
Properties of small rRNA methyltransferase RsmD: Mutational and kinetic study
- Olga V. Sergeeva1,
- Irina V. Prokhorova1,
- Yerdos Ordabaev1,
- Philipp O. Tsvetkov2,
- Petr V. Sergiev1,3,
- Alexey A. Bogdanov1,
- Alexander A. Makarov2 and
- Olga A. Dontsova1
- 1Lomonosov Moscow State University, Department of Chemistry and A.N. Belozersky Institute of Physico-Chemical Biology, Moscow 119992, Russia
- 2Engelhardt Institute of Molecular Biology, Russian Academy of Sciences, Moscow 119991, Russia
Abstract
Ribosomal RNA modification is accomplished by a variety of enzymes acting on all stages of ribosome assembly. Among rRNA methyltransferases of Escherichia coli, RsmD deserves special attention. Despite its minimalistic domain architecture, it is able to recognize a single target nucleotide G966 of the 16S rRNA. RsmD acts late in the assembly process and is able to modify a completely assembled 30S subunit. Here, we show that it possesses superior binding properties toward the unmodified 30S subunit but is unable to bind a 30S subunit modified at G966. RsmD is unusual in its ability to withstand multiple amino acid substitutions of the active site. Such efficiency of RsmD may be useful to complete the modification of a 30S subunit ahead of the 30S subunit’s involvement in translation.
Keywords
INTRODUCTION
Ribosomal RNA is modified in all living organisms. Modified nucleotides are clustered in the most functionally important centers of the ribosome (Urlaub et al. 1997). Complexity of an organism correlates with the number of modified residues in rRNA (Decatur and Fournier 2002). The majority of modified nucleotides in Escherichia coli rRNA are various types of base and ribose methylated residues (Sergiev et al. 2011a). Methyltransferases modify rRNA at certain stages of the assembly. Some of such enzymes could act on completely deproteinized rRNA in vitro (Gu et al. 1999; Tscherne et al. 1999; Lesnyak et al. 2006; Sergiev et al. 2006), while others could modify assembled subunits (Weitzmann et al. 1991; Lesnyak et al. 2007). There are some documented examples of modification enzymes that have preferences for assembly intermediates (Wrzesinski et al. 1995; Desai and Rife 2006; Connolly et al. 2008; Sergiev et al. 2008; Xu et al. 2008) and, among them, one protein that prefers associated 70S ribosomes (Ero et al. 2008). It is amazing how rRNA modification machinery is built into the general pathway of ribosomal subunits’ assembly and maturation. For the methyltransferases that should act at early assembly stages, it seems that the main problem is to complete modification before ribosomal proteins will bind. For modification enzymes that act on the late assembly intermediates or even on the assembled subunits, the principal problem might be to complete modification before the involvement of incompletely mature subunits in translation.
Our study is devoted to a very unusual enzyme—rRNA methyltransferase RsmD. The target of this enzyme, nucleotide G966 of the 16S rRNA, is located in a deep cleft of the 30S subunit, which is occupied by a P-site bound tRNA anticodon (Weitzmann et al. 1991; Lesnyak et al. 2007). The RsmD enzyme could methylate 16S rRNA only after binding S7 and S19 proteins to rRNA (Weitzmann et al. 1991), and modification proceeds up to a completely assembled 30S subunit (Lesnyak et al. 2007). In contrast to the majority of rRNA methyltransferases, which have separate target recognition and catalytic domains (Sergiev et al. 2007), RsmD is a minimalistic enzyme which has only a small β-hairpin extension (Kumar et al. 2011) in addition to the domain responsible for the catalysis. Nevertheless, it recognizes a single nucleotide among the 4500 nt of rRNA.
A knockout of the rsmD gene is viable, which is indicative of functional activity of the 30S subunits devoid of G966 modification (Lesnyak et al. 2007). However, 30S subunits lacking G966 modification are less effective in translation initiation and could not support some of the gene expression control mechanisms (D Burakovsky, I Prokhorova, O Sergeeva, P Sergiev, P Milon, A Bogdanov, O Dontsova, and M Rodnina, in prep.). Since 30S subunits unmodified at G966 could enter into translation, it is interesting how RsmD could ensure complete modification of all nascent ribosomes before the start of translating mRNAs. Since RsmD occupies nearly the same place on the 30S subunits as the P-site bound tRNA, it should also be important for methyltransferase not to affect the functioning of modified 30S subunits. In this work, we have demonstrated that RsmD tightly binds unmodified 30S subunits but not those after modification. The active center of RsmD could tolerate mutations of the conserved amino acids.
RESULTS
RsmD forms a stable complex with an unmethylated 30S subunit
Recombinant protein RsmD, containing a His6 tag on its N terminus, was prepared from the strain carrying the pCA24yhhF plasmid (Kitagawa et al. 2005). The resulting protein was purified according to SDS PAGE and was used in all of the following experiments. For substrate preparation, the strain lacking the rsmD gene on the chromosome (Baba et al. 2006) was used, while the isogenic wild-type strain was used to prepare the control sample. It was previously shown that assembled 30S subunits are a suitable substrate for pure RsmD (Lesnyak et al. 2007). To test RsmD·30S complex formation, a twofold excess of RsmD was mixed with either 30S subunits or 70S ribosomes in the presence of the unreactive S-adenosyl-L-methionine (SAM) analog, sinefungin. After incubation at 37°C, the complex was subjected to sucrose density ultracentrifugation (Fig. 1A). Gradient fractions were collected, and the amount of RsmD protein was estimated by immunoblotting (Fig. 1B). The amount of ribosomal protein S4 in the fractions was estimated for comparison (Fig. 1C). A nearly 1:1 stoichiometry of the RsmD and 30S subunit complex could be estimated. No complex formation with the associated 70S ribosomes could be detected (Fig. 1D).

Complex formation of RsmD with 30S subunits devoid of G966 methylation. (A) Fractionation of 30S·RsmD complex by sucrose density gradient ultracentrifugation. Shown is the optical density at 260 nm vs. fraction number starting from 30% to 10% sucrose. (B) Immunodetection of RsmD in the same fractions of sucrose density gradient. Lane K contains 1 pmol pure RsmD. (C) Immunodetection of ribosomal protein S4 in the same fractions of sucrose density gradient. Lane K contains 1 pmol total 30S proteins. (D) Efficiency of RsmD complex formation with 30S subunits and 70S ribosomes devoid of G966 methylation. Efficiency of complex formation was monitored by immunoblotting of the corresponding sucrose density centrifugation fractions.
To determine Kd values, 30S subunits were titrated with various concentrations of RsmD protein in the presence of sinefungin and S-adenosyl-L-homocystein (SAH) (Fig. 2). An almost similar binding of RsmD could be observed for 30S subunits unmethylated at G966 in the presence of SAH (Kd = 41 ± 15 nM) and sinefungin (Kd = 29 ± 5 nM). Small ribosomal subunits, methylated at G966, failed to bind RsmD protein with either cofactor.
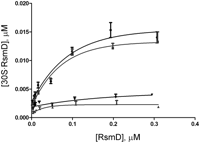
Titration curves of 30S subunits devoid of G966 methylation with the increasing concentration of RsmD. Unmethylated 30S subunits in the presence of sinefungin (squares), unmethylated 30S subunits in the presence of S-adenosyl-L-homocystein (SAH) (circles). Methylated 30S subunits in the presence of sinefungin and SAH (triangles pointing up and down, respectively).
To reveal 16S nucleotides that are in contact with the RsmD protein, we applied the chemical and enzymatic footprinting method. 30S ribosomes devoid of G966 modification and its complex with RsmD and sinefungin were subjected to treatment with DMS, kethoxal, and RNases T1 and T2 (Fig. 3). A short region of the 16S rRNA, proximal to the modification site, was found to be protected by RsmD in the complex.
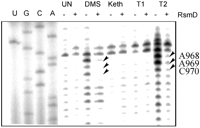
Interaction of 30S ribosomes devoid of G966 methylation with the methyltransferase RsmD in the presence of sinefungin monitored by chemical and enzymatic footprinting. Lanes A, C, G, U correspond to sequencing lanes in the presence of corresponding ddNTP; UN corresponds to untreated 30S subunits of 30S·RsmD complex; DMS, Keth, T1, and T2 each correspond to treatment with dimethyl sulfate, kethoxal, and RNases T1 and T2, respectively; +/− indicates the preincubation of unmethylated at G966 30S subunits with RsmD and sinefungin. Protected bases are marked by arrows.
The model of RsmD interaction with the 30S subunit suggested in our publication (Lesnyak et al. 2007) seems valid. In that model, nucleotides surrounding G966 fit into the small cavity on the RsmD surface near the SAM binding site. Nucleotides of the loop of helix 31 of the 16S rRNA 968–970 protected by the enzyme binding to the 30S subunit were also protected by the S19 protein (Powers and Noller 1995). The latter is essential for RsmD binding to the 30S subunit (Weitzmann et al. 1991).
Kinetic properties of RsmD
To measure kinetic characteristics of RsmD methyltransferase, we used multiple turnover reaction conditions. RsmD protein was taken in a catalytic amount, while unmethylated 30S subunits were taken in excess at varying concentrations. The reaction was started by addition of a RsmD and [3H]SAM solution to the 30S subunits. The reaction was stopped by the addition of RNA extraction buffer containing guanidine chloride at various time points, starting from 30 sec to 24 min. Methylated 16S RNA was extracted, and the amount of radioactive [3H]methyl groups included in the 16S RNA was measured by scintillation counting. The obtained data were used to calculate initial reaction rates. Lineweaver-Burk linearization allowed to calculate kinetic constants (Fig. 4A). It was found that the Michaelis constant of RsmD is 3.3 ± 0.6 nM, while kcat is 0.028 ± 0.004 min−1.
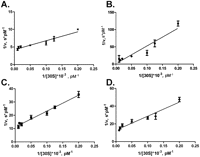
Lineweaver-Burk linearization of the Michaelis–Menten kinetics of the 30S methylation by (A) RsmD, (B) D58A, (C) PP128-129AA, (D) D58A/ PP128-129AA.
Thermodynamic characteristics of RsmD binding to SAM, SAH, and sinefungin
RsmD interacts with the methyl group donor, SAM, and after methyl group transfer to the 16S rRNA, SAH left in the active center of RsmD should exchange for another molecule of SAM. The SAM binding center of methyltransferase could be occupied with the competitive inhibitor, sinefungin, which could not serve as a source of a methyl group. To determine dissociation constants and thermodynamic characteristics of RsmD interaction with these small ligands, we applied isothermal titration calorimetry (ITC).
Ligand-free RsmD solution was titrated with SAM, SAH, or sinefungin solutions in the same buffer (Fig. 5A). SAM binding was shown to be tightest, with Kd = 0.58 μM, while SAH binds four times less tightly, with Kd = 2.5 μM. Abundance of SAM in E. coli was estimated to be 400 μM, while that of SAH was 1.3 μM (Halliday et al. 2010). Such a difference in binding and metabolite abundance ensures a rapid exchange of SAH to SAM following 30S methylation. The affinity toward sinefungin was shown to be half that of SAH and 8 times less than that of SAM (Fig. 5A).
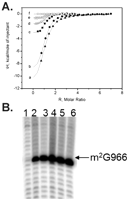
(A) ITC of RsmD interaction with small ligands. Shown are ITC profiles for SAM interactions with RsmD (c) and its mutants: PP129-129AA (d), D58A (e), and D58A/PP129-129AA (f), and of RsmD interactions with Sin (a) and SAH (b) at 25°C in the buffer 20 mM Hepes K pH 7.5, 5 mM Mg(OAc)2, 200 mM NH4OAc, 10% glycerol, 5 mM DTT. (B) Reverse transcription analysis of the total rRNA extracted from the knockout RsmD strain transformed with the plasmids coding for the following rsmD mutants: (1) PPF128-130AAS, (2) F130S, (3) PP128-129AA, (4) D58A, (5) D58A/PP128-129AA, (6) no mutation (control).
Binding of SAM is driven by both an enthalpy decrease and entropy increase (Table 1), indicating a significant contribution of all types of interactions, including hydrophobic, possibly provided by both adenine and methyl groups. Removal of the methyl group significantly affects the entropy change, making it negative, which could indicate the significance of the methyl group in hydrophobic interactions. The unfavorability of the binding change in ΔS was partially compensated by the gain in −ΔH (Table 1). Sinefungin binding appeared to be even more enthalpy-driven, with a large decrease in entropy (Table 1).
Properties of RsmD and its mutant forms
Mutational analysis of the RsmD methyltransferase
RsmD belongs to the family of Rossmann fold N-methyltransferases which have a conserved arrangement of active site residues (Sergiev et al. 2007). Two faces of the SAM binding cavity were formed by 127DPPF130 and 58DxFxGxG64 motifs. We introduced mutations into conserved positions 58D, 128P, 129P, and 130F. The following mutations were made: F130S, PP128-129AA, D58A, PPF128-130AAS, and D58A/PP128-129AA. Plasmids encoding mutant proteins were checked to complement knockout of the rsmD gene in vivo (Fig. 5B). Methylation of G966 of the 16S rRNA was monitored by reverse transcription of total RNA, extracted from the corresponding strains. Surprisingly, the majority of active site mutants retained the ability to form the m2G966 residue. The triple PPF128-130AAS mutation alone was found to abolish methylation.
Catalytic properties of RsmD mutants were tested in vitro in a multiple turnover assay (Fig. 4B–D) similar to that of the wild-type RsmD (Fig. 4A). The results of the experiment are presented in Table 1. Substitution of the conserved proline residues PP128-129AA present in all rRNA guanine-(N2)-methyltransferases (Sergiev et al. 2007), surprisingly, had only a marginal effect on the catalytic ability of the enzyme (Fig. 4C; Table 1). Mutation of D58A, both separately (Fig. 4B; Table 1) and in combination with PP128-129AA (Fig. 4D; Table 1), increased Km toward the 30S subunit by two orders of magnitude with only a marginal effect on kcat. Measurement of enzymatic properties of the F130S mutant in vitro was impossible due to its instability in solution.
The ability of mutant forms of RsmD to bind the SAM cofactor was tested by ITC (Fig. 5A). Mutations D58A and PP128-129AA decreased the enthalpic contribution to SAM binding, paralleled by an increase in the entropic contribution, which might indicate an increase in flexibility of SAM in its binding pocket. SAM binding to the enzyme with the combination of mutations D58A/PP128-129AA was impossible to measure due to a very small enthalpy of binding.
DISCUSSION
Ribosomal RNA methyltransferase RsmD operates on the late stage of the 30S subunit assembly (Weitzmann et al. 1991) and could modify a completely assembled 30S subunit (Lesnyak et al. 2007). The modification site of RsmD, nucleotide m2G966 of the 16S rRNA, is located in a deep cleft of the ribosomal decoding center (Berk et al. 2006). Modified m2G966 makes contact with the anticodon of the P-site bound tRNA (Moazed and Noller 1986). In the course of translation, the P-site is constantly occupied by tRNA, and it is hard to imagine how RsmD could modify G966 after the 30S subunit starts a round of translation. On the other hand, since G966 modification gives an advantage for 30S functionality (D Burakovsky, I Prokhorova, O Sergeeva, P Sergiev, P Milon, A Bogdanov, O Dontsova, and M Rodnina, in prep.), but the unmodified 30S subunit, nevertheless, could be involved in translation (Lesnyak et al. 2007), it is necessary to accomplish modification of the nascent small subunit before its involvement in translation initiation. After completion of modification, it is preferable to avoid RsmD interference with 30S subunit functioning.
In this work, we investigated functional properties of RsmD and found it to efficiently bind 30S subunits unmethylated at G966 nucleotide. Dissociation constants for RsmD complexes with unmethylated 30S subunits were found to be in the 30–40 nM range. This showed that binding is approximately two orders of magnitude better than those demonstrated for comparable rRNA methyltransferases RsmC (Sunita et al. 2007), Sgm (Husain et al. 2010), RlmI (Sunita et al. 2008), and ErmC (Maravic et al. 2003). The Michaelis constant of RsmD toward the 30S subunit is much better than that of comparable rRNA methyltransferases, while kcat is comparable to other enzymes of this type. RrmJ, which is responsible for methylation of the 2′-OH of nucleotide U2552 of the 23S rRNA, displays a Km of 0.7 μM relative to the 50S subunits and kcat of 0.064 min−1 (Hager et al. 2002). The methyltransferase ErmC, which has a kcat of 0.066 sec−1 and Km of 2.8 μM, provides erythromycin resistance (Maravic et al. 2003). RsmD methyltransferase has a kcat of 0.028 ± 0.004 min−1 and Km of 3.3 ± 0.6 nM. Given such tight binding of RsmD to the 30S subunits, one might consider the possibility that RsmD binding would interfere with translation initiation. However, we demonstrated how this problem is resolved, since after modification is complete, the 30S subunit loses its ability to bind RsmD. 30S subunit involvement in 70S ribosome formation makes an additional obstacle for RsmD binding.
Binding of SAM, SAH, and an inhibitor, sinefungin, to RsmD was studied with the help of ITC. Dissociation constants of RsmD with small ligands differed within the borders of one order of magnitude. SAM binding was tightest, about four times better than those of SAH. Given the two orders of magnitude prevalence of SAM in the cell, one can imagine that RsmD is predominantly complexed with SAM in vivo, with the rapid exchange of SAH to SAM after modification of the 30S subunit. The dissociation constant for SAM binding to RsmD, 0.58 μM, is comparable to or slightly better than that of other rRNA methyltransferases. The Sgm complex with SAM has a Kd of 18 μM (Husain et al. 2010), the Kd of the RsmC complex with SAM is 5 μM (Sunita et al. 2007), and the Kd of the RlmI complex with SAM is 3 μM (Sunita et al. 2008). This difference might not be very essential for functioning of methyltransferases in vitro, since the intracellular concentration of SAM—400 μM (Halliday et al. 2010)—is much greater than the Kd for all of these enzymes.
In our study, we performed mutagenesis of the RsmD active site (Fig. 6). The binding site for SAM is conserved in Rossmann fold N-methyltransferases (Sergiev et al. 2007). Peptide motifs (D/E)x(G/F)xGxG and (N/D/S)PP(Y/F/W/H), characteristic for all methyltransferases of this class, could be found in the RsmD primary structure (58DxFxGxG and 127DPPF). In a substantial number of examples described in the literature, even single amino acid substitutions of the conserved active site residues resulted in the complete loss of enzymatic activity (Hager et al. 2002; Maravic et al. 2003). However, conserved proline residues PP128-129 of RsmD could be substituted for alanines without catalytic activity loss. Moreover, the PP128-129AA mutation does not perturb Km and kcat to a significant extent. According to ITC experiments, SAM binding to the PP128-129AA mutant of RsmD was not perturbed either. There is a decrease in enthalpic contribution to the binding, while the entropy contribution was increased. Most likely, this reflects an increase in flexibility of SAM in its binding site. Mutation D58A in the conserved SAM binding site does not abolish methyltransferase activity in vivo and has almost no effect on Kd of SAM binding in vitro. This result was unexpected since similar substitution D202A in the RsmC active site abolished SAM binding and reduced catalytic activity by two orders of magnitude (Sunita et al. 2007). Mutation of an equivalent position in DNA methyltransferase EcoRV (Roth and Jeltsch 2001) also inactivated the enzyme. Similar to the PP128-129AA mutation, the D59A mutation increases entropic contribution to binding at the expense of enthalpic and is likely to increase SAM flexibility in its binding site. In contrast to the PP128-129AA mutation, the D58A mutation increased Km of RsmD toward 30S subunits by two orders of magnitude. It is, thus, likely that the D58 residue helps in 30S subunit binding. Another element essential for the 30S subunit binding was found recently in a study on Mycobacterium tuberculosis RsmD homolog Rv2966c (Kumar et al. 2011). This element is the β-hairpin located on the N terminus of the enzyme.

Overview of docked SAM inside the substrate binding site of RsmD protein. Shown are SAM molecule and amino acid residues subjected to site-directed mutagenesis in this work.
Even the RsmD triple D58A/PP128-129AA mutant retains some methyltransferase activity in vivo and in vitro, although it increases Km toward the 30S subunit by two orders of magnitude and displays insignificant enthalpy of SAM binding. Aromatic amino acid residue following the PP128-129 motif was known to be essential for methyltransferase activity of DNA and RNA methyltransferases, in particular EcoRV (Roth and Jeltsch 2001) and ErmC (Maravic et al. 2003). Only the triple PPF128-130AAS mutation resulted in the complete loss of RsmD methyltransferase activity.
Summarizing the results, we can state that RsmD methyltransferase evolved to possess an ability to bind the 30S subunit more tightly than comparable rRNA methyltransferases. This property might be essential for fast binding of unmodified nascent subunits to insure their modification prior to involvement in translation.
MATERIALS AND METHODS
Strains and plasmids
The BW25113 parental strain and JW3430 strain, carrying a kanR cassette inserted at the place of the yhhF gene (Baba et al. 2006), later renamed to rsmD, as well as the pCA24yhhF plasmid (Kitagawa et al. 2005) were kindly provided by Dr. H. Mori.
Protein RsmD preparation
Recombinant N-terminal hexahistidine-tagged RsmD protein was prepared from AG1 E. coli cells, harboring the pCA24N plasmid with the yhhF gene cloned under control of the T5lac promoter (Kitagawa et al. 2005). Cells were grown in LB media at 37°C until A600 0.5 and induced by IPTG, 0.5 mM f.c. After induction, cells were grown for an additional 2 h and lysed by sonication in a buffer of 20 mM Hepes K pH 7.5, 0.1% Triton ×100, 200 mM NH4OAc, 10 mM imidazole. Protein purification on Ni-NTA agarose (Qiagen) was done in the same buffer, containing 10 mM imidazole. After being washed three times with the same buffer containing 40 mM imidazole, the protein was eluted by increasing the imidazole concentration up to 500 mM. Eluted RsmD protein was dialyzed in the buffer containing 20 mM Hepes K pH 7.5, 0.1% Triton ×100, 200 mM NH4OAc, 1 mM Mg(OAc)2, 5 mM β-mercaptoethanol, 10% glycerol. The dialysis was repeated three times to get rid of all SAM, copurified with RsmD. Protein purity was proven by SDS-PAGE. Mutant variants of the RsmD protein were prepared similarly.
30S subunits preparation
The wild-type and JW3430 strain were grown to A600 0.6 in LB media at 37°C. Cells were harvested by centrifugation and lysed by grinding with 2× cell mass of aluminium oxide at 4°C in the cell-opening buffer, 20 mM Hepes K pH 7.5, 100 mM NH4Cl, 10.5 mM Mg(OAc)2, 3 mM β-mercaptoethanol. Cell debris and aluminium oxide were pelleted by 30-min centrifugation at 16,000 rpm in a JA20 rotor. Lysates were subjected to a 16-h ultracentrifugation in a Ti50 rotor at 33,000 rpm to pellet ribosomes. Ribosomes were resuspended in 20 mM Hepes K pH 7.5, 10.5 mM Mg(OAc)2, 500 mM NH4OAc, 7 mM β-mercaptoethanol buffer. Additionally, ribosomes were pelleted through the 30% sucrose cushion with the help of ultracentrifugation in Ti50 at 28,000 rpm for 13 h. 70S ribosomes were prepared by sucrose gradient centrifugation (10%–40%) in a Ti15 zonal rotor at 28,000 rpm for 19 h. After that, 70S subunits were pelleted in Ti50 at 50,000 rpm for 24 h. 30S subunits were isolated by sucrose gradient centrifugation (10%–40%) in a Ti15 zonal rotor at 28,000 rpm for 19 h in the same buffer but with 1 mM Mg(OAc)2. After that, 30S subunits were pelleted in Ti50 at 50,000 rpm for 24 h and dissolved in the buffer containing 50 mM Hepes K, 70 mM NH4Cl, 30 mM KCl, 7 mM Mg(OAc)2 (HAKM7). For rRNA preparation, we used phenol extraction in buffer with 300 mM NaOAc, 0.5% SDS, 5 mM EDTA, followed by ethanol precipitation. The activity of 30S subunits reassociated with 50S subunits was >95% in fMet-tRNAfMet binding.
Complex formation
For complex formation between 30S subunits and RsmD methyltransferase, we mixed 0.5 μM of 30S prepared from the strain lacking the rsmD gene with 1 μM of RsmD in the presence of 5 μM sinefungin in the buffer 50 mM Hepes K, 70 mM NH4Cl, 30 mM KCl, 7 mM Mg(OAc)2 (HAKM7). The mixture was incubated 10 min at 37°C. Following the formation of the complex, ultracentrifugation in a 10%–30% sucrose gradient in SW41 at 22,000 rpm for 19 h was used to isolate the complex. The gradient was fractionated with simultaneous monitoring of optical density at 260 nm. Gradient fractions were analyzed by the method of immunoblotting using anti-His6 antibodies (Qiagen) for RsmD protein detection, while anti-S4 antibodies, kindly provided by Dr. R. Brimacombe, were used for detection of ribosomal protein S4. Similar conditions were used to compare efficiencies of RsmD complex formation with unmethylated 30S subunits and 70S ribosomes, with the sole difference in ultracentrifugation speed being 18,000 rpm.
Calculation of dissociation constant
For the complex formation, we used 10 nM 30S and 5.3, 9.6, 12, 21, 53, 110, 210, and 320 nM methyltransferase RsmD concentrations in the presence of 10 μM SAH or sinefungin in HAKM7 buffer. Mixtures were incubated 40 min at 37°C, and then the complexes were isolated by ultracentrifugation in MLA-80 rotor at 50,000 rpm for the period of 18 h. The pellets were dissolved in HAKM7 buffer. After measurement of optical density at 260 nm, aliquots containing equal amount of 30S subunits were analyzed by ELISA using anti-His6 antibodies (Qiagen). “One site binding” analysis of the program GraphPad Prizm was used for the calculation of the dissociation constant.
Chemical and enzymatic probing
For chemical and enzymatic probing, dimethyl sulfate (DMS), kethoxal, and ribonucleases T1 and T2 were used. The complex of unmethylated 30S subunits (0.4 μM) with RsmD protein (0.8 μM) was formed in the buffer 20 mM Hepes K pH 7.5, 1 mM Mg(OAc)2, 200 mM NH4OAc for 30 min at 37°C. Following complex formation, 20 nM of either DMS or kethoxal 20% solution in ethanol was added, and incubation was continued for 10 min at 37°C. For the enzymatic probing, 1 unit of T1 ribonuclease and 0.1 unit of T2 ribonuclease were used. Reactions were stopped and RNA extracted as previously described (Sergiev et al. 2000). For the reverse transcription, [32P] 5′ end-labeled primer complementary to the 996–1012 nt of 16S rRNA was used.
Multiple turnover methylation of unmethylated 30S subunits by recombinant RsmD
To determine kcat and Km, recombinant RsmD in a 0.5 nM final concentration was rapidly mixed with unmethylated 30S subunits at 5 nM, 8 nM, 10 nM, 20 nM, 50 nM, 80 nM, and 100 nM final concentration in the presence of 1000× excess of [3H]SAM. The control sample was without 30S subunits. Reactions were mixed on a 96-well microtiter plate with the help of the Janus Extended workstation (Perkin-Elmer). Samples were incubated for 30 sec, 1.5, 3, 6, 12, and 24 min. Following the incubation, the reactions were stopped by adding the guanidine chloride-containing buffer for rRNA isolation (Qiagen). After rRNA isolation (RNeasy Mini Kit, Qiagen), all fractions were analyzed by scintillation counting. Obtained data were analyzed using the Michaelis-Menten equations and following linearization by Lineweaver-Burk. Km and kcat of RsmD mutant variants were measured and calculated similarly.
Functional activity of mutant RsmD in vivo
Mutations F130S, PP128-129AA, D58A, PPF128-130AAS, and D58A/PP128-129AA were introduced into the pCA24yhhF plasmid (Kitagawa et al. 2005) coding for RsmD with the help of the QuickChange kit (Stratagene). To monitor G966 methylation in vivo, total RNA was isolated from the cells devoid of rsmD gene on the chromosome, transformed with the pCA24yhhF plasmids containing specified mutations of the active site residues. For the reverse transcription, [32P] 5′ end-labeled primer complementary to the 996–1012 nt of 16S rRNA was used. Reverse transcription was done as previously described (Lesnyak et al. 2007; Sergiev et al. 2011b).
Isothermal titration calorimetry
ITC measurements were carried out on an iTC200 instrument (MicroCal) at 25°C in the buffer, 20 mM Hepes K pH 7.5, 5 mM Mg(OAc)2, 200 mM NH4OAc, 10% glycerol, 5 mM DTT. Aliquots of ligand solution (2 mL) were injected into the 0.2 mL cell containing the protein solution to achieve a complete binding isotherm. The protein concentration in the cell ranged from 10 to 20 μM, and the ligand concentration in the syringe ranged from 0.2 to 0.75 mM. The heat of dilution was measured by additional injections of the ligand after saturation; the values obtained were subtracted from the heat of the complex formation to obtain the effective heat of binding. We fitted the resulting titration curves using MicroCal Origin software (Fig. 5) and determined the dissociation constants (Kd), binding stoichiometry (N), and enthalpy (ΔH) using a nonlinear regression fitting procedure. Consequently, the entropy change (ΔS) was calculated according to the standard equation.
ACKNOWLEDGMENTS
We thank Dr. H. Mori, NIG, Japan, for providing us with the knockout strain JW3430 and pCA24yhhF plasmid, encoding RsmD. We also thank Dr. R. Brimacombe, MPIMG, Germany, for anti-S4 antibodies. This work was supported by grants from the Russian Foundation for Basic Research 10-04-01345-a, 11-04-01314-a, 11-04-01018-a, 11-04-12060-ofi from the Russian Ministry of Science 16.512.11.2108, Federal Agency for Science and Innovations 02.740.11.0706, Molecular and Cellular Biology Program of the Russian Academy of Sciences and Moscow State University Development Program PNR 5.13.
Footnotes
-
↵3 Corresponding author.
E-mail petya{at}genebee.msu.su.
-
Article published online ahead of print. Article and publication date are at http://www.rnajournal.org/cgi/doi/10.1261/rna.032763.112.
- Received February 17, 2012.
- Accepted March 6, 2012.
- Copyright © 2012 RNA Society








