
Modulating the cleavage and polyadenylation site: from research tools to therapeutic opportunities
- 1Graduate Program in Bioengineering, School of Engineering and Applied Sciences, University of Pennsylvania, Philadelphia, Pennsylvania 19104, USA
- 2Department of Bioengineering, School of Engineering and Applied Sciences, University of Pennsylvania, Philadelphia, Pennsylvania 19104, USA
- 3Genome Regulation and Cell Signaling Program, Ellen and Ronald Caplan Cancer Center, and Center for Systems and Computational Biology, The Wistar Institute, Philadelphia, Pennsylvania 19104, USA
- Corresponding author: btian{at}wistar.org
Abstract
Almost all protein-coding and long noncoding genes that are transcribed by RNA polymerase II employ cleavage and polyadenylation (CPA) for 3′ end maturation of their nascent RNAs. More than 70% of human mRNA genes display alternative polyadenylation (APA), resulting in expression of isoforms using different CPA sites (also known as poly(A) sites or PAS). APA isoforms often have distinct mRNA metabolism and/or contain variable coding sequences. PAS mutations and genetic variations have been implicated in a growing number of human pathological conditions, underscoring the importance of PAS for proper gene expression. Here we review approaches that modulate the usage of specific PAS and discuss some of their applications in the context of human diseases. We provide our perspectives on current challenges and future directions of strategies of PAS modulation for studying APA isoforms and perturbing gene expression as a therapeutic modality.
Keywords
- 3′ end processing
- antisense oligonucleotides
- CRISPR
- U7 snRNP
- alternative polyadenylation
- genetic diseases
INTRODUCTION
Cleavage and polyadenylation (CPA) are two coupled 3′ end processing steps required for maturation of most eukaryotic protein-coding RNAs (mRNAs) and long noncoding RNAs (lncRNAs) that are transcribed by RNA polymerase II (RNAPII) (Fig. 1A; Sun et al. 2020; Boreikaitė and Passmore 2023). While the endonucleolytic cleavage of nascent transcript also plays an important role in termination of transcription, the poly(A) tail added to the cleaved transcript has crucial functions in aspects of mRNA metabolism (Fig. 1A). The site for CPA, that is, the poly(A) site (PAS), is defined by surrounding RNA elements, referred to as PAS motifs herein (Fig. 1B). These motifs are recognized by the CPA machinery, a mega-Dalton complex composed of ∼20 core factors in human cells (Fig. 1B; Shi et al. 2009). Core CPA factors can form several subcomplexes, including the cleavage and polyadenylation specificity factor (CPSF), cleavage and stimulation factor (CstF), cleavage factor I (CFI), and cleavage factor II (CFII) (Fig. 1B; Sun et al. 2020; Boreikaitė and Passmore 2023). CPSF can be further divided into two functional modules, that is, the mammalian polyadenylation specificity factor (mPSF) and mammalian cleavage factor (mCF) (Fig. 1B).
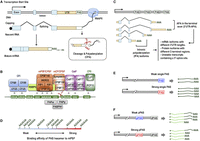
Aspects of cleavage and polyadenylation (CPA). (A) Schematic illustrating CPA during transcription. As shown, RNA polymerase II (RNAPII) makes a nascent RNA that is cotranscriptionally capped and spliced. PAS recognition triggers endonucleolytic cleavage of the nascent transcript, followed by poly(A) tail addition to generate a mature mRNA. (B) A canonical PAS recognized by the CPA machinery. The PAS is composed of two upstream UGUA elements bound by CFI, the PAS hexamer AAUAAA and adjacent U-rich motifs bound by mPSF, downstream U-rich and GU-rich motifs bound by CstF, and G-rich motifs bound by CFII. The cleavage site (CS) used by CPSF73 endonuclease cleavage is indicated by a scissor. Poly(A) tail synthesis is carried out by a poly(A) polymerase (PAPα and PAPγ are indicated), which is assisted by poly(A) binding protein PABPN1. (C) Multiple PAS within the same gene generate distinct mRNA isoforms. Cleavage within the 3′ terminal exon produces 3′ UTR-APA isoforms with different 3′-UTR lengths, whereas cleavage within introns yields intronic polyadenylation (IPA) isoforms that encode protein isoforms with different C-terminal regions or unstable transcripts. (D) PAS hexamers differ substantially in their binding to the mPSF complex as reported by Huang et al. (2025). The scale is approximately correlated with the Kd value for binding. Only the top 10 most frequent hexamers in human genes are shown (Tian et al. 2005). (E) Schematic showing that PAS strength is relevant to gene expression as demonstrated by human genetic mutations that strengthen or weaken the PAS. (F) Schematic showing APA isoform expression changes due to proximal PAS (pPAS) strength alterations. The strength of distal PAS (dPAS) remains constant in this example.
At least 70% of all human protein-coding genes harbor multiple PAS, leading to expression of mRNA isoforms with different 3′ termini, a phenomenon known as alternative polyadenylation (APA) (Shepard et al. 2011; Derti et al. 2012; Hoque et al. 2013). Most APA events take place in the 3′-most terminal exon of a gene, resulting in mRNA isoforms with different 3′UTR sizes (Fig. 1C). Owing to the many regulatory RNA motifs in the 3′UTR for mRNA metabolism (Mayr 2019), such as mRNA stability and localization, 3′UTR-APA can have substantial and cell type–specific impacts on posttranscriptional regulation of gene expression (Tian and Manley 2017; Gruber and Zavolan 2019; Mitschka and Mayr 2022; Tian et al. 2026). In addition, at least 20% of human genes contain PAS that are located upstream of the 3′-most exon (Fig. 1C; Tian et al. 2007; Wang et al. 2018b); CPA events at these PAS, mostly in introns and hence commonly known as intronic polyadenylation (IPA), lead to mRNA isoforms encoding protein isoforms with different C-terminal sequences as well as different 3′UTRs. Notably, IPA isoforms that contain a 5′ splice site (forming an internal/terminal composite exon, shown as the last case in Fig. 1C) are often unstable (Guvenek et al. 2022; Soles et al. 2025); as such, their activation generally leads to downregulation of gene expression. Studies over the last two decades have shown that APA isoform expression profiles are highly variable across cell types and conditions (Tian and Manley 2017; Gruber and Zavolan 2019; Mitschka and Mayr 2022; Tian et al. 2026).
In this review, we focus on molecular strategies that have been used to modulate CPA at specific PAS, including steric blocking of PAS motifs, genomic sequence editing of PAS, and regulation of PAS usage through perturbations of transcriptional elongation or splicing. We highlight key studies in which PAS modulation sheds light on APA isoform functions or leads to gene expression changes with disease implications. We discuss challenges in current methods and provide our perspectives on how PAS modulation strategies could be advanced as research tools as well as therapeutic modalities.
THE PAS AND CPA MACHINERY
The strength of a PAS, defined as its general efficiency of being recognized and used by the CPA machinery, is determined by its architecture, including the ensemble of PAS motifs and their relative placements to one another (Stroup and Ji 2023). Distinct interactions between the CPA machinery and PAS motifs could result in multiple cleavage sites (CS) for the same set of motifs (Stroup and Ji 2023). For clarity, we herein use the term PAS to refer to a group of CS that are located within a narrow window (typically ∼ ±24 nt) that are guided by the same set of PAS motifs.
PAS motifs
The hexamers AAUAAA or AUUAAA and their single nucleotide variants, typically located ∼20 nt upstream of the CS, play a central role for PAS strength (Fig. 1D; Sheets et al. 1990; Huang et al. 2025). Here we call this motif the PAS hexamer to avoid confusion with the term polyadenylation signal, which has also been used with the acronym PAS. The PAS hexamer is recognized jointly by CPSF30 and WDR33 in the mPSF module (Chan et al. 2014; Schonemann et al. 2014). For AAUAAA, the canonical hexamer, the A1, A2, A4, and A5 positions interact with CPSF30 and the U2 and A6 positions contact WDR33 (Boreikaitė and Passmore 2023). mPSF additionally contains CPSF160, a scaffold protein, and FIP1, which interacts with U-rich motifs adjacent to the PAS hexamer (Fig. 1B; Hu et al. 2005). Notably, two copies of FIP1 have been indicated to be present in the mPSF complex (Hamilton and Tong 2020; Muckenfuss et al. 2022), which is in accord with the fact that a PAS hexamer is generally flanked by both upstream and downstream U-rich motifs (Fig. 1B).
The binding affinity of a PAS hexamer to mPSF largely correlates with its usage activity in the CPA reaction in vitro (Sheets et al. 1990; Huang et al. 2025). For example, AAUAAA is approximately twofold more potent than AUUAAA in mPSF binding and CPA activity (Sheets et al. 1990; Huang et al. 2025), which is also in line with reporter assay data in vivo (Luo et al. 2013). Other variants of A[A/U]UAAA are substantially weaker than AAUAAA, by at least fivefold, in mPSF binding and CPA activity (Sheets et al. 1990; Huang et al. 2025). Importantly, whereas AAUAAA and AUUAAA are associated with ∼50% and ∼15% of all PAS in human genes, respectively, other PAS hexamers account for ∼20% of the total (Tian et al. 2005), indicating that many PAS have a suboptimal PAS hexamer.
The mCF module is composed of CPSF73, the endonuclease involved in the cleavage reaction; CPSF100, a pseudo-endonuclease with homology with CPSF73; and Symplekin, a scaffold protein. Human CS typically takes place before an adenosine (Wang et al. 2018b), even though the “CA” motif appears to be favored for some PAS (Sheets et al. 1990). Notably, it was found that JTE-607, a small molecule that inhibits CPSF73 through substrate competition, suppresses different CS sequences to variable degrees, indicating that CPSF73 could have some sequence preference (Liu et al. 2023). Together, the PAS hexamer, its surrounding U-rich motifs and the CS, constitute the core region of PAS, which interacts with the mPSF and mCF modules in the CPSF complex.
Within ∼30 nt downstream from the CS lie the U-rich and GU-rich (GUGU or UGUG) motifs that are recognized by CstF (Perez Canadillas and Varani 2003), a complex that is composed of CstF50, CstF64, and CstF77 and exists as a dimer in the CPA machinery (Boreikaitė and Passmore 2023). CstF64, or its paralog CstF64τ, is the RNA binding component of the complex. Also notable is the presence of the UCUG motif in the same region, which was found to be enriched for strong PAS (Hu et al. 2005; Stroup and Ji 2023). However, it is unclear whether the motif is also bound by CstF or another protein. Further downstream from the CstF binding site is the G-rich motif, often in the form of a stretch of guanosines. G-rich motifs are enriched for strong, 3′-most PAS (Wang et al. 2018b) and are likely to interact with PCF11 (Schafer et al. 2018) which, together with CLP1, constitutes the CFII complex.
PAS motifs located upstream of the PAS hexamer include UGUA and UAUA sequences. While the binding protein for the latter is unclear, UGUA interacts with the CFI complex, a tetramer composed of two molecules of CFI25 and two molecules of CFI68 and/or its paralog CFI59 (Yang and Doublié 2011). While CFI68 and CFI59 each contain an RNA recognition motif (RRM), the interaction with UGUA is mediated by the NUDIX domain of CFI25. Because of the tetramer configuration, two UGUA motifs are needed for strong interactions between CFI and its substrate RNA.
Since the PAS strength is determined by the ensemble of PAS motifs, a suboptimal PAS hexamer, that is, non-AAUAAA hexamers, can be assisted by the presence of other PAS motifs, such as downstream U-rich motifs (Nunes et al. 2010), to gain PAS strength. This compensatory relationship between PAS motifs is especially applicable to proximal PAS (pPAS), which, unlike the 3′-most PAS (often referred to as distal PAS or dPAS), are not typically associated with canonical PAS motifs (Hu et al. 2005; Yang et al. 2011). In addition, it should be noted that many nuclear RNA-binding proteins (RBPs) can influence PAS usage through binding to their cognate RNA motifs near the PAS, which is believed to play a key role in defining PAS usage profiles in a cell-specific manner (Tian and Manley 2017; Gruber and Zavolan 2019; Mitschka and Mayr 2022; Tian et al. 2026).
PAS mutations and genetic variants have been implicated in human disease conditions
Disruption of PAS usage by genetic mutations has long been shown to cause human diseases (Nourse et al. 2020). Not surprisingly, due to its central role in CPA, the PAS hexamer has been found to harbor most known disease-causing PAS mutations. In the first reported case, an A-to-G mutation, changing the PAS hexamer AAUAAA to AAUAAG, was found in the HBA2 gene (encoding hemoglobin A2) in α-thalassemia patients (Higgs et al. 1983). This CPA-suppressing (loss-of-function) mutation results in downregulation of HBA2 mRNA expression. Similarly, in the gene INS (encoding insulin), a mutation that changes AAUAAA to AAUAAG results in a >1000-fold reduction of INS mRNA levels and undetectable expression of insulin protein (Garin et al. 2010), causing neonatal diabetes in patients who carry homozygous mutations (Garin et al. 2010). Notably, the majority of known disease-causing PAS mutations have been found in genes with one predominant PAS (Nourse et al. 2020), suggesting that CPA inhibition is more detrimental to genes that rely on one major PAS for 3′ end processing as compared to genes that have multiple PAS. This phenomenon indicates that, for certain genes, CPA is a rate-limiting step for their mRNA production in some cell types or conditions. As such, alterations of PAS strength correlate with gene expression changes (illustrated in Fig. 1E). Along similar lines, recurrent mutations that disrupt canonical PAS hexamers appear widespread in cancers (ICGC/TCGA Pan-Cancer Analysis of Whole Genomes Consortium 2020; Kainov et al. 2024), which have been implicated in downregulation of mRNA expression of tumor suppressor genes (Kainov et al. 2024).
Besides the overt disease-causing PAS mutations, many genetic variants (GVs) affecting PAS motifs have been reported, some of which have clear clinical implications (Thomas and Saetrom 2012; Chen et al. 2021). For example, a germline single-nucleotide polymorphism (SNP) in the TP53 gene (encoding p53), resulting in either AAUAAA or AAUACA as the PAS hexamer, has an impact on its protein expression (Stacey et al. 2011). The AAUACA allele causes inefficient CPA of TP53 transcripts and hence decreased p53 protein expression, resulting in Li–Fraumeni-like cancer susceptibility in its carriers (Palmero et al. 2010). In addition, a growing number of GVs associated with APA isoform changes have been uncovered in the past few years (Mariella et al. 2019; Mittleman et al. 2020; Li et al. 2021; Tian et al. 2026). Many of these APA isoform expression-associated GVs, also called APA isoform quantitative trait loci or apaQTLs, have also been found to have associations with physiological traits and pathological risks (Li et al. 2023). A prominent example is the SNP (dsSNP ID: rs10954213) that affects the pPAS in the 3′UTR of IRF5 (Graham et al. 2007). An “A” allele leads to the strong PAS hexamer AAUAAA, whereas a “G” allele leads to a weaker PAS hexamer AAUGAA. As such, the A-allele-containing IRF5 gene preferentially expresses a shortened 3′UTR isoform that is more stable and produces a greater amount of IRF5 protein, a risk factor for the autoimmune disease systemic lupus erythematosus (SLE) (Graham et al. 2007). The relationship between pPAS strength and APA isoform expression is illustrated in Figure 1F.
STRATEGIES TO MODULATE CPA AT A SPECIFIC PAS
Three main approaches have so far been used to alter PAS usage (summarized in Table 1). First, steric hindrance of PAS motifs from their recognition by the CPA machinery leads to suppression of PAS usage. Second, genomic sequence editing of the PAS region through deletion or site-directed mutagenesis can permanently alter its usage. Third, blocking RNAPII elongation has proven effective in APA regulation. Here we discuss different techniques that have been used for these strategies.
Summary of current tools to alter PAS usagea
Antisense oligonucleotides (ASOs)
ASOs are typically composed of ∼20 bases that form duplex structures with their target RNAs (Liu et al. 2025). ASOs can sterically block interactions between mRNA and their cognate binding molecules, such as RBPs or microRNAs. RNA-based ASOs are usually modified for enhancement of stability and/or target affinity (Cakan et al. 2024), including 2′-O-methyl (2′-OMe) and 2′-O-methoxyethyl (2′-MOE) modifications of the ribose moiety and phosphorothioate (PS) and thiophosphoramidate structures that link adjacent riboses in place of the natural phosphodiester group. In addition, locked nucleic acid (LNA), which has a methylene bridge linking 2′-oxygen and 4′-carbon (Hagedorn et al. 2018), is commonly used in ASOs. Moreover, phosphorodiamidate morpholino oligomers (PMOs), or simply morpholinos, which consist of methylene morpholine rings linked through the phosphorodiamidate group, function similarly to ASOs. Antisense morpholinos (AMOs) have been widely used in vivo because of several advantages compared to other ASO chemistries, such as high target specificity (Summerton 2007). In addition, when DNA residues are used in an ASO, the duplex structure formed between the DNA and its target RNA recruits RNase H 1, leading to degradation of the target (Wu et al. 2004). DNA-containing ASOs often have the DNA part in the middle of the sequence and are also known as gapmers.
PAS-targeting ASOs have been shown to inhibit gene expression or alter APA isoform expression. An early study by Wu and Wu showed that a 21-mer DNA ASO with the PS backbone, which was designed to base-pair with a PAS in the hepatitis B virus, inhibited viral gene expression by over 80% in HepG2 cells (Wu and Wu 1992). While not explicitly shown, the effect is likely through RNase H1–mediated degradation of viral pre-mRNA or mature mRNA at the PAS region. Notably, this study also showed that conjugation of the ASO with an asialoglycoprotein-based carrier helped ASO delivery to cells bearing asialoglycoprotein receptors (Wu and Wu 1992). A more definitive demonstration of PAS regulation by ASOs came from the study by Vickers et al., in which ASOs were used to inhibit the usage of multiple PAS of the gene encoding E-selectin (Fig. 2A) (gene name: SELE) (Vickers et al. 2001). E-selectin is a cell adhesion molecule whose expression is mostly restricted to endothelial cells and is activated during inflammation. In the study, ASOs with 2′MOE and PS modifications, which make targeted RNA refractory to RNase H degradation, were designed to target three PAS in the 3′UTR through steric hindrance. Isoforms using the first two PAS have short 3′UTRs and are more stable than the isoform using the last PAS, presumably due to multiple copies of the AUUUA motif that promote mRNA decay (Vickers et al. 2001). It was shown that the ASOs targeting the last PAS, covering either the PAS hexamer or the CS, led to decreased expression of the longest 3′UTR isoform and a concomitant increase of two short 3′UTR isoforms. Because the two short 3′UTR isoforms are more stable than the longest 3′UTR isoform, the total mRNA level was also increased. Conversely, ASOs targeting the first two PAS led to increased expression of the longest 3′UTR isoform. This early work therefore not only demonstrates the feasibility of using PAS motif-blocking ASOs to inhibit PAS usage, but it also provides proof-of-principle evidence that perturbing one PAS in a gene that contains multiple PAS can alter relative expression levels of APA isoforms, thereby impacting overall gene expression.
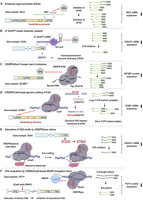
Current methods to modulate CPA at a specific PAS. (A) Antisense oligonucleotides (ASOs) lead to steric blocking of PAS motifs, physically hindering CPA factor binding. In the example gene SELE, ASOs targeting the distal PAS (dPAS) suppress CPA at the site but activate the usage of upstream, proximal PAS (pPAS) and expression of isoforms with shorter 3′UTRs and higher mRNA stability, thereby leading to increased SELE mRNA levels overall. (B) U7 SmOPT–based antisense cassette contains an antisense sequence complementary to its target RNA. In the example gene DUX4, a U7 SmOPT complex targets the disease-permissive PAS in exon 3, suppressing its CPA and leading to reduced DUX4 mRNA and protein levels. (C) CRISPR/dCas13-based steric hindrance of target RNA suppresses binding of CPA machinery. In the example gene IGF2BP1, targeting the dPAS shifts CPA to the pPAS, yielding a short 3′-UTR isoform that is more stable and efficiently translated than the long 3′UTR isoform. (D) CRISPR/Cas9-based genomic editing of PAS removes a PAS or modifies PAS motifs. The double-stranded breaks introduced by CRISPR/Cas9 are repaired by HDR or NHEJ. In the example gene CCND1, donor-mediated HDR inserts “AGGATCC” after the AATAA sequence, creating a canonical AAUAAA PAS hexamer. This much strengthened pPAS leads to substantially increased expression of a short, stable 3′-UTR isoform and hence elevates Cyclin D1 protein levels. (E) Alteration of PAS motifs by CRISPR/base editors. An adenine base editor (ABE) performs A-to-G conversion within the PAS hexamer to weaken or abolish its CPA efficiency. In the example gene DUX4, editing ATTAAA to ATTGAA vastly reduces CPSF recognition, suppressing CPA at the disease-permissive PAS in DUX4. (F) PAS modulation by CRISPR/dCas9-based RNAPII elongation block involves stalling of RNAPII between pPAS and dPAS, forcing the usage of pPAS. In the example gene PCF11, RNAPII stalling enhances intronic PAS, producing a truncated isoform with no apparent functions and hence downregulating gene expression of PCF11.
Several studies have used steric-blocking ASOs to target CPA events implicated in human diseases. Derepressed expression of the DUX4 gene in muscle owing to epigenetic changes causes cell death and muscle toxicity in patients with facioscapulohumeral muscular dystrophy (FSHD) (Arends et al. 2025). Importantly, the aberrant overexpression of the full-length DUX4 (DUX4-FL) mRNA is enabled by a PAS hexamer AUUAAA located in exon 3, which is present only in patients with a permissive 4qA haplotype (Lemmers et al. 2010). It was shown that AMOs targeting this PAS, covering either the PAS hexamer or CS, effectively inhibited the expression of DUX4-FL mRNA (Chen et al. 2016; Marsollier et al. 2016). Because inhibition of DUX4 protein expression has been the focus of FSHD therapies (Arends et al. 2025), PAS-blocking AMOs hold the promise of being a novel therapeutic modality for FSHD patients. Another notable study is the modulation of an intronic PAS in the gene encoding androgen receptor (AR). Usage of the PAS leads to expression of an IPA isoform AR-V7, which encodes a constitutively active form of AR protein. Importantly, AR-V7 expression has been implicated in resistance to AR-targeted therapies in castration-resistant prostate cancer (CRPC) patients (Van Etten et al. 2017). An AMO that targets the PAS hexamer of AR-V7 PAS was shown to suppress the expression of its mRNA, supporting the potential therapeutic use of the AMO for CRPC patients (Van Etten et al. 2017).
U7-small nuclear RNA–based antisense system
Similar in principle to ASOs, the modified U7 snRNP system with Sm-binding site optimized (termed U7 SmOPT) employs an antisense RNA sequence to base-pair with its target RNA, leading to steric hindrance (Fig. 2B; Gadgil and Raczynska 2021). Derived from the natural U7 snRNP, which is involved in the 3′ end processing of pre-mRNAs of replication-dependent histone genes (Ideue et al. 2012), the U7 SmOPT system has several changes in the U7 snRNA sequence that lead to inactivation of its activity in 3′ end processing of histone RNAs, higher transcriptional levels, and greater RNA stability (Wani et al. 2014). Because of its nuclear enrichment, the U7 SmOPT system is particularly suitable for targeting nuclear RNAs. In addition, due to its low cellular toxicity, the U7 SmOPT system has been employed in gene therapy studies against a wide variety of diseases, including perturbation of an exon skipping event in Duchenne muscular dystrophy (DMD) that is in clinical trials (Gushchina et al. 2023). The U7 SmOPT system has also been used to target the disease-permissive PAS of DUX4 gene (Rashnonejad et al. 2021). Using a plasmid expressing DUX4-FL in HEK293 cells, it was found that the PAS-targeting U7 SmOPT gave rise to similar DUX4-FL mRNA suppression to U7 SmOPT constructs targeting the start codon or splice sites (Rashnonejad et al. 2021). However, PAS-targeting U7 SmOPT was found to outperform other constructs in reducing DUX4 protein expression and DUX4-associated biomarker proteins in the myotubes derived from FSHD patients (Rashnonejad et al. 2021), raising the possibility that PAS-targeting might have additional effects on protein expression beyond suppression of CPA.
CRISPR-based steric hindrance
Based on the CRISPR/Cas13 system for targeted RNA degradation (Shi and Wu 2024), catalytically dead Cas13 (dCas13) offers sequence-specific binding of its target RNA, leading to steric hindrance effects. Using a reporter assay designed to evaluate the usage of a targeted PAS, Tian et al. compared eight types of Cas13 proteins from different bacterial species for their activities to suppress PAS usage, including dLbaCas13a, dLwaCas13a, dPspCas13b, dPguCas13b, dRanCas13b, dEsCas13d, dAdmCas13d, and dRfxCas13d (Tian et al. 2022). Of these, PguCas13b was found to be the most effective in suppressing PAS usage through targeting an upstream region containing UGUA motifs or the region containing PAS hexamer (Tian et al. 2022). This system, named CRISPR-iPAS, was used to modulate different types of APA events, including 3′UTR-APA and IPA events in skipped terminal exons or composite internal/terminal exons (Fig. 2C). Notably, targeting the PAS hexamer or the UGUA-containing region showed comparable efficiencies for PAS usage suppression. In addition, the CRISPR-iPAS system could be further enhanced when the dCas13 protein was tagged with three copies of enhanced green fluorescent protein (EGFP), suggesting that a bulkier dCas13 could lead to more effective steric hindrance (Tian et al. 2022).
CRISPR-based genomic editing of PAS
CRISPR/Cas9 enables targeted genome sequence removal through cutting double-stranded DNA by the Cas9 enzyme followed by direct DNA repair by the nonhomologous end joining (NHEJ) pathway or by homologous DNA repair (HDR) pathway with donor DNA (Doudna and Charpentier 2014). An early study by Wang et al. used the CRISPR/Cas9 system to mutate the PAS hexamer of pPAS (from AAUAAC to AAUAAA) in the 3′-most exon of CCND1 by the donor DNA/HDR method (Fig. 2D; Wang et al. 2018a). This change strengthened pPAS usage, leading to increased expression of a short 3′UTR isoform by ∼30% and a concomitant reduction of a long 3′UTR isoform expression by a similar amount. Because the short 3′UTR isoform of CCND1 is more stable, the overall transcript levels as well as the amount of protein increased substantially, leading to a higher cell proliferation rate caused by increased CCND1 activities.
Similar CRISPR/Cas9 strategies have been used to inhibit dPAS usage. Deletions of the dPAS in the gene Dscam1 in Drosophila (Zhang et al. 2019) and Calm1 in mice (Bae et al. 2020) have been shown to ablate long 3′UTR isoform expression in neuronal cells. In both cases, the long 3′UTR isoform to short 3′UTR isoform switch did not change the overall gene expression levels but led to impaired axon outgrowth, highlighting the importance of long 3′UTR isoforms for neuronal functions. In a similar vein, removal of an IPA site by CRISPR/Cas9 was used to validate the function of IPA isoform expression of PCF11 gene in mammalian cells (Kamieniarz-Gdula et al. 2019; Wang et al. 2019) and zebrafish (Kamieniarz-Gdula et al. 2019). It was found that the evolutionarily conserved IPA site of PCF11 plays a key role in maintaining the homeostasis of full-length PCF11 protein expression as well as the general CPA activity in the cell through IPA-mediated autoregulation of PCF11 expression.
The high efficiency of the CRISPR/Cas9 system makes it feasible to carry out high-throughput analysis of APA isoforms. In a study by Gabel et al., paired guide RNAs (pgRNAs) were used to delete 143 pPAS (first PAS in the 3′UTR) whose usage was dysregulated in cancer cells (Gabel et al. 2024). The −100 to +100 nt region of each PAS was used for pgRNA design, with the final library comprising eight to 10 unique pgRNAs for each pPAS. Using mouse melanoma cell lines B16-F10 and Melan-A, the authors found that removal of pPAS from the gene ATG7 forced usage of a distal PAS, effectively increasing the expression of its long 3′UTR isoform (Gabel et al. 2024). Because the long 3′UTR isoform has a lower protein output, pPAS ablation in ATG7 reduced ATG7 protein levels, slowing tumor growth and improving host survival (Gabel et al. 2024).
Alternation of PAS motifs by CRISPR base editors
While genomic mutagenesis through the donor DNA/HDR method has been widely used, CRISPR base editors have gained much traction in recent years for precise alternation of specific bases (Eid et al. 2018). CRISPR base editors employ a Cas9 nickase (nCas9) fused with a nucleotide deaminase enzyme, enabling direct conversion of targeted bases, such as C-to-U (cytosine base editor [CBE]) or A-to-I (adenine base editor [ABE]) changes (Komor et al. 2016; Gaudelli et al. 2017; Sikrova et al. 2021; Lee et al. 2025). The CRISPR base editor approach has been used to alter the disease-permissive PAS of DUX4-FL transcript in immortalized myoblasts derived from FSHD patients. It was found that in cells containing A-to-G base changes in the last three bases of AUUAAA, DUX4-FL expression was greatly diminished, corroborating the feasibility of editing PAS hexamer for CPA suppression and inhibition of mRNA expression (Fig. 2E). Interestingly, between the two cell lines that have different DUX4 expression levels due to the number of D4Z4 repeats, that is, baseline DUX4 mRNA levels being 100-fold higher in 3-unit D4Z4 repeat (FSHD13U) cells than 8-unit repeat (FSHD18U) cells, editing of the DUX4-FL PAS had a more pronounced impact on mRNA levels in FSHD18U cells (∼1000-fold downregulation) than in FSHD13U cells (∼10-fold downregulation). Notably, it was also found that the PAS editing redirected CPA toward some cryptic PAS located both upstream of and downstream from the targeted PAS, leading to residual DUX4-FL expression (Šikrová et al. 2021). Therefore, both baseline gene expression levels and surrounding PAS need to be taken into consideration when a PAS is targeted for modulation.
PAS modulation by CRISPR-based RNAPII elongation block
The CRISPR/dCas9 system has been used to inhibit gene expression by blocking elongation of RNAPII near the promoter region, an approach also known as CRISPR interference (CRISPRi) (Qi et al. 2013). CRISPR/dCas9 can also block RNAPII in the gene body. However, unlike in the promoter region, elongation block in the gene body is only functional when the dCas9 targets the nontemplate strand (Gilbert et al. 2014). This orientation dependence is due to the conformational difference between the sequences proximal versus distal to the protospacer adjacent motif (PAM) used by Cas9/dCas9 (Hall et al. 2022).
Shin et al. found that when the target site is located between two PAS (named pPAS and dPAS, respectively), CRISPR/dCas9 enhances CPA at the pPAS (Fig. 2F; Shin et al. 2022). Using this method, dubbed CRISPRpas, the authors demonstrated 3′UTR APA regulation in a reporter gene as well as in several endogenous genes, such as EIF1AD and TIMP2. Similarly, the method promoted IPA isoform expression in several tested genes, such as RAD51C, ANKMY1, and PCF11 (Shin et al. 2022). Variable effects on the overall gene expression were also observed, which appeared attributable to the distance between the CRISPR/dCas9 target site and the nearby PAS as well as mRNA stability levels of the resulting isoforms (Shin et al. 2022). PAS modulation by CRISPR/dCas9-mediated RNAPII elongation block was examined in more detail by the Proudfoot group (Zukher et al. 2023). It was found that CRISPR/dCas9 can cause RNAPII accumulation in the upstream region of target site and, consistent with the notion of elongation block, elicits transcriptional termination. The study also found that, unlike regulation of general elongation rate of transcription, the dCa9-based RNAPII elongation block impacts APA but not alternative splicing (Zukher et al. 2023). In addition, CRISPR/dCas9 targeting at the downstream region of 3′-most PAS alters RNAPII termination without discernable effects on gene expression. Therefore, CRISPR/dCas9-mediated elongation block appears specifically suitable for APA regulation.
Modulation of intronic PAS through splicing control
In addition to the direct PAS-suppressing methods mentioned above, technologies that modulate splicing can indirectly regulate IPA because of the kinetic competition between splicing and CPA. In line with this notion, global suppression of U1, U2, U4, or U6 snRNP by AMOs that target their snRNA sequences has been shown to activate IPA events globally (Kaida et al. 2010; Feng et al. 2024, 2025). Notably, ASOs that target the 5′ splice site region were found to switch transmembrane receptor tyrosine kinases (RTKs), such as the vascular endothelial growth factor receptor-2 (VEGFR2), to their secreted, soluble isoforms that have antagonistic functions (Vorlová et al. 2011). This approach holds the promise as a therapeutic modality for diseases that involve RTKs, such as cancer. Also notable is the method termed U1 adaptor, which entails a specific ASO that targets both a 3′UTR sequence and the 5′ region of U1 snRNP (called U1 domain in the ASO) (Goraczniak et al. 2009). It was shown that, through interactions with the U1 snRNP, U1 adaptors inhibit mRNA expression through U1-mediated inhibition of CPA (Goraczniak et al. 2009).
CURRENT CHALLENGES AND FUTURE OUTLOOKS
While an array of methodologies have been successfully used to regulate PAS usage in the past few years (summarized in Table 1), some challenges have emerged. Here we provide our perspectives on these challenges and potential solutions as well as considerations for further development of PAS-targeting technologies for therapeutic applications.
Targeting different PAS motifs
Most studies have focused on the PAS hexamer for steric hindrance or genomic editing. While the PAS hexamer is undoubtedly the most important signal for CPA, other PAS motifs, including the CS, can also play roles in defining the PAS strength. In addition, a PAS could be associated with multiple PAS hexamers, and some PAS hexamers can be located further upstream than their typical region, that is, ∼20 nt upstream of the CS. In the latter case, RNA structures have been shown to bridge a distal PAS hexamer to the CS (Wu and Bartel 2017). Therefore, for optional effects on PAS suppression, one needs to consider different parameters in designing target sequences for steric hindrance or genomic editing. To assist in the experimental design, PAS strength prediction tools, such as APARENT2 (Linder et al. 2022) and PolyaStrength (Stroup and Ji 2023), can shed light on how much PAS suppression can be achieved by changing PAS motif sequences. In addition, PAS conservation information could help understand the importance of targeted PAS motifs because of the negative selection pressure on functional sequences around the PAS (Wang et al. 2018b). Moreover, PAS reporter assays based on competition between two PAS can be used to corroborate the predicted effects (Shin et al. 2022).
Use of CPA inhibitors and activators
A set of small molecules have been developed in the past few years that inhibit CPA, such as JTE-607 and several benzoxaborole compounds (Kakegawa et al. 2019; Tao et al. 2024). These CPA inhibitors, or CPAi, suppress the function of CPSF73. In addition, several viral proteins have been found to have CPAi properties, such as the NS1 protein of influenza A virus (Bauer et al. 2018) and ICP27 of herpes simplex virus (Wang et al. 2020b). An unexplored area is whether these small molecules and viral proteins can be engineered to enhance specific PAS suppression. For example, conjugating CPAi small molecules or viral proteins to an ASO may provide additional CPA suppression activity and potentially enable inhibition of multiple PAS through a single ASO (Fig. 3A). Conversely, some protein domains of core CPA factors have been found to activate CPA, such as the intrinsically disordered regions (IDRs) of FIP1 (Lu et al. 2025) and subregions of RBBP6 (Boreikaite et al. 2022; Schmidt et al. 2022; Yoon et al. 2025). Whether these CPA activating domains can be employed by the CRISPR/dCas9 system to augment the usage of certain PAS is yet to be tested.
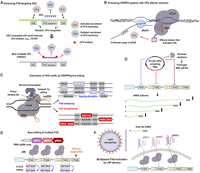
Potential strategies to enhance modulation of PAS usage. (A) PAS-targeting ASOs can be enhanced by conjugation with a CPA small molecule inhibitor, such as JTE-607, to achieve more complete inhibition of CPA for a gene. (B) CPA modulation domains tagged with CRISPR/dCas9 or dCas13 can help with PAS inhibition or activation. The effector domains can help recruit CPA machinery to the PAS or suppress the process (not shown). (C) Alteration of PAS motifs by CRISPR/prime editing. A prime editor (nCas9-reverse transcriptase fusion protein) precisely modifies PAS motifs. Colored boxes denote sequences inserted by prime editing, with red indicating insertions that strengthen PAS usage (e.g., conversion of weak PAS hexamers to stronger variants such as AAUAAA, or insertion of auxiliary UGUA-, GU-rich, or G-rich motifs), and blue indicating insertions that weaken PAS usage (e.g., insertion of random sequences that perturb PAS architecture). Cross symbols indicate sequence disruption by prime editing through deletion or mutagenesis of key PAS motifs. (D) Multiplexed PAS targeting by using a circular RNA containing multiple antisense sequences. As indicated, circular RNAs are quite stable because they are refractory to exonucleases. (E) Expression of multiple gRNAs from a tRNA–gRNA array for multiplexed PAS targeting. (F) Lipid nanoparticles (LNPs) can contain large cargos, enabling delivery of multiple gRNAs as well as the mRNA encoding a large Cas protein (dCas13b indicated in the picture).
Expanding CRISPR-based PAS editing
CRISPR/Cas9-based PAS engineering requires a PAM sequence near its target site, for example, NGG for Streptococcus pyogenes Cas9, the commonly used CRISPR/Cas9 system (Nishimasu et al. 2018). However, this requirement limits targetable sites in the surrounding regions of PAS, which are generally AT-rich (Tian et al. 2005). In light of this, CRISPR/Cas12a, which has emerged as a popular alternative to CRISPR/Cas9 (Jacobsen et al. 2019), uses TTTV as its PAM, making it more amenable to PAS targeting (Chen et al. 2020). As such, CRISPR/Cas12a and CRISPR/dCas12a systems are attractive alternatives for genomic editing of PAS and CRISPRpas-mediated APA modulation, respectively.
In addition, whereas mutagenesis of PAS motifs by CRISPR/base editors enables efficient single nucleotide modifications without introducing double-stranded DNA breaks, currently available base editors are restricted to specific base transitions, for example, C-to-T or A-to-G, limiting their target spectrum. In contrast, multiple base substitutions as well as short insertions and deletions can be readily achieved by using prime editing (Anzalone et al. 2019). By using a reverse transcriptase fused with nCas9 together with a prime editing guide RNA (pegRNA), CRISPR/prime editing allows precise rewriting of short cis regulatory elements (Fig. 3C; Anzalone et al. 2019). In addition, Cas9-based genomic editing relies on either HDR proteins whose expression is largely restricted to the S and G2 phases of the cell cycle or the NHEJ pathway, which generates mutations that require laborious characterization (Waters et al. 2014; Newby and Liu 2021). In contrast, prime editing does not require HDR proteins and ensures precise base changes, making it a desirable strategy to change short sequences in most cell types, including nondividing and primary cells (Anzalone et al. 2019).
Multiplexed PAS modulation
The ability to target multiple PAS is of importance in PAS modulation as accentuated by the studies of PAS suppression in DUX4 and SELE genes, in which targeting one PAS leads to “off-target” activation of alternative or cryptic PAS (see above). Therefore, targeting multiple PAS may be necessary in genes that display APA. Several methods are discussed here.
For ASO-based methods, circular RNAs that contain multiple antisense sequences have recently been found effective in simultaneous silencing of multiple target genes (Pan et al. 2025). Due to their closed-loop structure, circular RNAs are resistant to exonucleases and exhibit increased stability compared to linear RNAs, making them more long-lasting for in vivo applications (Fig. 3D; Enuka et al. 2016; He et al. 2021).
For multiplexed CRISPR-based approaches, tRNA–gRNA arrays have been used to express several gRNAs from a single transcript, which are processed by endogenous tRNA-processing enzymes (Fig. 3E; Yuan and Gao 2022). Akin to this, the CRISPR/Cas12a system has intrinsic CRISPR RNA (crRNA)–processing activities that can process multiple crRNAs from a single transcript (Campa et al. 2019). Notably, recent protein engineering of the dCas12a protein has substantially improved its DNA binding efficiency, further propelling CRISPR/dCas12a-based systems for in vivo applications (Guo et al. 2022). Also notable are the recent advances in using bacterial retrons for genome editing in human cells (Zhao et al. 2022). As part of the bacterial antiphage defense system, retrons generate copies of single-stranded DNA via self-primed reverse transcription (Buffington et al. 2025), which can potentially be harnessed for multiplexed PAS editing by CRISPR systems.
PAS modulation as a general tool to perturb gene expression
PAS modulation has been used primarily to target PAS with genetic mutations or regulate APA isoform expression. It is also conceivable that PAS modulation can be used as a general strategy to perturb gene expression. Because the PAS strength can differ greatly, as indicated by the large differences between different PAS hexamer variants in their binding to the mPSF complex (Huang et al. 2025), PAS modulation could up- or downregulate gene expression within a wide dynamic range. Conceivably, PAS modulation–based gene expression perturbation might be more straightforward for genes that use one predominant PAS than those having multiple well-expressed PAS. In contrast, selective modulation of alternative PAS would require detailed understandings of APA isoform properties, such as mRNA stability. In this regard, data from systematic analysis of mRNA stability at the APA isoform level could assist in PAS selection. Of note, IPA events might be of particular interest because of their marked effects on gene expression and protein products (Tian et al. 2007; Lee et al. 2018).
Considerations of therapeutic development
While detailed considerations of therapeutic development are not the focus of this review, we discuss some design issues that may have implications for future therapeutic applications. First, although ASOs can freely cross the plasma membrane, their localization into nucleus is not robust (Turner et al. 2005; Hill et al. 2023). Studies have shown enhanced nuclear enrichment of ASOs by using peptide- or small molecule–based nuclear importers (Kashyap et al. 2025). For example, cell-penetrating peptides have been shown to enhance both uptake and endosomal escape of AMOs, substantially increasing their nuclear localization and functions to regulate splicing in DMD and spinal muscular atrophy models (Järver et al. 2012). More recently, the bromodomain and extra-terminal (BET) inhibitor JQ1, known for its nuclear enrichment and chromatin engagement, has been demonstrated to improve the delivery of ASOs (ASO–JQ1 conjugates) to chromatin-associated RNA targets (Kashyap et al. 2025). This approach may also be attractive for PAS-targeting ASOs.
Second, one major concern for CRISPR-based gene therapies is the large size of the Cas protein versus the cargo limit of delivery systems. For example, the coding sequence of S. pyogenes Cas9 protein is ∼4.2 kb, which is close to the packaging limit of adeno-associated virus (AAV) vector, a commonly used delivery system in gene therapy that has a cargo limit of ∼4.7 kb (Dong et al. 1996). With the promoter region and guide RNA sequence included, the cargo size would exceed the limit (Wang et al. 2020a). To overcome this issue, recent studies have used smaller Cas9 orthologs and split-Cas9 systems (Levy et al. 2020; Davis et al. 2022). In this context, it is worth noting that the U7 Sm OPT system falls within the AAV packaging limit, making it an attractive approach for PAS-targeting gene therapies (Gadgil and Raczynska 2021).
Third, mRNA delivery by lipid nanoparticles (LNPs), which has shown clinical successes, for example, in mRNA vaccines (Hou et al. 2021), can accommodate up to 100 kb of total cargo (Miller et al. 2025). While achieving robust organ- and cell-type-specific delivery remains an active area of research for LNP-based therapeutics (Wei et al. 2024), LNPs appear to be particularly well-suited for multiplexed PAS-targeting CRISPR systems (Fig. 3F; Haque et al. 2024).
In summary, modulating the PAS has emerged as an effective strategy to correct genetic mutations affecting CPA, to examine APA isoform functions, and to perturb gene expression. Given the growing number of GVs that have been uncovered to affect APA isoform expression in various physiological and pathological conditions (Mittleman et al. 2020; Li et al. 2021), we envisage that modulation of PAS usage would play an instrumental role in understanding the significance of PAS mutations and GVs as well as affected APA isoforms. Moreover, because CPA can be a rate-limiting step for gene expression, PAS modulation has the promise of being a general tool to perturb gene expression in research and therapeutic settings alike.
ACKNOWLEDGMENTS
We thank members of the Tian laboratory for helpful discussions. Research in the Tian laboratory was supported by National Institutes of Health (NIH) grant R35GM153277.
Footnotes
-
Article is online at http://www.rnajournal.org/cgi/doi/10.1261/rna.080927.125.
-
Freely available online through the RNA Open Access option.
This article, published in RNA, is available under a Creative Commons License (Attribution-NonCommercial 4.0 International), as described at http://creativecommons.org/licenses/by-nc/4.0/.








