
Translational recoding signals between gag and pol in diverse LTR retrotransposons
Abstract
Because of their compact genomes, retroelements (including retrotransposons and retroviruses) employ a variety of translational recoding mechanisms to express Gag and Pol. To assess the diversity of recoding strategies, we surveyed gag/pol gene organization among retroelements from diverse host species, including elements exhaustively recovered from the genome sequences of Caenorhabditis elegans, Drosophila melanogaster, Schizosaccharomyces pombe, Candida albicans, and Arabidopsis thaliana. In contrast to the retroviruses, which typically encode pol in the −1 frame relative to gag, nearly half of the retroelements surveyed encode a single gag-pol open reading frame. This was particularly true for the Ty1/copia group retroelements. Most animal Ty3/gypsy retroelements, on the other hand, encode gag and pol in separate reading frames, and likely express Pol through +1 or −1 frameshifting. Conserved sequences conforming to slippery sites that specify viral ribosomal frameshifting were identified among retroelements with pol in the −1 frame. None of the plant retroelements encoded pol in the −1 frame relative to gag; however, two closely related plant Ty3/gypsy elements encode pol in the +1 frame. Interestingly, a group of plant Ty1/copia retroelements encode pol either in a +1 frame relative to gag or in two nonoverlapping reading frames. These retroelements have a conserved stem–loop at the end of gag, and likely express pol either by a novel means of internal ribosomal entry or by a bypass mechanism.
Keywords
INTRODUCTION
Retrotransposons and retroviruses (collectively referred as retroelements) have compact genomes ranging from 5 to 10 kbp. Despite their small size, retroelement genomes must produce a variety of gene products required for replication, and this is frequently accomplished through sophisticated translational recoding mechanisms. One frequent site for translational recoding is the boundary between gag and pol, two genes found in all retroelements. gag is the 5′-most gene and encodes structural proteins that form the virus (retroviruses) or viruslike (retrotransposons) particle. pol is located 3′ of gag, and encodes enzymes such as reverse transcriptase, which are required for replication. In most retroelements, there is no independent initiation of pol translation; rather, Pol is expressed as part of a Gag-Pol polyprotein. The production of Gag-Pol is likely important for packaging the Pol products within the particle. Furthermore, the level of Pol relative to Gag is critical for retroelement viability because particle assembly requires many more copies of Gag than Pol (Park and Morrow 1991; Karacostas et al. 1993; Shehu-Xhilaga et al. 2001; Telenti et al. 2002), and therefore Pol is typically expressed at the translational level through deviations from standard decoding mechanisms.
For most retroelements, ribosomal frameshifting is a common strategy employed to express Gag-Pol. In the majority of retroviruses such as HIV, pol is in the −1 frame with respect to gag and overlaps its 3′ end (Jacks et al. 1988). Standard translation results in the synthesis of Gag as the predominant protein; however, a proportion of the ribosomes shift to the −1 frame at the end of gag to produce Gag-Pol. Ribosomal frameshifting occurs at specific mRNA sequences known as frameshifting sites, and frameshifting efficiency is modulated by nearby or distant mRNA cis-elements known as stimulatory signals. Some retroelements, such as the Saccharomyces cerevisiae Ty1 and Ty3 retrotransposons, utilize +1 frameshifting to synthesize Gag-Pol (Belcourt and Farabaugh 1990; Farabaugh et al. 1993). In Ty1, the peptidyl-tRNA slips forward one base, and its ability to do so is strongly influenced by the availability of cognate aminoacyl tRNA for the A-site codon. This tRNA is sparse and corresponds to a hungry codon (Kawakami et al. 1993; Pande et al. 1995).
Stop codon readthrough is a second strategy utilized by retroviruses to synthesize Gag-Pol. In these cases, gag and pol are in the same frame and are only separated by a single stop codon. Whereas the majority of ribosomes terminate to produce Gag, a small proportion of the ribosomes incorporate a standard amino acid in place of the stop codon to produce Gag-Pol. In the case of Murine Leukemia Virus, a pseudoknot 3′ of the gag stop codon is critical for readthrough (ten Dam et al. 1990; Wills et al. 1991) such that approximately 5% of ribosomes insert an amino acid instead of terminating (Philipson et al. 1978; Yoshinaka et al. 1985).
In contrast to the cases of frameshifting and readthrough, some retrotransposons encode gag and pol on a single open reading frame (ORF). For these retroelements, both posttranscriptional and posttranslational mechanisms have been implicated in determining the ratio of Gag to Gag-Pol. copia, a retrotransposon from Drosophila melanogaster, uses alternative splicing to remove pol coding sequences from the mRNA, thereby allowing Gag to be synthesized at higher levels than Pol (Brierley and Flavell 1990). Posttranslational regulation of Gag and Pol has been suggested for Tf1 in Schizosaccharomyces pombe and Ty5 in Saccharomyces cerevisiae. For these elements, Pol is preferentially degraded, thus allowing an excess of Gag and the proper stoichiometry for replication (Levin et al. 1993; Atwood et al. 1996; Irwin and Voytas 2001).
A significant fraction of most eukaryotic genomes is comprised of retroelements. To understand how widespread the various recoding mechanisms are utilized for Pol synthesis, we surveyed retroelement sequences in the completed genomes of Caenorhabditis elegans, D. melanogaster, S. pombe, Candida albicans, and Arabidopsis thaliana. Other retrotransposons in GenBank were also analyzed. Using this data set, we describe the organization of gag and pol reading frames and putative recoding signals. We also report the discovery of a large lineage of plant retrotransposons in which the organization of gag and pol does not allow synthesis of Gag-Pol via any of the above-mentioned recoding mechanisms. Apart from the interest in the retroelements per se, the information provided by this study is relevant for future studies to determine the prevalence and types of recoding in nonmobile cellular genes from diverse organisms.
RESULTS AND DISCUSSION
The retroelement data set
Our retroelement data set has two components. The first is a group of core retroelements that includes annotated retroelements from GenBank. To generate the core data set, heuristic searches of GenBank were performed with keywords such as Ty1, Ty3, gypsy, copia, and retrotransposon. In addition, GenBank was screened with DNA sequences of several elements that represent the Ty1/copia (Pseudoviridae), Ty3/gypsy (Metaviridae), BEL, and DIRS groups. Sequences identified by these means were extracted and used to populate a retroelement database.
The second component of the data set was an expanded group of retroelements recovered from the completed genome sequences of C. elegans, D. melanogaster, S. pombe, C. albicans, and A. thaliana. These genomes were screened by BLAST, using reverse transcriptase (RT) amino acid sequences from each of the four retrotransposon clades (Ty1/copia, Ty3/gypsy, BEL, and DIRS). All BLAST hits were processed by a software package developed in our laboratory called RetroMap. The software compares the sequences upstream and downstream of an RT hit to identify flanking repeats, which are considered putative LTRs. A hit with putative LTRs is then parsed into the database for further analysis. RetroMap identified 478 potentially complete retrotransposons from A. thaliana, 281 from D. melanogaster, 19 from C. elegans, 6 from C. albicans, and 16 from S. pombe.
Most retroelements in eukaryotic genomes are replete with mutations, including spurious frameshifts and stop codons that may obscure translational recoding signals. We therefore limited our analyses to a subset of recently transposed elements. This was accomplished by eliminating from the data set those elements with less than 98% LTR identity. LTRs are typically identical at the time of insertion, and so elements with low LTR identity are more likely to have accumulated mutations. After removal of the degenerate elements, the expanded data set contained 162 retroelements from A. thaliana, 252 from D. melanogaster, 15 from C. elegans, 6 from C. albicans, and 14 from S. pombe.
To assess the effectiveness of our methods for retroelement identification, we compared the numbers of elements identified using RetroMap with the numbers reported in the annotated genome sequences. C. elegans was reported to have 20 full-length elements (Ganko et al. 2001); we found 19 in our original search, 15 of which were retained after removal of degenerate elements. In S. pombe, we identified 16 elements (14 were retained); 11 elements were reported in the annotated genome sequence (Wood et al. 2002). We previously annotated the retrotransposons of S. cerevisiae and identified 51 full-length elements (Kim et al. 1998); RetroMap identified 56 candidate elements (data not shown). Upon closer examination, the nonannotated retroelements identified by RetroMap often had internal deletions. In some cases, complex arrangements of flanking repeats caused RetroMap to give false positives or to miss some elements. Although RetroMap was quite effective in element identification, we recognize that this software tool has its limitations. Furthermore, our need to remove degenerate elements may have modestly lowered the diversity of sequences in the expanded data set.
Retroelement gene organization
Open reading frames were identified between the LTRs to characterize the gene organization of retroelements in the data set. Frameshifting or stop codon readthrough typically occurs between gag and pol, and so the readily identifiable zinc finger domain in gag and the active site of protease in pol were used to define the boundary of these two genes. Because a single mutation can break a reading frame, multiple, closely related elements in the data set were aligned. Related elements were defined as those sequences with near zero distances in RT phylogenetic trees (data not shown). A consensus sequence was then constructed to determine the most probable sequence of the ancestral element. Only one representative retroelement that most closely matched the consensus sequence was further studied.
ORFs were identified within the retroelements in the data set, and five different types of gag/pol ORF organization were identified: (1) a single gag-pol ORF; (2) gag and pol separated by a stop codon; (3) pol in the +1 frame relative to gag; (4) pol in the −1 frame relative to gag; and (5) gag and pol separated into three or more ORFs. The latter group was not considered further, because the multiple ORFs may have resulted from mutation. Figure 1 shows the phylogenetic relationships of 65 retrotransposons annotated in GenBank and the 56 new retrotransposons we identified that constitute the expanded data set. The tree was constructed using RT amino acid sequences, and the elements fell into clusters according to Ty1/copia, Ty3/gypsy, BEL, and DIRS group designations rather than according to the host species from which they originated. The gene organization was mapped onto the tree for each element.
Single ORF retroelements
One surprising observation was that in contrast to retroviruses, nearly half of the retrotransposons identified encode Gag and Pol in a single ORF. This is particularly evident for the majority of the plant retrotransposons and most retrotransposons in the Ty1/copia and BEL clades. For these elements, the required ratio of Gag to Pol may be achieved posttranslationally through preferential Pol degradation, as has been observed for the Tf1 and Ty5 yeast retrotransposons (Levin et al. 1993; Atwood et al. 1996; Irwin and Voytas 2001). It is also possible that a posttranscriptional mechanism, such as alternative splicing, is utilized to express an excess of Gag—a strategy employed by the Drosophila copia element (Brierley and Flavell 1990). There is still a formal possibility that the single-ORF elements use ribosomal frameshifting for Gag-Pol expression; a frameshift event that occurs at the end of gag would result in the synthesis of only Gag. Such a frameshift occurs in the Escherichia coli dnaX gene to synthesize a shorter form of a DNA polymerase III subunit (Larsen et al. 1997). We view such a mechanism to be unlikely for the retrotransposons, because the efficiency of frameshifting would need to be unusually high to produce excess Gag.
The possibility that plants lack frameshifting can be discounted because −1 frameshifting, and, to a lesser extent, +1 frameshifting, is utilized in plant viral gene expression (Baranov et al. 2001). An alternative explanation is that our survey was unable to identify plant retrotransposons that utilize −1 frameshifting because their sequences were highly degenerate. Furthermore, Arabidopsis is the only plant genome exhaustively surveyed, and we cannot rule out that −1 frameshifting may be utilized by retrotransposons in other plants. It should be noted that this study did uncover two related plant Ty3/gypsy elements in which pol is in the +1 frame relative to gag. These elements are discussed in greater detail later below.
gag and pol separated by a stop codon
This form of gag/pol ORF organization was very rare among the retroelements surveyed. Only the Ty3/gypsy RIRE2 element from rice (Ohtsubo et al. 1999) and the BEL element Kamikaze from Bombyx mori have a conserved stop codon between their gag and pol genes (Abe et al. 2001). Sequences surrounding a stop codon influence leakiness, and recent comparative analysis of viruses that utilize readthrough for pol expression identified a limited number of different sequence signatures surrounding the leaky stop codon in viruses (Beier and Grimm 2001; Harrell et al. 2002). Sequences downstream from the stop codon in gag of Kamikaze (CUAUCU) fall into one of the characterized retroviral groups and are similar to sequences used in Sindbis virus, in which efficient readthrough has been confirmed experimentally (Li and Rice 1993). However, sequences surrounding the stop codon in RIRE2 (UGUAAATAGGAAAGC) do not match any known viral readthrough sequence motif. Whether or not the Kamikaze or RIRE2 sequences mediate stop codon readthrough will need to be tested experimentally.
pol in the −1 frame
Retroelements with pol in the −1 frame relative to gag are limited to Ty3/gypsy and DIRS-type elements. Furthermore, none originates from plant hosts, whereas they are widespread in the animal kingdom. In fact, in the animal hosts, we found a −1 frameshift is the most common form of retroelement gene organization and was present in 28 of 51 animal retroelements surveyed; 20 elements had single ORFs, one element has a stop codon between gag and pol, and two have pol in the +1 frame relative to gag.
Classical −1 frameshifting involves slippage of two tRNAs on sequences that conform to the consensus X XXY YYZ (Jacks et al. 1988; Weiss et al. 1989; Dinman et al. 1991; Brierley et al. 1992). It has been suggested that both tRNAs slip backward 1 nt from XXY to XXX and from YYZ to YYY. tRNAs can be located in the A- and P- (Jacks et al. 1988) or the E- and P-sites (Horsfield et al. 1995) of the ribosome, and slippage involves at least two steps: dissociation of the tRNAs from the mRNA and reassociation with mRNA in the −1 frame. Thus, both stability of the initial codon:anticodon duplex and the stability of the final codon: anticodon duplex likely contribute to the overall efficiency of tRNA slippage. Consequently, if stability of the initial complex is relatively weak, strong base pairing in the new frame is not required. In addition, −1 frameshifting can be caused by a single tRNA slippage in the P-site, as has been suggested for CP/12K signal of potato virus M on sequence AAAAUGA (Gramstat et al. 1994).
Comparative analysis of overlapping regions between gag and pol among the −1 retrotransposons identified conserved sequence signatures among closely related elements in the phylogenetic tree (Fig. 1). Codon alignment of the overlapping regions is shown in Figure 2 with putative frameshift sites indicated. Most of the sequences conform to classical frameshifting cassettes (i.e., XXXYYYZ). Alignments shown in Figure 2 demonstrate additional conservation around heptamer frameshift sites. For example, the nucleotide 3′ of heptamer shifty sequence is conserved in the alignments in Figure 2A,B. This supports the recent suggestion that stacking potential between bases of the nucleotide in the wobble position and the 3′ nucleotide may sometimes contribute to the efficiency of A-site decoding (Ayer and Yarus 1986) and consequently frameshifting (Bertrand et al. 2002).
It should be noted that most of the retroelements with pol in −1 frame relative to gag originate from Drosophila and B. mori. No −1 retroelements were found in C. elegans. This differential distribution might be due to differences in cellular translational machinery in different animal hosts or differences in the types of retroelements that colonize certain hosts. Finally, an equal percentage of elements with gag and pol in a single frame or in −1 or +1 overlapping frames were found in fungi, which implies that no specific regulatory strategy confers an advantage for retrotransposons in fungal cells. Of course, confidence in conclusions regarding the propensity for certain forms of frameshifting in certain hosts is limited due to the small number of eukaryotic genome sequences available.
pol in the +1 frame
Only a few retroelements were identified with pol in the +1 frame relative to gag. These elements are distributed evenly throughout the phylogenetic tree and are present in animal, plant, and fungal hosts. In animals, the only characterized example of +1 frameshifting occurs in genes encoding antizymes (Matsufuji et al. 1995; Ivanov et al. 2000). Two examples of +1 frameshifting have been characterized in fungi, namely Ty1 (and its homologs, e.g., Ty4) and Ty3 from S. cerevisiae. For both Ty1 and Ty3, +1 ribosomal frameshifting has been confirmed and extensively characterized (Farabaugh 1995). Several insect elements and one fungal element also have pol in the +1 frame (i.e., Zam, Tv1, Grh). Although the D. melanogaster element 1731 was originally reported to have a +1 frameshift (Fourcade-Peronnet et al. 1988; Haoudi et al. 1997), the consensus 1731 element encodes a single gag-pol ORF. For the insect and fungal elements, close homologs were not available to compare sequences in the overlapping region to identify putative frameshifting sites. The two examples of plant retroelements with pol in the +1 frame are the only plant retroelements described to date in which gag and pol are in separate reading frames. One of the +1 retroelements from Arabidopsis (AtChr2_44644) shares 97% sequence identity with a second Arabidopsis element (AtChr2_4188838). The frameshift is located between gag and pol, and there are 310 nt in the overlap region between the reading frames. However, with only two closely related elements to compare, the data set is not sufficient to identify conserved sequences that may mediate frameshifting.
Unusual gene organization for the Opie-2 family of plant retrotransposons
Opie-2 of maize (U68408) is a second example of a plant retroelement with pol in the +1 frame relative to gag. The annotated Opie-2 sequence has three large ORFs, suggesting that it has accumulated mutations, and therefore additional Opie-2 and Opie-2-like retrotransposons were retrieved from GenBank. In total, 372 Ty1/copia group retrotransposons were extracted using the Opie-2 reverse transcriptase in BLAST searches. Most of these elements are from plant hosts, and the majority (191) are typical or “classic” Ty1/copia retrotransposons that encode a single gag-pol ORF (Fig. 3A). The remaining 181 elements include Opie-2 (SanMiguel et al. 1996) and SIRE-1 of soybean (Laten and Morris 1993) and are members of a proposed genus of Ty1/copia retrotransposons (referred to a Sireviruses; Peterson-Burch and Voytas 2002). The Sireviruses exhibit considerable polymorphism with respect to the organization of gag and pol. A number of Sireviruses from monocots and dicots have pol in the +1 frame, and these elements cluster together in the phylogenetic tree (indicated by shading in Fig. 3A), suggesting that they are derived from a common ancestor. Other Sireviruses encode gag and pol in a single ORF similar to the classic Ty1/copia elements.
The Sireviruses with pol in the +1 frame further define two groups. In the Type I elements, gag and pol overlap. The overlap region is 38 nt for Type I elements from monocots (e.g., Opie-2) and 90 nt in the dicot elements. gag and pol do not overlap in the Type II elements (e.g., Prem-2). Furthermore, Type II elements that originate from maize and rice have a conserved TAG stop codon in pol 10 nt downstream from the gag termination codon (Figs. 3B, 4A). A transition from the gag to pol reading frame does not appear possible by simple +1 frameshifting, unless the TAG stop codon is also read through. For those Type I elements in monocots and dicots that do not have the conserved stop codon in pol, a CAG codon (glutamine) is present at the corresponding position. We favor the hypothesis that CAG predated the stop codon, which resulted from a C-to-T mutation, because CAG is found in elements from more diverse hosts and therefore likely represents the ancestral state. In addition, because of their high copy number in rice and maize, the stop codon in Type II elements does not seem to compromise transposition.
Because conserved sequence motifs or RNA secondary structure characterize recoding sites, we further analyzed the sequences at the gag-pol boundary of the Sireviruses. A conserved sequence motif was found in Type I and Type II elements from both monocots and dicots; the single ORF elements did not have this motif (Figs. 3B, 4A). The motif spans 27 nt, is self-complementary, and likely forms a stem–loop structure (Fig. 4B; Walter et al. 1994). Moreover, in certain elements, covariant nucleotides are found that preserve self-complementarity. For example, in maize Type I elements, C-G pairing at the top of the stem structure is replaced by A-T pairing in maize Type II elements.
Because pol in the Type II elements cannot be expressed by standard ribosomal frameshifting, it is possible that the stop codon is removed by splicing; however, the splice site prediction software packages NetGene2 and SplicePredictor did not identify any conserved splice donor or acceptor sites around the frameshift region. A second possible expression mechanism is internal ribosome entry. However, no ATG or alternative TTG start codons are found in the interval between the conserved stop codon in gag and the protease active site in pol, although it is formally possible that another alternative start codon is used.
A final possibility for recoding is termed “bypass” and has only been observed for expression of the bacteriophage T4 gene 60 (Weiss et al. 1990). In this case, a 50-nt mRNA sequence is “skipped” by the translating ribosome (Herr et al. 2000). A bridge of RNA secondary structure and a 16 amino acid nascent peptide are required for bypassing to occur. Matching GGA codons separated by 50 nt are used as the “takeoff” and “landing” sites for the ribosome. A bypass mechanism may be used by the Opie-like elements, enabling the ribosome to bypass both the gag gene and pol gene stop codons in one “jump”. However, the signatures required for T4 gene 60 bypassing are not observed in Opie-like retrotransposons. Further investigation will be required to determine how the Opie-2-like retroelements express Pol. If expression is carried out by a novel translational mechanism, then it will be interesting to determine whether other cellular genes utilize this translational mechanism.
MATERIALS AND METHODS
Data collection
For elements in the core data set, element length was restricted to between 2 kb and 20 kb to approximate the length of most retrotransposons. Elements extracted for the core data set were assessed for structural integrity, namely for the presence of protease, integrase, and the RT sequence domains. In addition, any elements lacking an LTR were eliminated. The core data set consists of 126 elements.
To identify other retrotransposons from model organisms, we retrieved retrotransposons directly from the genome sequences. Reverse transcriptase amino acid sequences from Ty3/gypsy, Ty1/copia, DIRS, and BEL groups were used as electronic probes in BLAST searches against individual model organism genome sequences (Altschul et al. 1990). The probes included: BEL (U23420) and Pao (L09635) in the BEL group; DIRS-1 (M11339) and PAT (X60774) in the DIRS group; Athila4-1 (AC007209), Cer1 (U15406), Osvaldo (AJ133521), sushi (AF030881), Tf1 (L10324), and Ty3 (M23367) in the Ty3/gypsy group; and Art1 (Y08010), copia (M11240), Endovir1-1 (AB026651), SIRE-1 (AF053008), Tca2 (AF050215), and Ty5 (U19263) in the Ty1/copia group. Full-length retroelements were retrieved from the genome sequence by the software package RetroMap (B.D. Peterson-Burch and D.F. Voytas, unpubl.). RetroMap reads a BLAST output and identifies potentially complete elements by locating two repeated flanking sequences: the putative LTRs.
Data analysis
RT amino acid sequences were aligned with ClustalX (Higgins and Sharp 1988). MEGA2 was used to construct phylogenetic trees by the neighbor-joining distance method (Saitou and Nei 1987) and the Poisson correction model (http://www.megasoftware.net). The core and expanded data sets can be found, respectively, at the following websites: http://www.public.iastate.edu/~voytas/MSsupplimentary/Recoding/core and http://www.public.iastate.edu/~voytas/MSsupplimentary/Recoding/model_organism. ORF finder (http://www.ncbi.nlm.nih.gov/gorf/gorf.html) was used to characterize reading frame organization, and splicing signals were sought using the NetGene2 (http://www.cbs.dtu.dk/services/NetGene2/) and SplicPredictor software packages (http://bioinformatics.iastate.edu/cgi-bin/sp.cgi).
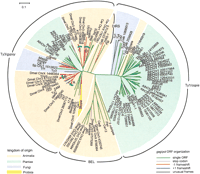
Phylogenetic relationships among retrotransposons and the distribution of putative translational regulatory mechanisms. The four major clusters of retrotransposons are labeled Ty1/copia, Ty3/gypsy, BEL, and DIRS. Retrotransposons in the core data set are labeled with their names at the end of the branches. Retrotransposons in the expanded data set begin with an abbreviation for the host organism: Ath, A. thaliana; Dmel, D. melanogaster; Sp, S. pombe; Calb, C. albicans; Cer, C. elegans. Following the host species abbreviation are the chromosome number and nucleotide position for given insertions. Branch coloring indicates the gene organization of gag and pol: green, single ORF elements; blue, pol in the +1 frame; red, pol in the −1 frame; pink, a stop codon between gag and pol. Shading identifies the kingdom from which the retrotransposons originate. The clades labeled A–D are described in Figure 2.

Codon alignments of retroelements with similar −1 frame cassettes in the overlap region between gag and pol. Panels A–D correspond to the four groups of retroelements depicted in Figure 1 that have pol in the −1 frame relative to gag. Sequences with greater than 75% identity are highlighted. Possible locations for tRNAs during frameshifting are marked by AAA (A-site), PPP (P-site), and EEE (E-site).
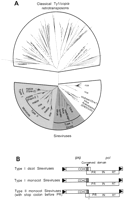
Unusual organization of gag and pol among Sireviruses. (A) Phylogenetic relationships of Ty1/copia retroelements. The top part of the tree depicts classical Ty1/copia retrotransposons (Pseudoviruses and Hemiviruses). Based on branch lengths, Sireviruses are more closely related than classical Ty1/copia retroelements, suggesting they recently colonized their host genomes. Retrotransposons in shaded areas have a potential frameshift between gag and pol. Host names for Sireviruses are labeled at the ends of branches, and the names in parentheses denote annotated retroelements in the core data set. (B) The ORF structure of retrotransposons in the shaded regions depicted in A. Only those elements with separate gag and pol reading frames have the conserved sequence motif. Type II elements in monocots likely utilize an unusual translation mechanism because a stop codon (asterisk) exists at the beginning of pol. A gag zinc finger (CCHC) and the protease active site of pol (PR) flank the conserved nucleotide motif in the frameshift region. (RT) reverse transcriptase; (IN) integrase.

Nucleotide alignment of the frameshift region in Sireviruses. (A) The first 31 columns in the alignment are highly conserved, self-complementary, and predicted to fold into a stem–loop structure. For the maize retrotransposons, covariant changes that preserve self-complimentary are marked by the arrows. The asterisk indicates the stop codon in gag. The diamond indicates the position of the stop codon in pol. The boxes mark the occurrence of the gag and the pol stop codons. (B) The stem–loop structure of the conserved motif.
Acknowledgments
We thank Mickey Tekippe for his work in annotating the core retrotransposon data set. This work was supported by NIH grants GM61657 to DFV and GM48152 to JFA. The publication costs of this article were defrayed in part by payment of page charges. This article must therefore be hereby marked “advertisement” in accordance with 18 USC section 1734 solely to indicate this fact.
Footnotes
-
Article and publication are at http://www.rnajournal.org/cgi/doi/10.1261/rna.5105503.
-
- Accepted September 2, 2003.
- Received June 13, 2003.
- Copyright 2003 by RNA Society








