
Regulation of mRNA decay and translation during the mammalian cell cycle
- 1Whitehead Institute for Biomedical Research, Cambridge, Massachusetts 02142, USA
- 2Department of Biology, Massachusetts Institute of Technology, Cambridge, Massachusetts 02142, USA
- Corresponding authors: icheese{at}wi.mit.edu; jly7{at}bidmc.harvard.edu
Abstract
Cell cycle progression requires cells to continually remodel their gene expression programs as they transition through distinct functional states. Although transcriptional and post-translational mechanisms have long dominated our understanding of this regulation, recent work additionally highlights the essential contribution of cell cycle–specific mRNA decay and translational control. Across G1, S, G2, and mitosis, cells dynamically modulate global and transcript-specific mRNA stability and translation to coordinate processes including DNA replication, growth, checkpoint signaling, and chromosome segregation. Mitosis presents a particularly striking challenge: Transcription is reduced, necessitating that cells rely on post-transcriptional mechanisms to sustain mitotic functions and preserve viability. In this review, we highlight how these coordinated layers of post-transcriptional regulation collectively contribute to cell cycle control.
Keywords
INTRODUCTION
The precise regulation of the cell cycle is crucial for cellular proliferation, organism development, and cancer treatment (Nurse 2000; Malumbres and Barbacid 2001). Each stage of the cell cycle—G1, S, G2, and mitosis—requires distinct and tightly controlled cellular processes and activities, which are enabled by differential gene expression and regulated protein activity (Jensen et al. 2006; Fischer et al. 2022). In cell culture, the mammalian cell cycle typically lasts ∼24 h, the majority of which cells spend in interphase (G1, S, G2), with mitosis lasting only ∼1 h (Alberts et al. 2002). During G1 phase, typically the longest phase of the cell cycle, cells grow and prepare for DNA replication. During S phase, cells replicate their DNA. During G2, cells continue growing and prepare for mitosis. Once cells enter mitosis, the nuclear envelope breaks down, and chromosomes condense and align at the metaphase plate before being segregated to daughter cells.
Progression between cell cycle stages in mammalian cells is controlled by positive regulators that promote cell cycle transitions by sensing cell size, mitogens, nutrient availability, cell–cell contacts, and other stimuli (Malumbres and Barbacid 2001). In particular, cyclin-dependent kinases (CDKs) act together with their cyclin partners to orchestrate cell cycle progression and stage-specific functions, phosphorylating substrates to promote key events such as DNA replication, nuclear envelope breakdown, and mitotic entry (Malumbres 2014; Pellarin et al. 2025). In addition, in the presence of errors such as DNA damage, replication stress, or mitotic chromosome misalignment, cell cycle checkpoints sense these defects and halt cell cycle progression (Barnum and O'Connell 2014). For example, during mitosis, the spindle assembly checkpoint detects defective kinetochore–microtubule attachments and arrests cells in mitosis until proper attachments can form (McAinsh and Kops 2023). These checkpoints can pause cell cycle progression for durations ranging from hours to even days, requiring cells to rewire their gene expression programs to maintain viability and persist during the extended arrest (Ly et al. 2024b; Khalizeva et al. 2026). Finally, exit from a cell cycle stage often involves regulated protein degradation. For example, at the end of metaphase, the anaphase-promoting complex/cyclosome (APC/C), a major E3 ubiquitin ligase complex, ubiquitinates multiple substrates such as securin and the mitotic cyclin B, marking them for proteasome-dependent degradation and promoting irreversible sister chromatid separation and mitotic exit (McAinsh and Kops 2023).
Regulated gene expression plays critical roles in cell cycle function (Whitfield et al. 2002; Fischer et al. 2022). Prior studies have focused primarily on the cell cycle–dependent control of transcription (Whitfield et al. 2002). On the transcriptional level, the transcription factor complexes (RB):E2F, MMB:FOXM1, and the DREAM complex enable periodic expression of two groups of transcripts (Fischer et al. 2022). The first group includes transcripts expressed in G1/S that are required for DNA synthesis, whereas the second group is comprised of transcripts that are most highly expressed in G2/M and are required for mitosis, such as cyclin B (Whitfield et al. 2002; Thiru et al. 2014). In addition to transcriptional changes, alternative splicing is also extensively remodeled across the cell cycle (Dominguez et al. 2016). At the global scale, transcription is halted during mitosis through changes at multiple levels (Contreras and Perea-Resa 2024); chromosomes are condensed (Antonin and Neumann 2016), and the transcriptional machinery is largely evicted from chromatin (Parsons and Spencer 1997; Perea-Resa et al. 2020), contributing to a strong reduction in transcription (Taylor 1960). Although transcription from most genomic regions is repressed during mitosis, centromere regions that retain cohesin localization and several other protein-coding genes continue to be transcribed (Palozola et al. 2017; Contreras and Perea-Resa 2024). However, the role of this ongoing mitotic transcription and these noncoding transcripts has been debated.
Notably, although stage-specific transcription has been well studied, the role of post-transcriptional gene processing, including mRNA modifications and decay, has only recently begun to be explored. In addition to the well-established global attenuation of translation during mitosis (Fan and Penman 1970; Pyronnet et al. 2001), recent studies have revealed new paradigms for cell cycle–regulated protein production and its importance for cellular function. These post-transcriptional mechanisms of gene regulation provide key insights into cell cycle control and division. Here, we review the regulation of mRNA stability and translational control during the mammalian cell cycle, highlighting both global regulation and mechanistic studies of the cyclic expression of individual genes.
DYNAMIC CONTROL OF mRNA DECAY DURING INTERPHASE
During interphase, cells progress through G1, S phase, and G2, each with different transcriptomes established by changes in the balance of new transcription and decay of existing mRNAs. Although the role of transcriptional control in cell cycle–dependent gene expression has been studied extensively, regulation at the level of mRNA stability has been largely overlooked. Recent studies using single-cell sequencing methods to measure global changes in mRNA synthesis and degradation rates across the cell cycle (Battich et al. 2020; Liu et al. 2023) found that both transcription and mRNA degradation play important roles in the expression kinetics of cell cycle–regulated genes. These findings suggest that genes with shared functions are likely to be regulated by similar turnover strategies to coordinate a given cell cycle process. For example, some S phase–expressed genes involved in DNA repair and G2-expressed genes involved in microtubule spindle assembly displayed coordinated transcription and mRNA degradation to maximize their gene expression (Battich et al. 2020). Thus, as discussed in detail below, cell cycle–dependent gene expression dynamics cannot be captured by transcription alone, and the contribution of mRNA degradation must also be considered.
G1-specific CCND1 mRNA stabilization promotes timely S-phase entry
To regulate cell cycle progression during interphase, a subset of genes is controlled at the level of mRNA decay. mRNA degradation is regulated through several mechanisms, including RNA modifications and RNA-binding proteins (Passmore and Coller 2022). These mechanisms can alter mRNA stability both globally and at a gene-specific level to tune gene expression throughout the cell cycle and provide a critical additional layer of control. For example, cyclin D1 (CCND1), which is highly expressed in G1, is a key regulatory component of CDK4, a kinase that phosphorylates the tumor suppressor protein RB to drive S-phase entry (Fischer et al. 2022). Importantly, the CCND1 mRNA displays differential stability across the cell cycle, with increased mRNA stability in G1 compared to S, G2, and mitosis (Fig. 1A; Hirayama et al. 2020). CCND1 mRNA stability is regulated by cell cycle–dependent m6A modifications, which increase after G1 to promote mRNA decay (Hirayama et al. 2020). The changes in CCND1 m6A levels are in part modulated by cell cycle–dependent localization of FTO, an eraser of the m6A modification. In G1, FTO localizes to the nucleus, which allows it to bind to and demethylate the CCND1 mRNA (Fig. 1A). In contrast, during S phase, phosphorylation of FTO induces its cytosolic localization, preventing its association with the nascent CCND1 mRNA. This results in increased CCND1 m6A levels and RNA degradation. Cell cycle–dependent m6A modifications are not limited to CCND1 and have been observed across hundreds of transcripts, particularly between G1 and S (Fei et al. 2020). This suggests that m6A modifications may play a larger role in regulating mRNA turnover and cell cycle progression.
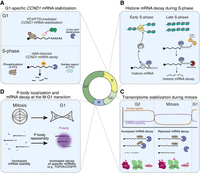
Regulation of mRNA decay throughout the mammalian cell cycle. (A) CCND1 mRNA is stabilized in G1 by two mechanisms: removal of m6A by FTO and enhanced binding by ubiquitinated PC4. After G1, FTO phosphorylation triggers its cytosolic export and PC4 phosphorylation reduces its RNA-binding, decreasing CCND1 mRNA stability. (B) Following DNA replication, replication-dependent histone mRNAs are rapidly degraded, ensuring histone levels match DNA replication. (C) Global mRNA stabilization upon mitotic entry to compensate for reduction in mitotic transcription. This correlates with PABPC1 toeprints and phosphorylation of regulators controlling poly(A)-tail length and mRNA decay. (D) mRNA decay and P-body dynamics at mitotic exit. During the M–G1 transition, specific mRNAs are degraded. Coinciding with decay, P-bodies reform during mitotic exit, facilitating rapid mRNA turnover.
The stability of CCND1 is also modulated by positive coactivator 4 (PC4), an RNA-binding protein that binds to CCND1 mRNA to promote its stability (Pan et al. 2024). PC4 is dynamically modified by phosphorylation and ubiquitination across the cell cycle to alter its affinity for the CCND1 mRNA (Fig. 1A). Importantly, PC4-mediated stabilization of CCND1 mRNA is required for the G1–S transition (Pan et al. 2024). Overall, the dynamic post-transcriptional control of CCND1 expression highlights the importance of mRNA decay for cell cycle progression.
Post-transcriptional regulation of histone mRNAs during S phase
During S phase, cells must both duplicate their genetic material and double the histones to package the replicated DNA. Following DNA replication, the cells stop histone production to limit excess unincorporated histones (Singh et al. 2010). Thus, cells need to rapidly induce histone production upon S-phase entry and then subsequently block it at the end of S phase. To achieve this regulation, a series of post-transcriptional processing events mediate the rapid degradation of histone mRNAs once DNA replication is complete (Fig. 1B; for a complete review, see Marzluff and Koreski 2017). Instead of the poly(A) tail typical for most mRNAs, nuclear processing of histone mRNAs results in an ACCCA mRNA tail immediately downstream (3′) of the stem loop, which is bound by the stem-loop binding protein SLBP (Marzluff and Koreski 2017). At the end of S phase, when DNA replication is complete, the histone 3′ exonuclease ERI1 removes the ACCCA mRNA tail from histone mRNAs, degrading into the stem loop (Fig. 1B). 3′ Polyuridylation by TUT7, accompanied by the removal of SLBP, targets the uridylated histone mRNAs for degradation by the exosome (Marzluff and Koreski 2017). This allows cells to rapidly degrade histone mRNAs following DNA replication, blocking new histone production.
REGULATION OF mRNA PROCESSING AND STABILITY DURING MITOSIS AND AT THE M–G1 TRANSITION
Transcriptional and mRNA processing changes during mitosis
Compared to interphase cells, mitotic cells face a distinct set of gene regulatory challenges. During mitosis, which typically lasts ∼1 h in cultured mammalian cells, overall RNA transcription is reduced to ∼25% relative to interphase (Prescott and Bender 1962; Palozola et al. 2017; Contreras and Perea-Resa 2024). Despite this global repression of mitotic transcription, Palozola et al. (2017) identified 3689 genes that undergo low but detectable levels of transcription during mitosis. In addition, the nuclear envelope breaks down during mitosis, leading to mixing of the nuclear and cytoplasmic contents within the cell. Given this drastic rearrangement of the cellular compartments, it may be important for cells to regulate the activity of nuclear RNA processing enzymes to prevent aberrant cleavage or processing of translationally competent cytoplasmic mRNAs. Indeed, recent work has revealed multiple pathways that prevent hyperadenylation of cytoplasmic mRNAs by nuclear polyadenylation machinery during mitosis. Specifically, the poly(A) polymerase (PAP) is hyperphosphorylated by CDK1/cyclin B in mitotic cells, which drastically reduces its enzymatic activity (Fig. 1C; Colgan et al. 1996). In addition, the nuclear poly(A)-binding protein PABPN1 is specifically phosphorylated in mitotic cells, which may contribute to preventing hyperadenylation and maintaining the transcriptome in cells lacking a nuclear envelope (Fig. 1C; Gordon et al. 2024). Overall, these findings suggest that there are several adaptations to mRNA processing during mitosis. Future studies will be needed to test how other nuclear proteins are differentially regulated during mitosis to contribute to mitotic cell physiology.
Global transcriptome stabilization during mitosis
Under normal circumstances, the short time (∼1 h) that cells spend in mitosis suggests that disruptions in gene expression might be tolerated as they would have minor effects on the steady-state proteome. However, if errors in chromosome attachments occur during cell division, mammalian cells can maintain the mitotic state for tens of hours to facilitate error correction before proceeding with chromosome segregation (Gascoigne and Taylor 2008; McAinsh and Kops 2023). Therefore, cells must regulate their gene expression post-transcriptionally to enable mitotic processes, safeguard cellular function during a prolonged arrest, and prepare for G1 entry.
Gene expression typically involves a constant balance of mRNA synthesis and degradation. In asynchronously proliferating mammalian cells, the median mRNA half-life is 2–4 h (Herzog et al. 2017; Lugowski et al. 2018; Eisen et al. 2020). When mRNA synthesis is significantly decreased during a prolonged mitotic arrest, cells may require compensatory mechanisms to maintain their transcriptomes. Our recent work demonstrates that, during a prolonged mitosis, cells compensate for the global reduction in transcription by inhibiting mRNA degradation to preserve their transcriptome (Fig. 1C; Khalizeva et al. 2026). Indeed, mRNA stability is globally increased during mitosis, with an approximately fourfold increase in the median mRNA half-life in M (>20 h) compared to G2 (5.6 h) or G1 (3.1 h) stages (Khalizeva et al. 2026). Such a global stabilization of mRNA supports a model in which cells reduce mRNA degradation to compensate for the decreased mRNA synthesis during mitotic arrest (Khalizeva et al. 2026).
Our work suggests that mitotic mRNA stabilization results from a reduction in deadenylation and potentially additional steps of the mRNA decay pathway, whereas downstream processes such as endonuclease- and exonuclease-dependent mRNA degradation may still occur during mitosis. In particular, we observed that poly(A) tail length distributions exhibit striking PABPC (cytoplasmic poly(A)-binding protein) toeprints in mitotic cells, but not in G2 cells (Fig. 1C). Such PABPC toeprints have also been observed following the downregulation of the CCR4–NOT complex, the primary deadenylase in eukaryotic cells (Slobodin et al. 2020), or following inactivation of CNOT6 (CCR4), one of the two catalytic subunits of CCR4–NOT (Yi et al. 2018). The appearance of PABPC toeprints during mitotic arrest suggests that the CCR4–NOT complex may be less efficient at clearing PABPC from poly(A) tails, resulting in slower deadenylation. Consistent with this model, data from a recent study suggest that CCR4–NOT may associate with RNA less efficiently during mitosis than during interphase (Rajagopal et al. 2025). Additionally, the median poly(A) tail lengths for most transcripts remain unchanged during a prolonged arrest in the transcriptionally reduced M phase (Park et al. 2016), indicating a lack of tail shortening in the absence of new mRNA synthesis. Importantly, depletion of PABPC1&4 restores mRNA degradation in mitotic cells and results in premature mitotic exit, highlighting the role of PABPC in protecting mRNA from deadenylation-dependent degradation during mitosis, as well as the importance of mitotic mRNA stabilization for the cells’ ability to maintain a mitotic arrest.
In addition to altered deadenylation during mitosis, other contributions to mRNA degradation are also affected. For example, nonsense-mediated decay (NMD) is repressed during mitosis and restarts as cells enter G1 (Ryu et al. 2019). NMD is typically initiated upon the first round of translation of a newly exported transcript when an exon junction complex (EJC) deposited during cotranscriptional splicing is not removed from the CDS by translating ribosomes (Kervestin and Jacobson 2012). In such cases, the EJC serves as a signal for both NMD-specific and general mRNA degradation machinery to remove the faulty transcript. However, phosphorylation of the core EJC component eIF4A3 at Thr163 by CDK1&2 during mitosis leads to a reduction in its association with intron-containing mRNA molecules, a failure to form an EJC, and consequently a reduction in EJC-dependent NMD (Fig. 1C; Ryu et al. 2019). This may represent an additional layer of protection that cells have developed to prevent transcriptome depletion during mitosis.
Overall, multiple mechanisms contribute to mRNA stabilization during mitosis, enabling cells to prevent transcriptome loss when mRNA synthesis is globally reduced.
mRNA decay and P-body localization at the M–G1 phase transition
Upon the completion of mitosis, cells “reset” their gene expression profiles for the new cell cycle, including major chromatin reorganization, resumption of transcription and cotranscriptional RNA processing, and widespread protein degradation (de Castro et al. 2016; Goel et al. 2025). mRNA degradation plays a key role in this resetting, with two distinct phases of mRNA decay during mitotic exit and in early G1 (Krenning et al. 2022). Several long noncoding RNAs, which are no longer chromatin-associated during mitosis, also appear to be degraded as cells reenter G1 (Bury et al. 2020; Blower et al. 2023).
A recent study revealed waves of mRNA degradation during mitotic exit as a mechanism to clear mitotic transcripts and reset the transcriptome. Based on degradation timing and kinetics measured by single-cell RNA sequencing in RPE1 cells, Krenning et al. (2022) identified 220 genes for which mRNA levels decline at the M–G1 transition. One subset of transcripts, termed the “immediate decrease” group, had rapid rates of degradation coinciding with mitotic exit and continued to decline during the first hour of G1. This group included transcripts such as CDK1 (mitotic kinase, Solomon et al. 1990), TOP2A (mitotic chromosome organization, Nielsen et al. 2020), UBE2C (mitotic E2 ligase, Townsley et al. 1997), and others with established mitotic functions. The second subset of transcripts was degraded gradually, with slower kinetics during the M–G1 transition, highlighting the possibility of multiple distinct mechanisms of mRNA decay at this transition. The regulation of deadenylation likely plays a role in orchestrating this mRNA degradation at the end of mitosis (Krenning et al. 2022), as transcripts undergoing rapid decay at the M–G1 transition have shorter poly(A) tails during mitosis than the average mRNA (Park et al. 2016; Krenning et al. 2022). This observation is consistent with the possibility that despite a global reduction in deadenylation, a subset of transcripts, which are targeted for immediate degradation upon mitotic exit, may still continue to be deadenylated during mitosis to be primed for rapid decay in G1. Overall, these findings point to a model in which the targeted degradation of transcripts for mitosis-specific proteins drives the transition back to an interphase-like transcriptome.
Mitosis also coincides with changes in P-bodies—ribonucleoprotein granules that are enriched in RNA decay machinery and contribute to rapid mRNA decay (Blake et al. 2024). During interphase, P-bodies are constitutively present in the cytoplasm. However, during mitosis, P-bodies dissolve and then reassemble upon entry into G1 (Fig. 1D; Aizer et al. 2013; Rai et al. 2018). A recent study suggested that the reassembly of P-bodies may provide an additional layer of cell cycle–dependent regulation of mRNA degradation, particularly at the M–G1 transition (Safieddine et al. 2024). The mRNAs that undergo precisely timed degradation during mitotic exit encode proteins that function primarily in G2 and mitosis. These transcripts show high accumulation in P-bodies in G1 (Fig. 1D; Safieddine et al. 2024). For example, TOP2A and CENPF, which play roles in mitosis, specifically localize to P-bodies at the M–G1 transition and undergo rapid mRNA degradation (Nielsen et al. 2020; Navarro and Cheeseman 2021). This suggests that mRNA localization to P-bodies during mitotic exit may silence transcripts that are no longer needed, resetting the cellular transcriptome for the new cell cycle. The molecular mechanisms that drive the G1-specific P-body localization of these transcripts remain unclear, although longer mRNAs appear to be enriched in these P-bodies (Safieddine et al. 2024).
Together, these studies highlight mRNA decay as an important layer of the regulation of gene expression during the cell cycle, both as a global strategy for transcriptome maintenance and as a means of regulating stage-specific transcripts.
TRANSLATIONAL CONTROL IN MITOSIS
As described above, mRNA production and stability influence cell cycle progression. In addition, recent work has highlighted translational control as another critical layer of gene expression regulation during the cell cycle, particularly during mitosis. Changes in translational control can enable rapid and dynamic proteome remodeling from the existing transcriptome without changes in steady-state mRNA levels. However, ribosome profiling across interphase has revealed only limited translational regulation, with only 83 genes differing between G1 and S phases and just 13 between G1 and G2 (Stumpf et al. 2013). This contrasts sharply with the widespread transcriptomic and proteomic remodeling across G1, S, and G2 (Jensen et al. 2006; Fischer et al. 2022). These studies indicate that translation plays a comparatively minor role in shaping the interphase proteome. In contrast, mitosis represents a fundamentally different regulatory landscape. During mitosis, transcription is globally reduced (Fan and Penman 1970; Tarnowka and Baglioni 1979; Pyronnet et al. 2001), and translational control becomes essential and highly specialized. In this section, we describe the mitotic translational landscape, emphasizing the mechanisms that establish this program and the consequences of disrupting mitosis-specific translational control.
Global translational repression during mitosis
Compared to interphase, mitosis is marked by dramatic changes to translational control, including a global reduction in protein synthesis. The observation of decreased translation during mitosis dates back to the 1970s, with studies finding that the incorporation of radioactive amino acids declined to 20%–30% of interphase levels in cells synchronized in mitosis using the microtubule poison nocodazole (Fig. 2A, bottom; Fan and Penman 1970; Pyronnet et al. 2001). This work received some criticism as long-term treatment with nocodazole is toxic and can alter translation (Sivan et al. 2011; Coldwell et al. 2013; Tanenbaum et al. 2015). Thus, it was unclear whether translation is actually reduced in mitosis or whether the reduced protein output was a by-product of nocodazole treatment. However, more recent studies have tested other mitotic inhibitors such as the microtubule poison paclitaxel, the kinesin-5 inhibitor STLC, the proteasome inhibitor MG-132, and the APC/C inhibitor proTAME (Miettinen et al. 2019). For example, our work found that STLC treatment results in a potent mitotic arrest without inducing the integrated stress response (Ly et al. 2024b). In these cells, ribosome profiling shows that translation is reduced to ∼25% of interphase levels during a 16 h STLC-induced mitotic arrest (Ly et al. 2024b), consistent with other metabolic labeling experiments in mitotically synchronized cells (Miettinen et al. 2019).
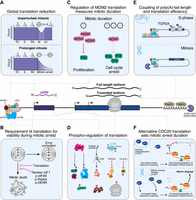
Translational control in mitotic cell physiology and viability. (A) Global mRNA translation is strongly repressed during mitosis, with repression increasing over time. (B) Mitosis-specific translational pathways are essential for survival, error correction, and G1 reentry after prolonged mitosis. (C) MDM2 levels decline with mitotic duration due to global repression and uORF-mediated inhibition; postmitotic cells entering G1 show elevated p53 and cell cycle arrest. (D) Multiple translation factors are phosphorylated, reprogramming the translational machinery. (E) Poly(A) tail length correlates with translational efficiency in mitosis, especially for short-tailed mRNAs, further attenuating translation. (F) Nuclear release of eIF1 globally enhances leaky ribosome scanning, producing truncated CDC20 isoforms that promote mitotic slippage.
As cells treated with antimitotic inhibitors remain arrested for multiple hours—whereas mitosis typically lasts for only about ∼1 h—subsequent studies sought to measure translation during an unperturbed mitosis. Tanenbaum et al. (2015) tested the extent of translation during an unperturbed mitosis in diploid RPE1 cells. Using metabolic labeling and single-cell imaging to track translation rates in real time, these studies revealed a more moderate reduction in global translation to ∼70% of interphase levels (Fig. 2A, top; Tanenbaum et al. 2015; Miettinen et al. 2019). These results indicate that during a normal, unperturbed mitosis, global translation is only modestly reduced. Consistent with this, further analyses found that the extent of translation inhibition scales with the duration of mitotic arrest: The longer cells remain in mitosis, the stronger the reduction in translation (Fig. 2A, bottom; Miettinen et al. 2019).
Overall, these data show that translation is globally reduced during mitosis—particularly under mitotic arrest—reflecting dampened, rather than abolished, protein production. In the sections below, we discuss the physiological significance of this decrease, while also highlighting the requirement for ongoing protein synthesis and the selective remodeling of translation for specific mRNAs.
Mitotic translation is required for mitotic cell viability
Given the well-established global reduction in translation during mitosis, new protein synthesis was assumed to be largely dispensable for mitotic progression or viability. However, accumulating evidence demonstrates that continued protein synthesis is essential for mitotic physiology, particularly during an extended mitotic delay (Fig. 2B). Inhibiting translation globally using cycloheximide or related inhibitors during a mitotic arrest triggers premature mitotic exit (Tsang and Cheeseman 2023) and apoptosis (Ly et al. 2024b). Similarly, the RNA helicase eIF4A1, which unwinds 5′ UTR secondary structures to promote translation initiation, is essential during mitosis: Inhibition of eIF4A1 by hippuristanol treatment markedly increases cell death in mitotically arrested cells (Fig. 2B; Moustafa-Kamal et al. 2020). Inhibition of eIF4B, a cofactor that enhances eIF4A1 activity, also compromises mitotic viability (Fig. 2B; Wang et al. 2016).
Together, these findings demonstrate that new protein synthesis, even at reduced levels, is essential for mitotic cell viability and suggest that combining translation inhibitors with mitotic poisons may enhance antimitotic chemotherapy. Consistent with this idea, cotreatment with homoharringtonine and paclitaxel has been proposed as a synergistic strategy to treat triple-negative breast cancer (Plett et al. 2022). Further work will be needed to systematically explore translation inhibitors in combination with antimitotic agents as a therapeutic strategy.
Mitotic translational repression of MDM2 is required for sensing mitotic duration
As ongoing translation is essential for mitotically arrested cells, the biological significance of the widespread translational repression that occurs during a prolonged mitosis has remained enigmatic. One simple possibility is that mitotically arrested cells, which are not growing or increasing in cell size, may naturally downregulate protein production. In contrast, recent studies have suggested that mitotic translational repression can also play an active role by creating a sensor that monitors mitotic duration (Fig. 2C). The duration of mitotic arrest often reflects the severity of the mitotic errors, with a prolonged arrest signaling potentially problematic cells. Eliminating cells that have experienced severe mitotic errors serves as an effective mechanism to select for healthy cells within a proliferating population. Indeed, cells that experience extended mitotic delays can trigger a p53-dependent arrest in the subsequent cell cycle (Meitinger et al. 2024; Fulcher et al. 2025).
Sensing mitotic duration is mediated, at least in part, by the global translational repression that occurs during a prolonged mitosis (Fulcher et al. 2025). As cells enter mitosis and overall protein synthesis decreases, proteins with shorter half-lives will be depleted over time. This includes MDM2, which has a half-life of ∼30 min (Fulcher et al. 2025). During an extended mitotic arrest, MDM2 levels decline rapidly, with <10% remaining after 4 h in mitosis (Fig. 2C; Fulcher et al. 2025). As described below, in addition to the global reduction in protein synthesis during mitosis, the presence of upstream open reading frames (uORFs) in MDM2 further reduces its translation during a mitotic arrest (Ly et al. 2024b). Consequently, daughter cells emerging from a prolonged mitosis inherit lower MDM2 levels than those that complete mitosis quickly. Because MDM2 is the E3 ligase for the tumor suppressor p53, reduced MDM2 leads to elevated p53 and triggers a cell cycle arrest (Fig. 2C; Fulcher et al. 2025). Thus, global translational repression during mitosis functions to promote a molecular “timekeeper,” linking mitotic duration to postmitotic MDM2 levels and cell cycle control. Similarly, multiple other proteins with short half-lives are also expected to decline during a prolonged mitotic arrest. Whether other cellular processes depend on this mitotic translational reduction remains an important open question.
Phospho-regulation of mitotic translation
The global reduction of translation in mitosis is regulated through several mechanisms, including phosphorylation. Multiple studies have highlighted the central role of phosphorylation of ribosomal proteins and translation factors in establishing mitosis-specific translational control (Fig. 2D). In mitosis, the master regulatory cyclin-dependent kinase CDK1/cyclin B orchestrates a wide range of mitotic events. CDK activity has been linked directly to the global remodeling of translation that occurs during mitotic entry. During mitosis, CDK1 phosphorylates RAPTOR, a regulatory subunit of mammalian target of rapamycin (mTOR). This inhibits canonical mTOR activity (Moustafa-Kamal et al. 2020), which may allow for increased cap-independent translation during mitosis (Pyronnet et al. 2000; Le Breton et al. 2005). mTOR is the major driver of cap-dependent translation during interphase, suggesting that new translation would be eliminated upon mTOR inhibition. However, given the requirement for ongoing protein production during mitosis, translation must still occur even without mTOR activity. Indeed, studies indicate that CDK1 itself functionally substitutes for mTOR under these conditions (Shuda et al. 2015). Although CDK1 suppresses mTOR activity, it also phosphorylates many, but not all, of mTOR's target proteins, thereby sustaining the mitotic translational program (Shuda et al. 2015).
Beyond mTOR, several key initiation factors—including eIF4EBP (Pyronnet et al. 2001), eIF4G1 (Dobrikov et al. 2014), eIF4A (Moustafa-Kamal et al. 2020; Sahoo 2022), eIF4B (Wilker et al. 2007), and eIF2α (Kim et al. 2014; Sparago et al. 2026)—are phosphorylated during mitosis, contributing to the global reduction in translation initiation (Fig. 2D). The translation elongation factors eEF1 (Sivan et al. 2011) and eEF2 (Sparago et al. 2026), as well as the ribosomal protein RPL12 (Imami et al. 2018), are also phosphorylated in a CDK-dependent manner, and these modifications alter translation production during mitosis (Fig. 2D). In addition, the translation reinitiation factor DENR, which promotes translation reinitiation downstream from uORFs, undergoes mitosis-specific phosphorylation that stabilizes the protein and promotes its activity during mitosis (Clemm von Hohenberg et al. 2022). Preventing this mitosis-specific phosphorylation alters the translation of DENR targets. Importantly, phosphorylation of DENR is required for accurate chromosome segregation and mitotic cell viability (Fig. 2B; Clemm von Hohenberg et al. 2022). In most cases, perturbing any single phosphorylation event only modestly alleviates mitotic translational repression, suggesting that these modifications act collectively to mediate this effect. Overall, these data highlight the physiological importance of mitotic phosphorylation of translation factors.
5′UTR-mediated translational control during mitosis
Beyond the global reduction in translation that occurs during mitosis, translation is also controlled differentially at the level of individual genes and even at specific translation start sites. For example, specific mRNAs—and even coordinated groups of transcripts—undergo selective translational regulation during mitosis. Translation initiation is recognized as the rate-limiting step in protein production and is tightly regulated during mitosis. Much of this control is mediated by features in the 5′ untranslated region (UTR), including RNA-binding protein motifs, secondary structures, and uORFs (Hinnebusch et al. 2016).
Several groups of transcripts appear to be co-regulated at the level of translation initiation through their 5′ UTR sequences. For example, mRNAs encoding ribosomal proteins and translation factors display a high level of translation during mitosis (Park et al. 2016; Haneke et al. 2020). Notably, many of these transcripts contain a terminal oligopyrimidine (TOP) motif, characterized by a polypyrimidine stretch that is directly proximal to the 5′ cap (Philippe et al. 2020). The translation of TOP-motif mRNAs is inhibited by the binding of LARP1 to the 5′ cap and the TOP-motif (Philippe et al. 2020; Saba et al. 2024). Phosphorylation of LARP1 inhibits this binding, promoting the translation of TOP-motif mRNAs (Haneke et al. 2020). During mitosis, CDK1 phosphorylates LARP1, resulting in the activation of TOP-motif mRNAs (Fig. 2D; Haneke et al. 2020). Although enhanced TOP-motif mRNA translation during mitosis is consistently observed across multiple studies, its biological significance remains unresolved. One proposed model suggests that upregulation of the translation machinery in mitosis may prepare cells for robust protein synthesis in the upcoming G1 phase (Park et al. 2016), but this hypothesis requires direct experimental validation.
In addition to TOP motifs, uORFs are also regulated during mitosis to alter translation. Translation of AUG uORF usually causes the ribosome to terminate before reaching the main ORF such that uORFs are generally considered to act as translational repressors (Brar et al. 2012; Hinnebusch et al. 2016). The differential translation of uORFs between conditions can serve to remodel translational programs (Brar et al. 2012; Cheng et al. 2018). Based on ribosome profiling, proteomics, and reporter-based experiments, recent studies suggest that uORF translation is globally enhanced during mitosis relative to interphase (Kowar et al. 2025). For example, several cohesin subunits contain uORFs, and reporter assays together with ribosome profiling suggest that increased uORF initiation during mitosis represses the production of cohesion subunits (Stumpf et al. 2013). Similarly, MDM2 mRNA contains multiple uORFs (Jin et al. 2003; Hollerer et al. 2019) that are preferentially translated during mitosis, thereby repressing MDM2 protein production during mitosis (Ly et al. 2024b). Notably, MDM2 uORFs have strong Kozak start codons, and weakening them abolishes mitotic uORF activation in reporter assays (Ly et al. 2024b). Kowar et al. (2025) found that enhanced translation of uORFs during mitosis results in the increased presentation of uORF-derived peptides on the cell surface specifically in mitotically arrested cells. Given that frontline chemotherapies arrest cells in mitosis, the authors leveraged the increased uORF presentation in mitotically arrested cells to preferentially target mitotic cells for immunotherapies. For example, they found that CD8+ T cells can preferentially target surface uORF peptides in taxol-treated mitotic cells compared to interphase cells, providing a potential combination treatment for antimitotic chemotherapies (Kowar et al. 2025).
Together, these results highlight the potent role of 5′ UTR elements in the mitosis-specific regulation of translational efficiency.
3′UTR-mediated translational control during mitosis
In addition to the role of 5′ UTR-mediated regulation of translation, several 3′ UTR elements also contribute to translational control during mitosis. Perhaps the most striking example of 3′ UTR control is the mitotic regulation of the EMI1 3′ UTR (Tanenbaum et al. 2015). To ensure proper mitotic progression, the APC/C must be activated to trigger the degradation of key mitotic substrates, including cyclin B and securin (McAinsh and Kops 2023). Because EMI1 functions as an APC/C inhibitor, the EMI1 protein must be present at the onset of mitosis but eliminated prior to APC/C activation. Multiple layers of regulation achieve this dynamic control. Upon mitotic entry, EMI1 is inactivated by CDK1-mediated phosphorylation and subsequently degraded by E3 ubiquitin ligases. In parallel, the EMI1 mRNA is subject to mitotic translational repression. Tanenbaum et al. (2015) showed that this repression is mediated by the 3′ UTR of EMI1. Eliminating EMI1 mitotic translational repression by replacing the EMI1 3′ UTR impaired APC/C activity during mitotic exit, underscoring the importance of translational regulation for orderly cell cycle progression (Tanenbaum et al. 2015).
The 3′ UTR also plays a key role in translation in oocytes and early embryos. In particular, poly(A) tail length shows a strong positive correlation with translational efficiency (Subtelny et al. 2014; Xiang and Bartel 2021; Xiang et al. 2024). In contrast, this coupling between tail length and translation is much weaker during interphase of somatic cells (Subtelny et al. 2014). Interestingly, during somatic cell mitosis, a subset of mRNAs appears to display poly(A) tail length–dependent translational coupling (Fig. 2E; Novoa et al. 2010; Park et al. 2016). For instance, the poly(A) tail of TOP2A shortens during mitosis, coinciding with reduced translational efficiency (Fig. 2E; Park et al. 2016). Interestingly, the TOP2A mRNA is also actively degraded at mitotic exit, such that tail shortening may serve a dual role in repressing translation during mitosis while priming the transcript for destruction upon mitotic exit (Krenning et al. 2022). Despite this intriguing coupling, the mechanism of poly(A) tail length–dependent translation during mitosis remains unclear.
Start codon selection during mitosis
The mechanisms described above focus on cases that modulate the overall translational efficiency of an mRNA during mitosis. However, a single mRNA can produce multiple different protein products through the use of alternative start codons such that changes in start site selection can have dramatic impacts on protein function (Ly et al. 2025b, 2026). Due to leaky ribosome scanning during translation initiation, ribosomes can bypass the first start codon to initiate translation at a downstream start site, yielding an N-terminally truncated isoform (Wright et al. 2022; Ly et al. 2026). In addition, ribosomes can initiate at non-AUG codons in the 5′ UTR, generating N-terminally extended protein isoforms (Wright et al. 2022). Our recent work revealed that start codon selection is dynamically remodeled during mitosis, resulting in the differential usage of thousands of translation initiation sites compared to interphase cells (Ly et al. 2024b). Strikingly, mitotic entry triggers a global shift toward more stringent start-codon selection, characterized by enhanced translation of optimal Kozak context AUG start codons coupled with the repression of nonoptimal initiation sites, including non-AUG codons and AUGs in weak Kozak contexts (Fig. 2F; Ly et al. 2024b).
The key factors that control alternative start codon selection in mitosis are the translation stringency factors eIF1 and eIF5 (Grosely et al. 2025). eIF1 enforces the stringent control of start codon recognition, with higher eIF1 levels causing ribosomes to bypass poor Kozak context AUG and most non-AUG sites (Ivanov et al. 2010). In contrast, increased eIF5 levels relax start codon selection, such that eIF5 overexpression promotes initiation at nonoptimal AUG and non-AUG codons (Loughran et al. 2012). During mitosis, eIF1 activity is increased, resulting in stringent start codon selection. This change in eIF1 activity during mitosis is due to an elegant regulatory mechanism based on the differential localization of eIF1 in interphase and mitosis (Fig. 2F). During interphase, a substantial pool of eIF1 is sequestered in the nucleus where it is unable to interact with the cytosolic ribosome or alter translational control. However, upon mitotic entry and nuclear envelope breakdown, the nuclear pool of eIF1 is released into the cytosol where it is able to associate with the cytosolic ribosome (Fig. 2F). This nuclear release of eIF1 during mitosis acts to rapidly increase available eIF1 levels, mimicking an overexpression of eIF1 in the cytosol to increase the stringency of start codon selection (Ly et al. 2024b). Importantly, specifically eliminating the nuclear population of eIF1 abolishes the changes in start codon selection that occur during mitosis, induces mitotic apoptosis, and prevents mitotic slippage (Ly et al. 2024b) (see below).
Beyond leaky ribosome scanning, internal ribosome entry sites (IRESs) provide an additional mechanism for the cell cycle–specific production of protein isoforms. A well-characterized example is CDK11, which is expressed as two alternative N-terminal isoforms, CDK11p110 and CDK11p58 (Cornelis et al. 2000). Production of the shorter p58 isoform is driven by an IRES and is preferentially used during mitotic arrest. Notably, this shorter CDK11p58 isoform has many key roles during mitosis, such as sister chromatid cohesion (Hu et al. 2007) and bipolar spindle formation (Petretti et al. 2006). Although the molecular mechanisms governing this translational switch remain incompletely understood, these findings highlight the importance of mitotic cap-independent translation in generating cell cycle–specific alternative N-terminal protein isoforms. Collectively, these results implicate start codon selection as a key layer of mitotic translational control.
Alternative start codon selection tunes mitotic timing
The dramatic change in alternative start codon selection during mitosis shapes multiple aspects of mitotic cell physiology. In particular, the control of alternative translation is critical to mediate the balance between mitotic death and premature exit from mitosis. In the presence of mitotic errors, the spindle assembly checkpoint inhibits the APC/C, an E3 ubiquitin ligase that drives mitotic exit. At the center of this regulation is the APC/C substrate adapter CDC20, which plays a dual function as the key target of the spindle assembly checkpoint and as a promoter of APC/C activity (McAinsh and Kops 2023). When CDC20 is bound by checkpoint proteins, it acts as an inhibitor of APC/C, preventing premature chromosome segregation. Conversely, in the absence of checkpoint proteins, CDC20 can activate the APC/C, triggering anaphase and mitotic exit. The balance between APC/C inhibition and activation dictates the duration of the mitotic arrest in the presence of errors, with some cells ultimately “slipping” out of mitosis even in the presence of persistent damage. Thus, mechanisms that regulate the ability of CDC20 to associate with checkpoint proteins versus APC/C play a critical role in tuning mitotic arrest duration.
Recent work from our laboratory demonstrated that the single CDC20 mRNA undergoes alternative translation initiation to produce distinct N-terminal protein isoforms, which differentially regulate APC/C activity (Tsang and Cheeseman 2023). The CDC20 mRNA undergoes leaky ribosome scanning through the annotated CDC20 start codon, which is under a weak Kozak context. This allows for the production of a truncated CDC20 isoform that lacks 43 aa at its N terminus. Critically, this N-terminal truncation removes its Box 1 motif, a short linear motif that promotes the interaction of CDC20 with checkpoint proteins. Therefore, the truncated CDC20 isoform preferentially interacts with the APC/C. The relative ratio of the full-length and truncated CDC20 isoforms and their differential protein stability act as a timer to dictate the amount of time a cell with mitotic errors will remain in mitosis before undergoing mitotic exit to “slip” into interphase (Fig. 2F). An increased amount of full-length CDC20 relative to truncated CDC20 allows for prolonged mitotic arrest, whereas increased truncated CDC20 relative to full-length CDC20 induces premature mitotic exit (Fig. 2F; Tsang and Cheeseman 2023).
Importantly, CDC20 isoform ratios can also be tuned through regulated alternative start codon selection. When cytoplasmic eIF1 levels are elevated in mitosis, the increased stringency of start site selection promotes bypassing of the weak Kozak annotated CDC20 start codon, allowing increased translation of truncated CDC20. Over the course of a mitotic arrest, the truncated CDC20 isoform gradually accumulates until it can activate the APC/C even in the presence of mitotic errors, driving mitotic slippage (Fig. 2F). Importantly, eliminating the nuclear pool of eIF1 and the mitotic change in translation stringency blocks the accumulation of the truncated CDC20 isoform and suppresses mitotic slippage (Ly et al. 2024b). Therefore, mitotic translational control of CDC20 is critical to define the duration of a mitotic delay when cells experience errors.
Overall, these results suggest that the mitotic release of nuclear eIF1 enhances leaky scanning to alter CDC20 isoform ratios and potentially regulates the alternative translation of other factors, ultimately shaping the duration of mitotic arrest.
CONCLUSION AND FUTURE OUTLOOK
The precise temporal regulation of the cell cycle is essential for cellular life. Cells achieve this control by changing gene expression in a cell cycle–dependent manner, with precise global and gene-specific regulation at every step of gene expression, including transcription, mRNA processing, mRNA stability, translation, and protein modifications and decay. Recent studies highlight the key contributions of mRNA decay and translational regulation to cell cycle–dependent control of gene expression, and consequently cell cycle progression and checkpoint fidelity.
Although significant progress has been made in understanding post-transcriptional control of the cell cycle, important open questions remain. The studies described in this review provide insights into the regulation of mRNA stability of cell cycle–regulated transcripts and the mitosis-specific control of key translation factors. Future studies should address the physiological significance of this regulation by examining the consequences of disrupting the precise timing of these events.
A major event for mammalian mitosis is nuclear envelope breakdown. In this review, we highlighted the functional significance of eIF1 release from the nucleus during mitosis. The release of other nuclear RNA processing components may enable them to function in the cytosol at this stage (Aviner et al. 2017; Somma et al. 2020). Similarly, nuclear envelope breakdown also disperses many nuclear RNAs into the cytoplasm (Tarnowka and Baglioni 1979; Bury et al. 2020; Blower et al. 2023). Future work should explore potential cytoplasmic functions of nuclear RNAs during mitosis.
In addition to post-transcriptional regulation during the somatic cell cycle, parallels and distinctions emerge when comparing mitosis and meiosis (Ohkura 2015; Maier et al. 2021; Ly et al. 2024a, 2025a). For example, in both mitosis and meiosis, poly(A) tail length correlates with translational efficiency, indicating a shared regulatory logic. However, the range of this coupling differs between mitosis and meiosis. During mitosis, the coupling between poly(A) tail length and translational efficiency is largely restricted to transcripts with short poly(A) tails, whereas in meiosis this coupling extends across a much broader spectrum of tail lengths. Defining the molecular basis of such mitotic and meiotic differences in gene expression will be an important future direction.
In addition to uncovering the molecular mechanisms of cell division, studies of cell cycle–specific post-transcriptional control are relevant for cancer treatment. Our work and the other studies described here highlight the targeting of mitosis-specific translational control and mRNA decay machinery as a potentially effective therapeutic strategy in combination with antimitotic drugs. Future work will continue to uncover cell cycle–specific regulation of gene expression that may be leveraged in combination with frontline chemotherapies that affect the DNA damage checkpoints or other stages of the cell cycle.
ACKNOWLEDGMENTS
Work in the Cheeseman laboratory is supported by grants from the National Institutes of Health (NIH)/National Institute of General Medical Sciences (R35GM126930), and the Chan Zuckerberg Initiative Rare as One Project grant. J.L. and Y.F.T. are supported in part by the Natural Sciences and Engineering Research Council of Canada. E.K. is supported by the MIT HEALS graduate fellowship. We thank Tom Cech for critical reading of the manuscript and helpful suggestions.
Footnotes
-
↵3 Co-first authors
-
Article is online at http://www.rnajournal.org/cgi/doi/10.1261/rna.080910.125.
-
Freely available online through the RNA Open Access option.
This article, published in RNA, is available under a Creative Commons License (Attribution-NonCommercial 4.0 International), as described at http://creativecommons.org/licenses/by-nc/4.0/.








