
Identification of 3D motifs in Rfam with JAR3D
- 1Department of Computer Science, University of Findlay, Findlay, Ohio 45840, USA
- 2Department of Mathematics and Statistics, Bowling Green State University, Bowling Green, Ohio 43403, USA
- Corresponding author: rollj{at}findlay.edu
-
Handling editor: Adrian Ferre-D'Amare
Abstract
Many non-protein-coding RNAs are being discovered each year. At first we know them only by their sequences in a few organisms, but to understand their function and interactions, we need to understand what 3D structures they may form, in whole or in part. Many hairpin and internal loops are known to form recurrent structured 3D motifs, for example, kink turn and sarcin-ricin internal loops, and GNRA and T-loop hairpin loops. A new non-protein-coding RNA may have one or more known structured 3D loop motifs. Here we introduce a tool which identifies loops in Rfam seed alignments that match known 3D loop motifs and makes those identifications easily accessible. JAR3D was developed to map sequences of hairpin and internal loops to known 3D motifs, and was extended for this work to three-way and four-way junction motifs. We applied JAR3D to 4166 Rfam seed alignments from Rfam 15.0 and made the results accessible on the JAR3D web page, making it easy to evaluate the possible matches for each loop in each Rfam family. We provide several examples which validate JAR3D's ability to identify the correct loop motif, using 3D structures of RNAs outside of the training set. We created a page to search for instances of a particular loop motif across all Rfam families, to study how widespread the occurrence of each motif is. We provide statistics on how many Rfam loops appear to match well to a known 3D motif. Match rates are much higher for internal loops than for hairpins or multihelix junctions.
Keywords
INTRODUCTION
A large number of non-protein-coding RNAs of different types have been discovered in recent years (Chen and Kim 2024; Nemeth et al. 2024; Yadav et al. 2024). When a new noncoding RNA is discovered, it is important to learn about common features across different organisms. A first step is to gather sequences of a particular RNA from different organisms, make alignments using conserved regions, and use the fact that most RNAs have helices of Watson–Crick base pairs with AU ↔ GC ↔ GU covariation. In so doing, one can identify most Watson–Crick base pairs, known as the secondary structure (Gutell 2014). Alignments and secondary structures of many RNA molecule types are available at Rfam, a well-known and widely used database of noncoding RNA families (Ontiveros-Palacios et al. 2025). In this article, we work with Rfam release 15.0 from September 2024, which has 4178 RNA families.
Many noncoding RNA molecules function by virtue of the 3D structure(s) they form (Fabbri et al. 2019; Vicens and Kieft 2022). Sometimes, these 3D structures are stable enough that their 3D structures can be solved using X-ray crystallography, cryo-electron microscopy (cryo-EM), or nuclear magnetic resonance and deposited in the Protein Data Bank (Bonilla and Jang 2024; Burley et al. 2024). Well-known examples are ribosomes and tRNA. Determining the full atomic-resolution 3D structure of a noncoding RNA is difficult and usually lags behind knowledge of the secondary structure by several years. Of the 4178 Rfam families in Rfam 15.0, only 168 have associated 3D structures in the PDB (see Materials and Methods). Some Rfam families have very many associated PDB chains, from different organisms and/or solved under different experimental conditions. The representative sets of RNA 3D structures produced weekly by RNA 3D Hub organize the PDB chains by organism and molecule and now indicate the associated Rfam family, where available (Leontis and Zirbel 2012).
When an atomic-resolution 3D structure of an RNA molecule is not yet available, additional understanding can be gained by predicting the 3D structure from sequence. Recent advances have been made in predicting the full 3D structure at the level of xyz coordinates, with tools like AlphaFold3 and others participating in recent RNA Puzzles and CASP competitions (Kryshtafovych et al. 2023; Roll and Zirbel 2023; Abramson et al. 2024; Bu et al. 2025). In many cases, these tools are able to correctly predict the overall shape of the molecule and the Watson–Crick helices, and are making progress on modeling RNA–protein interactions. However, these tools still often struggle to predict non-Watson–Crick base pairs (Bu et al. 2025). We note that in order to be a target in a 3D structure prediction challenge, the molecule must have an experimentally solved 3D structure. Some noncoding RNAs may not adopt a stable 3D structure, or may adopt multiple semistable 3D structures, and yet smaller regions of the molecule may still adopt a stable 3D structure.
In this paper, we concentrate on the 3D structures of “loop” regions between the Watson–Crick helices, which are where the majority of non-Watson–Crick base pairs occur. As noncoding RNA molecules fold back on themselves to form helices and 3D structures, other structured regions are also formed. Hairpin loops (HLs) occur at the end of a helix where the chain turns back on itself, internal loops (ILs) occur between two helices, and junction loops join three (J3) or more (J4, J5, …) helices together. These regions are often referred to as loops due to how they are drawn in secondary structure diagrams, as if the nucleotides repel one another, but in fact the nucleotides in loops are often well structured by stacking, non-Watson–Crick base pairs, and base–backbone interactions (Leontis et al. 2006; Stombaugh et al. 2009; Zirbel et al. 2009; Parlea et al. 2016). The pairwise interactions in loops and the 3D structure tend to be conserved across organisms, and as such, they constrain the sequence variability in those regions of the RNA (Lescoute et al. 2005; Stombaugh et al. 2009; Parlea et al. 2016). In many cases, the loops make contacts outside the secondary structure to help form the full 3D structure and/or make critical contacts with other molecules. As such, identifying the loops correctly can help to predict the 3D structure and the function of the RNA.
Many hairpin and internal loop motifs are recurrent, meaning that the same arrangement of nucleotides occurs in different types of RNA molecules in nonhomologous locations (Lescoute et al. 2005; Petrov et al. 2013; Parlea et al. 2016). Examples of common recurrent loop motifs are kink turn and sarcin-ricin internal loops, and GNRA, UNCG, and T-loop hairpin loops, but there are many more. Instances of loops from representative high-resolution structures are organized by geometry and base pairs into “motif groups” in the RNA 3D Motif Atlas, which is updated every 4 weeks (Petrov et al. 2013). The RNA 3D Motif Atlas provides broad coverage of RNA loop motifs, well beyond the most common ones like sarcin-ricin, kink turn, or GNRA. On each motif group page, the instances are labeled according to the molecule that they occur in, making it easy to see when they occur in nonhomologous locations. Most known loop instances come from ribosomes, and some from multiple locations in the ribosome. Recurrent motifs also occur in the 3D structures of nonribosomal noncoding RNAs. Known 3D motifs are thus likely to occur in noncoding RNAs for which no 3D structure is yet known.
A number of tools use sequences to identify the presence of a recurrent loop motif, without making specific predictions of the xyz coordinates. They use probabilistic models to map novel RNA loop sequences to the known 3D structures they are likely to form. RMDetect (Cruz and Westhof 2011) builds Bayesian network models for IL motifs using multiple sequence alignments, and is distributed with models for four recurrent IL motifs. JAR3D makes stochastic context-free grammars (SCFGs) for all of the motif groups in the RNA 3D Motif Atlas based on the geometry of observed 3D loop structures (Zirbel et al. 2015; Roll et al. 2016). BayesPairing builds Bayesian network models for HL, IL, and junctions, searches a single RNA sequence with regular expressions, and accounts for the compatibility with secondary structure (Sarrazin-Gendron et al. 2019, 2020). CaCoFold-R3D uses covariation in multiple sequence alignments to predict secondary structure and pseudoknots as well as over 50 different RNA loop motifs, including HL, IL, J3, and J4 (Karan and Rivas 2025).
RMfam is a tool for the annotation of RNA structural motifs, with results available in Rfam since Rfam 12.0 (Gardner and Eldai 2015; Nawrocki et al. 2015). RMfam provides covariance models for RNA motifs that Rfam curators can use to scan sequence alignments for regions where a motif is likely to occur and then use that information accordingly when creating Rfam sequence alignments and consensus secondary structures. RMfam has a broader definition of motif than the tools listed above, which are only concerned with loop motifs. RMfam motifs will usually cover more cWW base pairs around loop motifs to allow for variability in flanking base pairs, and may even contain multiple loop motifs, or none at all. For example, a model for a sarcin-ricin motif in JAR3D would be concerned just with the internal loop and flanking cWW base pairs, whereas the RMfam models for sarcin-ricin motifs cover the internal loop plus an adjacent hairpin loop from the canonical example of the sarcin-ricin motif in Helix 95 of the large ribosomal subunit.
In previous work, JAR3D was used to identify recurrent loops in the potato spindle tuber viroid (PSTVd), an infectious RNA agent with 27 loops (Wu et al. 2019). JAR3D was used to predict that loop 27 of the viroid was likely to have a structure similar to the loop of a conserved hairpin located in the 3′ untranslated region of histone mRNAs in animal cells. JAR3D was then also used to predict which mutations to loop 27 were likely to disrupt the structure of the loop. Experiments with the mutations revealed that the structure of loop 27 was vital to the function of PSTVd, as mutations that were predicted to disrupt the structure of loop 27 disrupted the viroid's ability to replicate in infected plants. PSTVd does not have an Rfam family, so the work was done by manually inputting sequences into the JAR3D website. Similar work was done with loops 1, 6, and 15, with JAR3D being used to score the ability of mutated sequences to form the motif likely to be present in the wild-type molecule (Wu et al. 2024). The same approach could be used with other ncRNAs.
In this paper, we use JAR3D to map the sequences of all hairpin loops, internal loops, three-way junctions, and four-way junctions in each Rfam family to the best-matching motif groups in the RNA 3D Motif Atlas. We use the Rfam seed alignments to obtain the sequences and the Rfam consensus secondary structures to find the columns of the alignment that contain each loop. The motif matches for each Rfam family are precomputed and posted on the JAR3D website; the results page helps the user evaluate the quality of each match to a motif group. When a 3D structure is associated with the Rfam family, for each loop in the Rfam family we provide a link to visualize the corresponding nucleotides in 3D, making it possible to validate the motif identifications. On a separate page, one can search across all Rfam families for matches to a given motif group, so that one could, for example, find all good matches for a kink turn IL motif across all Rfam families. We give examples of the capabilities described above and note the cases where they provide independent validation of JAR3D's motif identification. We also give summary statistics about the matches of loops to known motif geometries across all Rfam families for which no 3D structure is available.
RESULTS
When a user types an Rfam family identifier in the JAR3D input page https://rna.bgsu.edu/jar3d/ or visits a URL of the form https://rna.bgsu.edu/jar3d/result/RF03064-3.48, the web server shows all hairpin loops (HLs), internal loops (ILs), three-way junction (J3), and four-way junction (J4) loops in the Rfam family and their best matches to motif groups. In the URL above, RF03064 is the identifier of an Rfam family, and 3.48 is the release of the RNA 3D Motif Atlas that it is scored against. Scoring against Motif Atlas release 3.98 is also available. The 12 ribosomal and tRNA Rfam families are excluded from this study because they are already well represented in the 3D structure database.
The JAR3D results page lists the loops in the Rfam family and the starting and ending columns of each strand of the loop in the seed alignment. Loops are numbered according to the lowest column number. For each loop, the sequences from the seed alignment are listed, along with their multiplicities, from highest to lowest count. For each loop, up to 10 matching motif groups are listed, along with numerical scores that indicate the quality of the match, as described in the RAGATH-18 example below. When the Rfam family is associated with a 3D structure from PDB, a link is provided for each loop to visualize the corresponding nucleotides in 3D, if any. This makes it possible to validate JAR3D motif identifications in 156 out of 4166 Rfam families.
The Results section has three subsections. In the first, we work through the JAR3D results for one Rfam family to illustrate how to interpret the matches for each loop. The family is chosen because some loops match known motifs well, and others have no good match. Many Rfam families are similar. Also, for this family there is a 3D structure available, which we can use to validate the predictions from JAR3D. In the second subsection, we illustrate the ability to search across Rfam families for a specific loop motif, which makes it possible to explore just how recurrent common motifs are. This subsection gives more opportunities to illustrate the process of interpretation and more opportunities for validation against 3D structures. In the third subsection, we give an overview of all loops in Rfam families for which no 3D structure is yet available, to get an idea of how many Rfam loops have good matches to known geometries and what known geometries match most often. This gives some insight into what percentage of Rfam loops are already known, and what percentage are novel.
RF03064 RAGATH-18 example
RAGATH-18 is a molecule that has been shown to have applications for RNA-guided DNA endonucleases (Ren et al. 2024). In this section, we discuss the motif matches for RAGATH-18 as they would have appeared when JAR3D release 3.48 was first available, which provides a retrospective validation of JAR3D's ability to identify the correct motif.
RAGATH-18 sequences appear in Rfam group RF03064. Results for the loops in RF03064 are available at https://rna.bgsu.edu/jar3d/result/RF03064-3.48. Figure 1 shows the secondary structure of RAGATH-18; there are four internal loops and one hairpin loop, numbered as in the JAR3D output. Figure 2 shows the top JAR3D matches to each of the loops, using the same numbering scheme. Four scores are given to evaluate the quality of matches: acceptance rate, mean cutoff score, full edit distance, and interior edit distance. More information on how these scores are calculated and how to interpret them may be found in Materials and Methods. Loop 1 is a very good match to a kink turn motif group, with 99.3% of the sequences from Rfam matching the family, and a mean cutoff score of 73, where 100 is the best possible. The full and interior edit distances make clear that the closest sequence match differs in one of the two flanking Watson–Crick base pairs; otherwise, there is an exact sequence match, which is another strong indicator of a good match. Loop 2 does not have a good match, as indicated by all of the numerical measures. That can be interpreted to mean that the loop makes a geometry that has not been seen before. With many loop nucleotides on one side of the stem and none on the other, a leading possibility is that this loop makes Watson–Crick pairs with another RNA, at least temporarily, which leaves no particular sequence or structure signature in RAGATH-18. Loop 3 is a good match to a sarcin-ricin internal loop; such loops occur in a variety of RNAs with the same geometry and sequence signature as the internal loop in Helix 95 of the large ribosomal subunit that is attacked by the sarcin and ricin toxins, but that does not imply that those toxins have anything to do with RAGATH-18. Loop 4 is an internal loop with highly variable sequence length from 4 to 11 nt. The top potential matches have no conserved base pairs beyond the flanking WC base pairs, and the 3D motif instances shown exhibit a variety of geometries of bulged and stacked bases. Mean cutoff scores are all negative, indicating that no one motif group is consistent with a substantial subset of the sequences. Loop 5 is a hairpin, which does not have a strong sequence match. Looking at the sequences of Loop 5 on the JAR3D site gives an indication why; some hairpin sequences are very long, and others are short, indicating that the end of this stem is of variable length. As such, it is unlikely to have a common 3D structure after the last Watson–Crick pair.
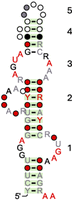
Secondary structure of RF03064 from https://en.wikipedia.org/wiki/File:RF03064.svg with loops numbered as in the JAR3D results in Figure 2.

Top JAR3D results for each of the five loops in RAGATH-18, listed in the same order as on the secondary structure in Figure 1.
Rfam group RF03064 first appeared in Rfam 14.1 with a release date of February 12, 2019. The Rfam seed alignment is the same in Rfam 14.1 as in Rfam 15.0, which we processed with JAR3D for this paper. RNA 3D Motif Atlas release 3.48 was based on RNA 3D structures from August 18, 2021. No 3D structures containing RAGATH-18 were available to JAR3D at this time. A 3D structure containing the kink turn from RAGATH-18 was released earlier on March 7, 2021 with PDB id 7EAG; this structure was added to the RNA 3D Motif Atlas later in version 3.77, and it is too small to be mapped to RF03064 (Huang et al. 2021). A full 3D cryo-EM structure of RAGATH-18 was released on April 17, 2024, with PDB id 8IAZ, chain E (Ren et al. 2024). Note that 8IAZ was solved by electron microscopy, so its loops were not used to build release 3.98 of the RNA 3D Motif Atlas or the JAR3D models based on release 3.98. 8IAZ chain E nucleotides 51–123 correspond to the RAGATH-18 structure in RF03064. The 3D structure clearly shows a kink turn at the base of the stem and a sarcin-ricin loop near the hairpin, corresponding to loops 1 and 3 in Figure 1. The regions of the 3D structure corresponding to loops 2 and 5 are missing nucleotides, and so we get no further insight about them. Thus, this example shows how JAR3D could have been used in August 2021 to process RF03064 and identify the kink turn and sarcin-ricin loops in RAGATH-18, providing two examples of retrospective validation.
Search across Rfam for a specific motif
We built a new web service that searches across Rfam for a specified motif to understand how frequently it may occur. The Rfam search page is at https://rna.bgsu.edu/jar3d/rfam/. After selecting the desired motif atlas release, the user may type one or more motif group identifiers in the input box. The examples on the page have prepopulated lists of all motif groups for selected common motifs such as kink turn, sarcin-ricin, and GNRA. Direct URL input is also enabled by following the format produced by the examples. See Materials and Methods for the criteria used to filter potential matches.
We illustrate by discussing the results of a search for a sarcin-ricin, or G-bulge, internal loop motif group across Rfam using JAR3D version 3.98. We use motif group IL_26307.2, a sarcin-ricin motif with 7 nt on one strand and 6 nt on the other. The search results can be found at https://rna.bgsu.edu/jar3d/rfam/3.98/IL_26307.2. Selected columns from the search results are shown in Table 1. The links in the “loop number” column direct to the matching loop on the results page for the indicated Rfam family. There are 22 loops in Rfam 15.0 that match IL_26307.2. Of the 22 matches, eight of the Rfam families were associated with 3D structures at the time of this writing, as indicated in the “PDB count” column in Table 1.
Results from searching Rfam for possible instances of IL_26307.2, a 7x6 sarcin-ricin internal loop motif.
For the eight matches to Rfam families having a 3D structure, we now discuss and interpret the strength of the JAR3D match and review the 3D structure data, as a guide to interpreting JAR3D results. In six cases, the 3D structure resolves the region of the molecule with the match and exhibits a sarcin-ricin motif; in two cases, the 3D structure does not cover the loop in question.
-
RF03064, RAGATH-18. Loop 3 was discussed above using JAR3D version 3.48, and the results with version 3.98 are similar. The acceptance rate of 98.24 and the mean cutoff score of 70.41 indicate a strong match. One can see the corresponding nucleotides in a 3D structure by clicking the link in the loop number column, then clicking “View nucleotides in PDB file(s).” The 3D structure shows a sarcin-ricin motif.
-
RF03022, RT-10. Loop 8 is a strong match, with an acceptance rate of 96.46 and a mean cutoff score of 79.74. Using the link in the loop number column, we see that the seed alignment for Loop 8 is very consistent, with little length variation in the sequences. The 3D structures show a motif very similar to a 7 × 6 sarcin-ricin, but with the typical AGUA base sequence replaced with AUUA and the first U bulged out and an arginine from a nearby protein where a G makes a base triple in the sarcin-ricin motif. The sequence AUUA occurs in some of the Rfam seed sequences; this is a clear example where some species have a sequence substitution, which alters the local geometry of the motif.
-
RF03072, raiA RNA. The acceptance rate is only 81.06, and the mean cutoff score is 43.26. The lower scores are due to many sequences in the seed alignment having too few bases in this portion of the alignment to form the sarcin-ricin motif, so they score poorly. Loop 11 has many sequences that match the sarcin-ricin motif well, and this is confirmed by the 3D structures as a 7 × 6 sarcin-ricin motif. Still, the JAR3D match to sarcin-ricin is not as clear-cut as the examples above. Aside from that, 55 sequences in the raiA RNA seed alignment have no bases in this portion of the alignment, and so JAR3D filters out those sequences without scoring them. Further investigation of this RNA and its alignment would be needed to understand why some sequences lack bases in this part of the alignment.
-
RF03087 ROOL RNA. The majority of sequences in loop 28 are a good match for IL_26307.2, but the alignment has a large number of sequences with fewer nucleotides in this region, resulting in a middling mean cutoff score of 51.6. The 3D structure shows a 7 × 6 sarcin-ricin motif.
-
RF00061 Hepatitis C virus internal ribosome entry site. Loop 6 has a 100% acceptance rate but a relatively low mean cutoff score of 15.60. The 3D structure containing the loop shows the geometry of a sarcin-ricin motif. Viewing Loop 6 on the RF00061 results page reveals that the best match for the loop is to another sarcin-ricin motif group, IL_88269.4, with an acceptance rate of 100% and a mean cutoff score of 87.99. IL_88269.4 is a 6 × 5 sarcin-ricin motif, which matches the size of the sequences seen in the hepatitis C virus internal ribosome entry site. Thus, in this case, the Rfam search identifies that a sarcin-ricin motif is present, but more steps are needed to find the best-matching sarcin-ricin motif group. We note that the loop results page only lists the top 10 matches; an Rfam search match might not be among the top 10. This is more likely to happen in smaller loops, which may have many possible geometries.
-
RF00011 Bacterial RNase P class B. Loop 9 is a weak match to sarcin-ricin, with an acceptance rate of 61.82% and a mean cutoff score of 14.95. RF00011 maps to four 3D structures, but only the PDB file 1NBS resolves the nucleotides in the corresponding positions; it shows a sarcin-ricin motif (Burmeister et al. 1992). The low mean cutoff score and acceptance rate can be explained as follows. From the RF00011 results page, we can click the “Align sequences to group” button for IL_26307.2 to view sequence-level results, including individual sequence scores and how the sequences align to IL_26307.2. The sequence-level results reveal a large number of likely UA cWW base pairs and missing nucleotides in the sequences that do not meet the cutoff for IL_26307.2. This seems to indicate that not all organisms in the seed alignment have a sarcin-ricin motif in this position, but many do. None of the available 3D structures are from organisms that failed to meet the cutoff, so further investigation would be required to know what is happening in this region in these other organisms.
-
RF03125, Sarbecovirus 3′UTR. The JAR3D match for the Sarbecovirus 3′UTR loop has a 100% acceptance rate, but a very low cutoff score of 0.34. Looking at the loop results page for RF03125, the best match for Loop 14 is IL_29198.2, a 7 × 7 sarcin-ricin motif with an intercalated A, but with a mean cutoff score only somewhat higher at 13.33. The seed alignment for RF03125 has 11 sequences, and they show no sequence variability in Loop 14, just one loop sequence 11 times over. The sequence lacks the “AGUA” subsequence typically seen in sarcin-ricin motifs, and the cutoff scores for the matches to IL_29198.2 and IL_26307.2 are low enough that the loop may not form the typical sarcin-ricin geometry, but may share some similarities. There is no mapping from RF03125 to a PDB chain, but RF03125 is part of an Rfam clan, which has a family that maps to a PDB chain. However, the 3D structure contains only part of the RNA, and does not cover Loop 14, so we have no direct evidence about the 3D structure of this loop.
-
RF03121 Alphacoronavirus 3′UTR. Loop 6 is a weak match to the sarcin-ricin motif group. Judging by the position of the characteristic AGUA sequence motif in the second strand, it is possible that some modifications to the alignment would improve the score, but such changes are outside the scope of this paper. The JAR3D website reports one 3D structure associated with RF03121, and here is how it works: There is no PDB chain that we map directly to RF03121, but Rfam family RF03121 is part of an Rfam clan, CL00117, Coronavirus 3′UTR. That clan contains seven Rfam families representing different coronaviruses, and one of them, RF00164, Coronavirus 3′ stem–loop II–like motif, maps to a PDB chain. In this particular clan, there is great variability in the size and the secondary structures of the Rfam families, with RF00164 being a short stem–loop and RF03121 having multiple internal and hairpin loops. There is no clear correspondence between these secondary structures, and so we simply do not have any 3D structure information for RF03121 Loop 6.
Of the six matches above where the loop geometry can be confirmed by a 3D structure, only the structure associated with RF00011 is used to build release 3.98 of the RNA 3D Motif Atlas. Thus, the other five examples are independent validations of JAR3D's ability to identify the correct motif.
The remaining 14 matches in the IL_26307.2 search listed in Table 1 have no 3D structures in the PDB at time of writing. We will briefly discuss some of these matches here. Most of these Rfam families were discovered through bioinformatics and have not been extensively studied. RF01703, Dictyoglomi-1 RNA, is one such Rfam family, though it is notable for having four conserved sarcin-ricin motifs (Westhof 2010), all of which appear in Table 1: Loops 4, 12, and 15 have IL_26307.2 as their top match, and all look like very strong candidates for a 7 × 6 sarcin-ricin motif, while the best match to Loop 9 is IL_41756.4, a motif group for a larger variant of the sarcin-ricin motif. Other strong matches to 7 × 6 sarcin-ricin are the loops in RF03535 and RF03021. The loops in RF02921 and RF03019 are stronger matches to other sarcin-ricin motif groups. The loops in RF02014, RF02686, RF0320, and RF03073 have a large number of sequences with single nucleotide changes to the AGUA sequence typically seen in sarcin-ricin motifs that result in high acceptance rate but lower mean cutoff scores. The loop in RF01446 has a fairly strong match to a non-sarcin-ricin motif group, IL_47346.2. The loop in RF00468 has a number of sequences that are a very good match to a 7 × 6 sarcin-ricin motif, but the majority are not; however, they were close enough to result in a positive mean cutoff score.
The SCFG models built by JAR3D for loop motifs are based on the sequence, consensus base pairs, and other interactions in each motif group in the RNA 3D Motif Atlas. The Motif Atlas makes relatively fine distinctions between motif instances that have different numbers or types of base pairs. As a result, JAR3D can identify different subclasses of common motifs such as kink turns and sarcin-ricin motifs. There are 17 motif groups annotated as sarcin-ricin, differing mostly in the number and type of base pairs away from the core base triple. The sarcin-ricin example on the Rfam search page provides all 17. If we had searched for all 17 sarcin-ricin motif groups at the same time, we would have seen multiple results for each of the eight loops in Rfam families associated with 3D structures listed above, with IL_26307.2 being the best match for the loops in ROOL RNA, raiA RNA, RT-10, Bacterial RNase P class B, and RAGATH-18, and with better-matching alternatives in the other families as discussed above. When one searches for only one motif group, JAR3D sometimes finds “partial” hits for motif groups, for which most of the structure of the motif is matched, but a base or base pair needs to be added or deleted. For common motif groups like sarcin-ricin motifs and kink turns, such variants have likely been seen in 3D structures and will already have JAR3D motif groups, but for others, a partial match is better than none.
The Rfam motif search results page has a feature that facilitates validation of JAR3D motif identifications. On the full search results page, there is a column (not shown in Table 1) that indicates the number of PDB chains associated with the Rfam family, among the PDB structures that contributed loops to the specified motif atlas release. When the number is zero, but the “PDB count today” column is nonzero, there is the opportunity to independently validate a JAR3D motif identification. As of this writing, only a few scattered examples present this opportunity. The instances that we checked matched the geometries predicted by JAR3D, but examining them comprehensively is outside the scope of this paper.
Statistics across Rfam families
In this section, we address the question: “How often do known 3D motifs occur in Rfam families that do not have a solved 3D structure?” We restrict our attention to the 4010 Rfam families that are not associated with a solved 3D structure in PDB. We also exclude some trivial internal loops: Many internal loops in Rfam appear because one or a few sequences have a short insertion, and these ILs generally map to motif groups of single-base insertions. To reduce the impact of such simple cases on the statistics, we only keep internal loops in which the average number of bases in the loop is 6 or higher; this excludes 4526 ILs, which are mostly single base bulges. We keep consistent bulges of size 2 or isolated noncanonical (such as UU cWW) or non-Watson–Crick (such as GG cWH) base pairs in the middle of a helix.
Table 2 shows the total number of each type of loop across these Rfam families, after the restrictions imposed above. For each loop, we scored the loop sequences against all JAR3D models in release 3.98 and counted how many loops had at least one motif group with a mean cutoff score above the threshold as indicated in the column headers. Because acceptance rate is always greater than mean cutoff score, the counts in the last two columns indicate the number of loops that have particularly good matches to JAR3D models for motif groups.
Counts of loop matches across Rfam families
Using a mean cutoff score of 60, we see that ∼15% of HL have a good match, and ∼50% of IL, but only ∼2% of J3 and J4 have a good match. For HL, the small percentage may be due to small noncoding RNAs often having stems that can accommodate insertions without disrupting important interactions, leading to no particular constraint on the length of the stem or the nature of the hairpin, and so no strong motif matches. For J3 and J4, the RNA 3D Motif Atlas also shows that multihelix junctions rarely occur again in nonhomologous locations; it is possible that they evolve anew each time to fit the needs of the RNA, with few junction sequence–structure geometries being preferred strongly enough to evolve independently.
In Table 3, we list the motif groups that have the largest number of strong matches to Rfam loops, among Rfam families that are not mapped to a 3D structure. HL and IL appear first, using a mean cutoff score threshold of 80, and listed in decreasing order of number of loops that match the motif group at or above that threshold. To conserve space, the listing ends with the well-known GNRA hairpin for HL and with tandem noncanonical cWW base pairs for IL. For J3 and J4, we use a lower mean cutoff score threshold of 60 and list all matches. Because more than one motif group may have a good score against the same Rfam loop sequences, there is double counting in the rows of Table 3.
Motif groups that have the largest number of Rfam loops that match them with a cutoff score above set thresholds, among Rfam families that are not mapped to a 3D structure
The HLs with the most matches are small, have no base pairs, and thus have little constraint on the base sequence. For IL, small loop motifs also dominate, with all but the last group being right at the minimum size that we consider for IL. Symmetric loops with one base on each strand account for most matches; these often make AG cWW or UU cWW or GG cWH base pairs. With J3, three of the four motif groups are recurrent, in the sense that the 3D motif instances come from nonhomologous positions in different RNAs, suggesting that these three-way junction geometries have evolved independently multiple times. Notably, the recurrent J3 loop motifs are larger than the minimal number of core nucleotides, bucking the trend with HL, IL, and J4. With J4, two of the three motif groups are recurrent, and all three have the minimal possible size, 8 nt, all of which make cWW base pairs to flank the four helices that are connected at the junction.
DISCUSSION
Identifying 3D loop motifs from sequences is challenging in many cases, for a variety of reasons, and often requires careful manual analysis. We touched on some of the considerations in the first two subsections in Results, where we had the opportunity to compare the quality of JAR3D matches to 3D structures. We provide a list of general considerations to keep in mind here, drawing from our experience with JAR3D and with the RNA 3D Motif Atlas:
-
Some loops have consistent sequence length and sequence variation that is compatible with a single known motif group, yielding a high acceptance rate and mean cutoff score, and then 3D motif identification is the most reliable.
-
Some loops have some sequences that match a motif group well and others that do not, often because of sequence length variation. This brings down the JAR3D acceptance rate and mean cutoff score. The motif may occur in some organisms but not others; there could be a sequencing problem, or there could be an alignment problem. More investigation and human judgment will be needed. In the JAR3D results, it may help to click “Align sequences to group” to evaluate the matches at the level of individual sequences.
-
Hairpin loops are particularly prone to sequence length variation, when the stem that they cap varies considerably in length between species. All sequence positions beyond the last common Watson–Crick base pair then appear in the HL sequence. JAR3D scores will tend to be poor. The hairpin turn itself may not have a consistent interaction partner in these cases, and so may not form a consistent 3D geometry.
-
Some loops have good matches to many motif groups. For example, single base bulges are the most common internal loop, and the 3D geometry of the bulged base is impossible to predict in isolation. When the bulge is always the same base, like A, the best JAR3D match will be to the motif group for that bulged base, and viewing that motif group will show a variety of 3D conformations for the bulged base. When the bulged base varies, the best-matching motif group is often a “major-groove platform” motif, which can be formed by a single bulged base of any type, but that is still just one of the possible geometries for a single base bulge. Similarly, small internal loops are often consistent with many different 3D geometries and could even change geometry dynamically.
-
Some loops have no good match to any known 3D motif group. There are several potential explanations, which are worth keeping in mind:
-
New 3D motif: The sequences may form a well-structured 3D motif that we have not seen yet. This is particularly likely for J3 and J4, because they seem to adopt new geometries often.
-
Flexibility: The loop may need to avoid folding into a stable 3D geometry to provide flexibility; a variety of sequences can accomplish this, so there is no clear sequence signature. Similarly, to avoid the attention of the immune system of the host, a helix may need to be interrupted occasionally by some internal loop, with no specific sequence requirement.
-
Pseudoknot: Some loops make long-range Watson–Crick base pairs with other parts of the RNA (pseudoknot) or with other RNA molecules. This happens with HL, with ILs that have one long strand and one short, and with J3 and J4. The strand that makes the WC base pairs has no particular sequence signature, although it may exhibit sequence conservation.
-
Ad hoc interaction: With HL and IL, the loop sequences may have evolved to form an ad hoc interaction with distant nucleotides in the same RNA or with a protein interaction partner. We see such loops often in the RNA 3D Motif Atlas, in loops that have few base pairs, little base stacking, and have the nucleotides bulged out to make a variety of interactions. The sequence may not match any known 3D motif.
-
Underfolding: Rfam can be underfolded, with fewer WC base pairs identified than are present in most organisms, with the result that the loop sequences are longer than the 3D motif that is present in the molecule, and then does not match well (Schneider et al. 2023). RMdetect and CaCoFold may work well here because they have the ability to identify additional Watson–Crick base pairs and focus on the unpaired nucleotides for motif matching (Cruz and Westhof 2011; Karan and Rivas 2025).
-
We note that identifying sarcin-ricin and kink turn internal loops, as we have done in examples above, is relatively easy because they are highly recurrent and have distinct sequence signatures. These motifs offer good guidance for interpreting the JAR3D loop results. They seem to occur in Rfam families with associated 3D structures more often than other common IL and HL motifs.
A key virtue of the tool described here is that it connects loops in Rfam structures to motif groups and thereby to other instances of the same motif in the same molecule or in other RNA molecules. That makes it possible to learn about interaction partners, functional roles, mutations that are seen and not seen, specific organisms whose loop sequences do or do not match the 3D motif, etc. Such knowledge can be more useful than simply getting a prediction of the xyz coordinates of the entire molecule.
With the broad coverage of loop motifs provided by the RNA 3D Motif Atlas, the results in Table 2 on the matches to known HL, IL, J3, and J4 indicate that few HL, J3, and J4 match the expected sequence variability of known 3D loop motifs. This will limit fragment assembly methods for predicting 3D structures that rely on matching entire loops to known geometries. We note that in some Rfam families, all loops may be novel, in the sense that they do not appear in the RNA 3D Motif Atlas, and so cannot be identified with JAR3D.
Here we compare the JAR3D-Rfam resource to the motif annotation tool RMfam. The output provided by the tools is similar; RMfam provides a fraction of hits measure, which is similar to JAR3D's acceptance rate, and a sum of bits measure, which can provide information similar to JAR3D's mean cutoff score. Both RMfam and JAR3D may match a region of an RNA to multiple possible motifs. JAR3D focuses on loop motifs, while RMfam's models are used to scan alignments for a broader range of RNA structural motifs. This means that JAR3D results are reliant on a correct secondary structure, whereas RMfam results are not. Because of this, RMfam will likely be able to detect and annotate more recurrent hairpin loop structures like GNRA and UNCG loops than JAR3D can, because of variability in the length of the stems that they cap. RMfam covariance models are limited to a range of 100 nt, which means that RMfam is unable to annotate internal loops for which the two strands are far apart, a limitation which is not shared by JAR3D. RMfam covers a limited number of well-known motifs, while JAR3D also covers less well-known motifs. For sarcin-ricin/G-bulge internal loops, RMfam includes only the canonical LSU H95 version together with an adjacent hairpin, whereas JAR3D includes these loops without the hairpin, which occur in many other contexts. Finally, motif hit locations in RMfam are only available in graphical form on an automatically generated secondary structure diagram, making it very difficult to compare the tools directly on a large scale. We note, however, that in RF03064 RAGATH-18, RMfam does not identify a kink turn or sarcin-ricin motif, while JAR3D does and the 3D structure confirms them.
While this paper was under review, the paper on CaCoFold was published (Karan and Rivas 2025). We processed all Rfam families (except tRNA and ribosomes) using the CaCoFold predicted secondary structures from the supplementary materials in that paper. JAR3D motif identifications within those secondary structures are now available following the format of these examples: type RF03064-CCF in the JAR3D input box, choose CaCoFold secondary structure on the Rfam motif search page, or get direct URL access using links such as https://rna.bgsu.edu/jar3d/result/RF03064-CCF-3.48 and https://rna.bgsu.edu/jar3d/rfam/CCF-3.98/IL_02349.4. The CaCoFold secondary structures solve some underfolding problems, introducing additional Watson–Crick base pairs to more accurately frame loop motifs. In Figure 5 of the CaCoFold paper, the kink turn motifs in Rfam families RF00012 and RF00015, plus the loop annotated K-turn in RF00017, are now identified by JAR3D and confirmed by 3D structures, where they were not identified by JAR3D using the original Rfam secondary structures. The K-turn-b in RF00017 and the K-turn in RF00018 are also newly identified by JAR3D, but 3D coordinates show a different IL in RF00017, and no 3D coordinates are available for RF00018. On the other hand, with RF03064 for RAGATH-18, where the original Rfam secondary structure correctly frames the sarcin-ricin motif discussed above, the CaCoFold secondary structure adds an additional Watson–Crick pair, shortening the loop sequences so that the outer bases do not show the usual GC, AU, GU base combinations, and the sequences are not well recognized by JAR3D. Comparing the sarcin-ricin motif query in Results to results with CaCoFold secondary structures using the URL https://rna.bgsu.edu/jar3d/rfam/CCF-3.98/IL_26307.2, 15 loops are the same, 15 are new (three that are confirmed by 3D structures, eight that appear to be strong matches, and four that appear to be weak matches), and seven loops were lost (one that had been confirmed by 3D structures, three that had good matches, and three that had weak matches). Repeating the study that produced Tables 2 and 3, CaCoFold secondary structures have fewer IL, but the counts are broadly similar, and the most commonly found motif groups are essentially the same. On the whole, CaCoFold secondary structures show clear improvement over the original Rfam secondary structures. To make the most complete set of motif identifications for an Rfam family, one should check the JAR3D results for both secondary structures.
The JAR3D motif matches for Rfam families will continue to improve due to the weekly updates on the RNA 3D Hub server. Rfam families that are already mapped to PDB chains will have additional chains added automatically. Novel matches of PDB chains to Rfam families will be reviewed manually between Rfam releases. When Rfam makes a new release, we will process the loops in all families with JAR3D versions 3.48 and 3.98 and will generate a new version of JAR3D with the RNA 3D motif atlas release available at that time. New Rfam families will have their loops processed by JAR3D, and any improved seed alignments and secondary structures in existing Rfam families will be used. Finally, if additional secondary structures become available, through other prediction methods or through modeling of alternative secondary structures, we will endeavor to include them as separate views of the Rfam family. Thus, over time, the JAR3D-Rfam resource will cover more noncoding RNAs, and more completely.
In conclusion, we used JAR3D to score all hairpin and internal loops and three-way and four-way junctions in substantially all Rfam families in release 15.0, to find the best matches in the RNA 3D Motif Atlas. We present the results in a user interface, which shows the strength of the matches and which links out to matching motif groups. Where available, we list 3D structures associated with the Rfam family and link to visualizations of nucleotides corresponding to each loop, to enable verification of the JAR3D motif identifications. We provide a separate interface to search for a given motif across all Rfam families. In this paper, we use examples to illustrate the evaluation and interpretation of JAR3D results. We provide a retrospective validation of motif identifications in RAGATH-18 and validate five sarcin-ricin motif identifications outside of the JAR3D training set. We describe potential new occurrences of the sarcin-ricin motif. Finally, we give statistics about the counts of HL, IL, J3, and J4 across Rfam families with no associated 3D structure and the rates at which they match known 3D geometries. Internal loops are most likely to match a known geometry, while HL, J3, and J4 have solid matches to known geometries less frequently.
MATERIALS AND METHODS
SCFG models for motif groups in the RNA 3D Motif Atlas
We followed the procedure detailed in Zirbel et al. (2015) to construct SCFG models for HL, IL, J3, and J4 motif groups in the RNA 3D Motif Atlas. An SCFG models the generation of new sequences subject to parameters set from 3D instances of the motif group. Nested base pairs are modeled by SCFG nodes that generate two bases at once on opposite sides of the sequence, and these are specific to the Leontis–Westhof base pair type. Thus, when a GC cWW is observed in a 3D instance, higher probability is given to generating GC, CG, AU, UA, medium probability to GU and UG, and lower for other base combinations. When an AG tHS is observed, the highest probability goes to AG and CA, according to the isostericity of the base pairs. Parameters are averaged over observed base combinations in the 3D instances. Base triples and larger networks are modeled by Markov random fields (MRFs). Conserved bases that stack on other bases but do not base-pair are modeled with low deletion probability. Bulged bases, which neither stack nor make base pairs, are modeled as variable length insertions, with length and base probabilities set according to the observed base distribution.
Creating JAR3D models for a release of the RNA 3D Motif Atlas is computationally intensive, as is scoring and storing all loops from Rfam with JAR3D, so we limit ourselves to two Motif Atlas releases, namely release 3.48 from August 18, 2021 and release 3.98 from June 18, 2025. We note that the code to extract loops from 3D structures was improved between releases 3.48 and 3.98 of the RNA 3D Motif Atlas, with the consequence that 3.98 has more J3 and J4 compared to 3.48. The Matlab code used to produce the SCFG models is available at https://github.com/BGSU-RNA/JAR3D. The data files for the releases and the JAR3D Java .jar files are available online at https://rna.bgsu.edu/data/jar3d/models/. The data files and .jar files make it possible for a user to run JAR3D locally on individual loops.
Scoring sequences against JAR3D models
The JAR3D SCFG models produce a probabilistic score for sequences based on how much the backbone of the RNA molecule would have to change compared to sequences seen in 3D if the sequence was folded into the same structure. We also calculate the minimum edit distance from each input sequence to the sequences seen in 3D, excluding the flanking cWW base pairs, which we call the interior edit distance. The probabilistic score is combined with the interior edit distance to create a cutoff score. The best possible cutoff score is 100; scores from 0 to 100 are said to be accepted by the model, and scores lower than 0 are said to be rejected by the model. There is no lower bound for the cutoff score. The combinations of probabilistic score and interior edit distance that make the cutoff score >0 are called the acceptance region. The acceptance region is specific to the motif group and is chosen to keep the false positive rate to 4% or lower, when tested against certain randomly generated sequences. Sequences from the motif group itself typically have cutoff scores above 90. The JAR3D output provides mean cutoff score across the input loop sequences and also acceptance rate, which is the percentage of sequences from the input loop sequences that have cutoff scores >0. The JAR3D output also provides the median interior edit distance and median full edit distance (including flanking cWW base pairs). More details and validation studies on novel sequences are available in Zirbel et al. (2015).
Extracting loops from Rfam secondary structures
Rfam release 15.0 has 4178 families with seed alignments. We excluded the 12 Rfam families for ribosomal 5S, 5.8S, SSU, LSU, and tRNA because they are well understood already from 3D structures in PDB and are not similar to current noncoding RNA 3D structure prediction targets. We downloaded the remaining 4166 Rfam seed alignments on July 24, 2025, using release 15.0 of Rfam. Pseudoknot symbols from the Rfam SS_cons secondary structure line were removed to produce a “reduced” secondary structure. The JAR3D web server extracted HL, IL, J3, and J4 sequences according to the reduced secondary structure. We find that 347 Rfam families have no predicted Watson–Crick base pairs in the seed alignment, and thus no loops to identify. JAR3D expects loop sequences to include flanking Watson–Crick base pairs, so extracted sequences with too few bases to form flanking pairs are noted but omitted from the scoring. The remaining sequences of each loop were scored against JAR3D models for each motif group in RNA 3D Motif Atlas releases 3.48 and 3.98, and these scores were stored in the JAR3D database.
We used the Rfam data in the JAR3D database to build an additional service which allows the user to input one or more motif group identifiers and receive back a list of all loops in the 4166 Rfam families that match the motif group. We filter for acceptance rate ≥80%, or mean cutoff score >0. This casts a wide net.
Mapping PDB chains to Rfam families
On the Rfam FTP site, the file Rfam.pdb lists associations between PDB chains and Rfam families. When a PDB chain has multiple associations, we keep the one with the highest bit score. The RNA 3D Hub weekly pipeline augments the associations in Rfam.pdb as follows. When a new PDB chain has the same experimental sequence as a chain already mapped, it is given the same association. For each PDB chain that has no association, we use cmsearch from Infernal (Nawrocki and Eddy 2013) to score the experimental sequence against all covariance models in the Rfam.cm file on the Rfam FTP site, keeping the match with the highest bit score. For established Rfam families, if the bit score of a match is better than the minimum seen for that family, the association is recorded. Matches to Rfam families beyond those listed in Rfam.pdb are reviewed manually, looking for a bit score above 30, a match length above 20, a matching molecule name, the PDB sequence fitting well with the seed alignment, and an adequate proportion of nucleotides observed with xyz coordinates. Applying the procedure to Rfam release 15.0, Rfam.pdb maps to 130 Rfam families, and 38 additional families were added manually. For the 168 Rfam families that have a PDB chain mapped to them, we combined the PDB chains with the Rfam seed alignment using cmalign and esl-alimerge from Infernal (Nawrocki and Eddy 2013). We used the combined alignment to map column numbers in Rfam seed alignments to the combined alignment and then to unit ids in PDB files; unit ids for nucleotides in PDB structures are described at https://www.bgsu.edu/research/rna/help/rna-3d-hub-help/unit-ids.html. The complete set of associations between Rfam families and PDB chains, the combined alignments, and the column number to unit id mappings are available online at https://rna.bgsu.edu/data/alignments/rfam/.
ACKNOWLEDGMENTS
This work was supported by the National Institute of General Medical Sciences of the National Institutes of Health, award number R01GM085328 to C.L.Z. The content is solely the responsibility of the authors and does not necessarily represent the official views of the National Institutes of Health.
Footnotes
-
Article is online at http://www.rnajournal.org/cgi/doi/10.1261/rna.080764.125.
-
Freely available online through the RNA Open Access option.
- Received September 12, 2025.
- Accepted December 11, 2025.
This article, published in RNA, is available under a Creative Commons License (Attribution-NonCommercial 4.0 International), as described at http://creativecommons.org/licenses/by-nc/4.0/.









{kind=link}