transcription templates for mutant P RNAs (pUC19 derivatives) were constructed using standard PCR techniques as described in Li et al. (2009) or using the Stratagene Quikchange site-directed mutagenesis protocol; DNA sequences were verified by dideoxy sequencing (e.g., custom service by Eurofins Genomics).
In vitro transcription and labeling of RNA 5′ or 3′ ends
RNAs were produced by run-off transcription with T7 RNA polymerase and subsequent gel purification as described in Busch et al. (2000). The substrate, Tth pre-tRNAGly, was transcribed from plasmid pSBpt3′HH linearized with BamHI (Busch et al. 2000), Eco P RNA from plasmid pJA2′ (Vioque et al. 1988) linearized with FokI; Tth, Dra and Ptr WT and mutant P RNAs were transcribed from the respective pUC19 derivative plasmids linearized with EheI, BbsI, and FokI, respectively. 5′-endlabeling of pre-tRNAGly and 5′ as well as 3′-endlabeling of P RNAs were performed as detailed in Heide et al. (1999).
Probing experiments
Partial hydrolysis of T. thermophilus P RNA by RNase T1
Limited digestion by RNase T1 was performed by incubation of 3′- or 5′-endlabeled T. thermophilus P RNA (20,000 Cherenkov cpm, also containing 60 ng/µL A. aeolicus 6S RNA as carrier) for 10 min at 37°C or 55°C in buffer A (20 mM sodium citrate/HCl, 0.2 mM EDTA, 7 M urea, pH 5.0) for denaturing conditions or buffer B (50 mM Hepes pH 7.0, 4.5 mM Mg(OAc)2, 0.1 M NH4OAc, pH 7.5 at 37°C) for native conditions. Hydrolysis was started by addition of RNase T1 (Thermo Fisher Scientific) to a final concentration of 0.16 U/µL followed by 10 min incubation at 37°C and ethanol precipitation to stop the reaction.
Iodine-induced hydrolysis of phosphorothioate analog-modified P RNA
For generation of U or A ladders, modified T. thermophilus P RNA was used. Incorporation of ATP-αS or UTP-αS nucleotides (to ∼5% modification) was achieved by run-off transcription (Busch et al. 2000; Heide et al. 2001) using the T7 Y639F mutant RNA polymerase (Sousa and Padilla 1995). Iodine hydrolysis was performed in the presence of 10 mM Tris-HCl pH 7.5, 60 ng/µL of A. aeolicus 6S RNA as carrier RNA and 0.1 mg/mL I2 for 20 min at 37°C.
Probing gels
All probing samples were loaded onto 12% PAA/8 M urea gels (thickness: 0.4 mm; width: 16 cm; length: 40 cm); gels were run at 6–10 mA until xylene cyanol had migrated ∼36 cm from the slot.
Complementation analyses in the E. coli rnpB complementation strain BW
In vivo complementation assays were performed in the E. coli rnpB mutant strain BW with derivatives of plasmid pACYC177 as previously described (Wegscheid and Hartmann 2006); plasmid-borne P RNA coding sequences to be tested were embedded between the native E. coli rnpB promoter and 3′-precursor/terminator region using standard PCR techniques.
Preparation of recombinant RNase P proteins
E. coli and B. subtilis RNase P proteins carrying an amino-terminal His-tag (His-tagged peptide leader: MRGSHHHHHHGS, encoded in plasmid pQE-30 in E. coli JM109) were expressed and purified essentially as described (Rivera-León et al. 1995).
Enzyme kinetics
For single-turnover RNA-alone reactions (Table 1), trace amounts (≤1 nM) of 5′-32P-endlabeled T. thermophilus pre-tRNAGly substrate were preincubated in reaction buffer C (100 mM Mg[OAc]2, 100 mM NH4OAc, 0.1 mM EDTA, 50 mM MES, pH 6.0 at 37°C) for 5 min at 55°C and 25 min at 37°C. P RNAs (varied in the range of 0.1 to 15 µM) were preincubated in the same buffer for 5 min at 55°C and 55 min at 37°C. Processing reactions were started by combining enzyme and substrate solutions and assayed at 37°C.
Multiple turnover reactions catalyzed by Tth P RNA were performed in buffer D (20 mM Mg[OAc]2, 100 mM NH4OAc, 50 mM Hepes, pH 7.0), using 20 nM P RNA and 200 mM pre-tRNAGly, if not stated otherwise, with P RNA preincubation conditions specified in the figure legend. For reaction conditions in Supplemental Figures S2, S3, see the respective figure legends.
For cleavage assays with reconstituted RNase P holoenzymes, buffer KN (20 mM Hepes-NaOH, 2 mM or 4.5 mM Mg[OAc]2, 150 mM NH4OAc, 2 mM spermidine, 0.05 mM spermine, and 4 mM β-mercaptoethanol, pH 7.4) (Dinos et al. 2004) was used to closely mimic physiological conditions. In vitro reconstitution of RNase P holoenzymes was performed as follows: P RNAs were incubated in buffer KN for 5 min at 55°C and 50 min at 37°C, after which RNase P protein was added, followed by another 5 min at 37°C before addition of substrate. Cleavage assays were performed at 37°C. Aliquots of the cleavage reactions were withdrawn at various time points and analyzed by electrophoresis on 20% polyacrylamide/8 M urea gels. Data analysis and calculations were performed essentially as previously described (Busch et al. 2000). Briefly, gels were subjected to phosphorimaging and radioactive bands representing substrate and 5′-cleavage product were quantified with the AIDA (raytest) image analysis software. First-order rate constants of cleavage (kobs) were calculated by fitting the data to the equation for a single exponential: fcleaved = fendpoint × (1 − e−kobs×t), where fcleaved = fraction of substrate cleaved, t = time, fendpoint = maximum cleavable substrate (Grafit version 5.0.13, Erithacus Software). For determination of single turnover Vmax (kreact) and KM [KM(sto)] values, kobs values measured at five different enzyme concentrations (based on at least three replicate experiments each) were plotted against the enzyme concentration; kreact and KM(sto) were obtained by fitting the data to a “Michaelis–Menten-like” enzyme kinetics model: kobs = kreact × [P RNA]/(KM(sto) + [P RNA]). For experimental conditions in Supplemental Figure S4, see figure legend.
SUPPLEMENTAL MATERIAL
Supplemental material is available for this article.
ACKNOWLEDGMENTS
This work was supported by the German Research Foundation (DFG), grant HA1672/19-1 to R.K.H.
Footnotes
-
Article is online at http://www.rnajournal.org/cgi/doi/10.1261/rna.078735.121.
-
Freely available online through the RNA Open Access option.
- Received March 2, 2021.
- Accepted June 30, 2021.
This article, published in RNA, is available under a Creative Commons License (Attribution-NonCommercial 4.0 International), as described at http://creativecommons.org/licenses/by-nc/4.0/.








