
RNA aptamers as tools for the purification and analysis of in vivo assembled ribonucleoproteins
- Corresponding author: ute.kothe{at}umanitoba.ca
Abstract
A large number of ribonucleoprotein (RNP) complexes are being discovered mediating numerous cellular functions. To investigate the composition, structure, and functional mechanism of RNP complexes, it is advantageous to isolate an RNP that was assembled in vivo. This review provides a systematic overview of a versatile and highly effective method to accomplish this task, namely, the purification of RNPs from cells using genetically encoded RNA aptamers. Inserting an RNA aptamer into the RNA of an RNP enables binding of the tagged RNP with high affinity and specificity to a ligand as an effective affinity chromatography purification strategy. Therefore, the purification of RNPs using aptamers has been used successfully to identify heterogenous populations of RNPs forming around a single RNA as well as to characterize intermediates in the formation of complex RNPs such as the ribosome. Here, we discuss in detail the selection of an appropriate RNA aptamer based on the properties of both the aptamer and its ligand, and we describe critical considerations in designing RNP purifications.
Keywords
INTRODUCTION
In addition to the well-known roles in storing and transmitting genetic information, RNA can serve a variety of structural and functional roles in many cellular processes. While some functional RNAs can operate as stand-alone RNA enzymes (ribozymes) (Esteban et al. 1997; Perreault et al. 2011; Harris et al. 2015), many RNAs form functional complexes together with proteins known as ribonucleoproteins (RNPs) (Collins et al. 2009; Krasilnikov 2011; Manikandan et al. 2023). As understanding of the many roles played by RNPs within cells is growing quickly, techniques to study the numerous interactions RNAs form within cells become more and more important.
Whereas insights into the function of well-known RNPs can be gained from in vitro reconstitution experiments, a comprehensive understanding of RNP's roles and mechanisms requires the examination of RNPs assembled in vivo. This goal can be achieved, for example, through the targeted extraction of the RNA and associated biomolecules or through use of reporters that can be visualized in the cell. In particular, large RNPs (such as the ribosome or spliceosome but also some smaller RNPs) have been studied using the targeted purification of RNPs to allow for analysis of specific states of their assembly or function as well as to identify novel components of the RNPs (Hartmuth et al. 2002; Zhang et al. 2016). Common strategies for RNP purification include immunoprecipitation using antibodies or the incorporation of affinity tags into the protein components of an RNP.
As the RNA is often the central and functional component of RNPs, there are advantages in inserting a target for purification, namely, an RNA aptamer, into the RNA component of the RNP (Hogg and Collins 2007; Chaker-Margot et al. 2015; Zhang et al. 2016). RNA aptamers are RNA structural elements that bind with high affinity and specificity to a given ligand. By incorporating RNA aptamers into an RNA, the RNA and associated components of the RNP can be purified by immobilizing the ligand to a chromatography resin. For instance, the Klinge and Ye labs used aptamer-tagged ribosomal RNA (rRNA) truncations to select specific stages of small subunit ribosome assembly (Chaker-Margot et al. 2015; Zhang et al. 2016). This approach allowed them to identify the ribosomal proteins, assembly factors, and RNA components of each of these ribosome precursors using a combination of mass spectrometry, gel electrophoresis, and blotting assays (Chaker-Margot et al. 2015; Zhang et al. 2016). Due to the unknown composition of these preribosomes and the dynamic nature of ribosome assembly, identification of preribosome components would be practically impossible in an in vitro system. Subsequently, a similar approach allowed purification and characterization of large ribosomal subunit precursors (Chaker-Margot and Klinge 2019). Notably, the Klinge lab also used aptamer-tagged rRNA truncations to purify ribosome precursors for structure determination by cryo-electron microscopy (cryo-EM) (Sanghai et al. 2023). Likewise, the Collins lab applied RNA aptamer-tagging to show that the 7SK RNA, a regulator of the P-TEFb kinase, forms a diverse set of RNPs, each containing a different population of proteins (Hogg and Collins 2007). In this case, the purification of tagged RNAs, combined with the analysis of protein content by mass spectrometry, identified an unexpected set of additional protein interaction partners for 7SK RNA. This finding enabled the characterization of the interaction of these newly discovered proteins with 7SK RNA, providing further insight into the biological role of 7SK RNPs. These examples illustrate how it is useful to incorporate the aptamer into the RNA at the genetic level to tag the target RNA without disrupting the natural processes of the cell, such as RNP assembly pathways. In this review, we will provide an overview of different RNA aptamers and the design of tagging an RNA with an RNA aptamer for affinity purification. Subsequently, we will discuss critical considerations for RNP purifications using RNA aptamers as a versatile tool to study in vivo assembled, functional RNA–protein complexes.
DEVELOPMENT OF RNA APTAMERS
The earliest RNA aptamers were developed in 1990 to bind a protein (Tuerk and Gold 1990) and a set of organic dye molecules (Ellington and Szostak 1990). Both RNA aptamers were developed using similar methodologies, wherein a randomized pool of RNA is applied to a ligand-derivatized chromatography resin, and the bound RNAs are used as an RT-PCR template for the next round of randomization. This technique is known as systematic evolution of ligands by exponential enrichment (SELEX) (Tuerk and Gold 1990). Since then, a wide array of RNA aptamers has been developed targeting different ligands either from randomized RNA libraries or from existing RNA structures that are further optimized by SELEX. RNA aptamers are generally short RNAs with a length in the range of tens of nucleotides. While some SELEX experiments use RNA sequences larger than 100 nt, the resulting aptamers are often used as a basis for determining smaller RNA aptamers that maintain binding activity (Wang and Rando 1995; Hamasaki et al. 1998).
Other RNA aptamer design considerations can be incorporated as either modifications to the basic SELEX pipeline or as refinements to RNA structures uncovered by SELEX. For example, SELEX pipelines can include a counter-selection step. In cases where an RNA aptamer must specifically bind a target ligand with much higher affinity compared to its affinity for similar molecules, selection for sequences that show strong binding to the ligand is not sufficient—it is also necessary to perform a second selection step wherein RNAs that show a strong affinity for undesirable ligands are removed from the pool. The counter-selection step increases specificity of the aptamer even within sets of highly similar ligands such as theophylline and caffeine, which differ only by one methyl group (Jenison et al. 1994). This approach is also useful in removing RNA structures that display nonspecific binding to a class of molecules, such as proteins.
Beyond optimizing the affinity and specificity of an RNA aptamer for its ligand, it is also beneficial to select RNA aptamers with distinct kinetic binding parameter. In particular, a slow dissociation (off) rate from the ligand is desirable as it ensures that the aptamer remains stably bound to a chromatography resin during washing steps. In addition, a fast association (on) rate can help with capturing the RNA aptamer when incubating the cell lysate with the chromatography resin. To achieve slow off and high on rates, the selection step in SELEX can be performed under stringent conditions such as short binding times, extensive washes with free ligand in the wash buffers, and long elution times. The free ligand competitively binds to RNA aptamers, that dissociate from the resin during SELEX, and prevents rebinding such that RNA aptamers with high off rates are removed during the selection (Davis and Szostak 2002; La et al. 2024). For a small number of RNA aptamers, the on and off rates for ligand binding have also been determined through biophysical characterization (Davis and Szostak 2002; Latham et al. 2009; Moschen et al. 2015), but most RNA aptamers have not been kinetically characterized preventing a systematic comparison.
The RNA structures generated by SELEX often represent an optimal binding aptamer that can be used as a basis for so called “minimal binding structures” by reducing the size of the RNA aptamer while only minimally altering its ligand affinity. Due to the ease of inserting these smaller RNA aptamers into other RNAs as affinity tags without disturbing the RNA function, these smaller RNA aptamers are often selected for use in pulldowns and other aptamer-based techniques over the larger original structure. For instance, the original tobramycin-binding RNA aptamer generated through SELEX binds with a KD of 0.77 nM to tobramycin but consists of 109 nt (Wang and Rando 1995). Subsequently, several smaller related RNA aptamers were created with a reduced length of only 40 nt, which still bind tobramycin with a KD in the low nanomolar range (5.15 nM) (Hamasaki et al. 1998). By now, one of these smaller tobramycin-binding RNA aptamers, the J6F1 variant, is more widely used than the original J6 RNA aptamer (Hartmuth et al. 2002; Hogg and Collins 2007).
OVERVIEW OF RNA APTAMERS
Aptamers are typically categorized by their ligand, with RNA aptamers binding protein ligands classified separately from RNA aptamers that bind to small molecules. Within sets of aptamers targeting a single ligand, there is often a great diversity in size and structure. Comparison of protein-binding RNA aptamer structures reveals a variety of interactions facilitated by hydrogen bonds, ionic interactions, and van der Waals interactions (Bjerregaard et al. 2016). In small molecule–binding RNA aptamers, the aptamer often forms a binding pocket that surrounds the ligand, binding it through hydrogen bonds and ionic interactions similar to ligand binding by proteins (Jiang and Patel 1998). In the case of small molecule ligands with aromatic groups, the ligand can also sometimes be seen to intercalate into RNA helices or quadruplexes forming stacking interactions with the nucleobases (Trachman et al. 2018).
While RNA aptamers require a great diversity in tertiary structure to successfully achieve their inherent affinity and specificity, many aptamers have conserved secondary structure regions that can be altered without changing function so long as compensatory mutations are made to maintain base-pairing. In these cases, small alterations are often made to the RNA aptamer depending on the requirements of the application. In particular, base pairs between the 5′ and 3′ ends of the aptamer forming a closing helix are useful when integrating the aptamer into a target RNA sequence. For instance, the S1 streptavidin aptamer is one of several similar aptamers designed based on a larger RNA structure that was evolved to bind streptavidin in a manner that competes with the interaction of streptavidin with biotin (Table 1; Fig. 1; Srisawat and Engelke 2001). This RNA aptamer was successfully used by the Dragon lab to purify the proteins associated with an essential small nucleolar RNA (snoRNA), snR30 (Fig. 1C). For this purification, the aptamer was incorporated into the loop of a nonessential stem–loop structure of snR30 (Lemay et al. 2011). The Sephadex binding aptamer D8 features a similar bulged stem–loop structure (Fig. 1E) and was evolved to bind to the common size exclusion chromatography resin, which consists of dextran B512 (Srisawat et al. 2001). This aptamer was not used for a full RNP purification but was shown to be successfully purified by chromatography as part of its design (Srisawat et al. 2001). Later, variants of the S1 and D1 RNA aptamers with altered secondary structures were compared by purifying poliovirus RNAs after transfection of the RNA aptamers into a human cell line (Flather et al. 2016). By extending the stem–loop and removing mismatched base pairs in both S1 and D8, increased purification effectiveness could be achieved (Leppek and Stoecklin 2014; Flather et al. 2016). The resulting aptamers, denoted as S1m and D8m, are larger overall than the original RNA aptamers, but contain similar loop and bulge regions (Fig. 1, compare C and E; Leppek and Stoecklin 2014; Flather et al. 2016).
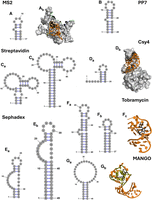
Comparison of aptamer structures used in RNP pulldowns. The secondary structures of aptamers are shown as predicted in the respective publications (Table 1). (A) (a) MS2 protein binding aptamer and (b) a 3D representation of the aptamer with the stem in orange, the bulged nucleotide in gold, and the loop region in yellow bound to the protein in gray (PDB ID 5MSF); (B) PP7 protein-binding aptamer; (C) variants of the S1 streptavidin binding aptamer, (a) S1 and (b) S1m; (D) (a) the Csy4 protein-binding hairpin, (b) a 3D representation of the aptamer (PDB ID 4AL5); (E) variants of the Sephadex binding aptamer (a) D8 and (b) D8m; (F) variants of the tobramycin-binding aptamer (a) j6f1, (b) J6, and (c) a 3D representation of the aptamer in orange bound to tobramycin in black (PDB ID 2TOB); (G) (a) the MANGOII aptamer and (b) a 3D structure of the aptamer in orange showing the G-quadruplex in gold and the ligand (thiazole orange-biotin) in black (PDB ID 6C65). Three-dimensional structures were visualized in The PyMOL Molecular Graphics System (Version 2.7, Schrödinger, LLC).
RNA aptamers used in RNP purifications
In other aptamers, a single bulged nucleotide is highly important to the efficacy of the aptamer (Fig. 1A,B; Johansson et al. 1997; Rowsell et al. 1998; Lim and Peabody 2002). The MS2 aptamer and the PP7 aptamer are two RNA aptamers that bind to specific viral coat proteins. The MS2 aptamer was developed by SELEX to bind the coat protein of the MS2 phage. Interestingly, the MS2 aptamer bears marked similarities to the RNA sequence that is responsible for the assembly of the viral capsid, especially in the loop regions and a conserved bulge within the loop, which are essential for its interaction with the protein (Fig. 1A; Johansson et al. 1997). This similarity to naturally evolved systems is common in aptamers with a biological counterpart, such as the aptamer which binds the T4 viral DNA polymerase, one of the earliest aptamers developed (Tuerk and Gold 1990). Reliance on a bulged nucleotide for binding is also seen in the similar PP7 aptamer, which was developed to bind the coat protein of the PP7 phage. Like the MS2 aptamer, it consists of a stem–loop with a single bulged adenine base that is required for binding to the coat protein (Fig. 1B). Removal of the bulged region results in an almost complete loss of the ability of the aptamer to bind its target protein (Lim and Peabody 2002). The PP7 aptamer is used as part of the RNA affinity in tandem (RAT) system (Hogg and Collins 2007). In the RAT system, two RNA aptamers are attached to the 5′ or 3′ end of a target RNA, which is then purified by tandem purifications. This system uses the PP7 aptamer along with a tobramycin-binding aptamer and has successfully been applied with a variety of RNAs including the 7SK RNA, the telomerase RNA component (TERC), and a C/D box snoRNP, SNORD27 (Hogg and Collins 2007; Falaleeva et al. 2016; Roake et al. 2019). Another RNA, that very strongly binds to a protein, is a 16 nt hairpin interacting with the Cys4 endoribonuclease (Fig. 1D; Lee et al. 2013). This naturally occurring RNA has been used as an RNA aptamer together with an inactive Cys4 variant to purify and characterize microRNA–protein complexes. Due to its short-length, the Cys4-binding hairpin RNA is particularly well suited for the analysis of small RNAs.
The tobramycin aptamer used in the RAT system is one of two RNA aptamers that are most commonly used for affinity purifications. In the initial evolution of the tobramycin aptamer, a large (>100 nt) structure was discovered that binds to tobramycin with high specificity and affinity. A specific stem–loop in this region, the J6 stem–loop, was found to be the minimal binding region of the aptamer, which exhibits strong tobramycin binding despite its reduced length (Wang and Rando 1995; Hamasaki et al. 1998). This extended stem–loop, J6F1, has been used to successfully purify precursors of the spliceosome by incorporating it into a pre-mRNA that binds the spliceosome (Fig. 1F; Hartmuth et al. 2002).
The fluorogenic RNA aptamer Mango has also been used for RNP purification as it offers several advantageous features. RNA Mango was developed with the purpose of creating an RNA aptamer that bound to the weak fluorophore thiazole orange thereby causing a fluorescence increase in the ligand (Fig. 1G; Dolgosheina and Unrau 2016). Due to the weak fluorescence of the unbound thiazole orange and the >1000 increase in fluorescence quantum yield when bound to the RNA aptamer, it is therefore possible to visualize and observe the thiazole orange–RNA Mango complex by fluorescence (Autour et al. 2018). The RNA Mango aptamer can also bind to biotinylated and desthiobiotinylated version of the thiazole orange ligand. By coupling these ligands to a streptavidin derivatized resin, the Mango aptamer has been used as an affinity tag for the purification of the U1 RNA and its associated RNP (Panchapakesan et al. 2017). In addition to the established ability of the Mango system to purify RNP complexes, a wealth of other fluorescent aptamers, such as Pepper and o-Coral offer potential for usage in purification based on their high affinity as reviewed by the Unrau and Ren labs (Lu et al. 2023; Lei et al. 2025). In these cases, the ability to use the RNA aptamer for RNP purification depends on the derivatization off the ligands to bind a resin. A clever approach to use the coPepper RNA aptamer for purifications was recently reported by first designing the cognate HBC ligand (mimicking the GFP fluorophore) to include an electrophilic handle that reacts with the RNA aptamer forming a stable covalent bond. Secondly, the authors included a vinyl handle in the ligand, which can react with desthiobiotin-tetrazine allowing purification for the coPepper aptamer and associate proteins by streptavidin affinity chromatography (Bereiter et al. 2025).
INTRODUCTION TO RNP PURIFICATIONS USING RNA APTAMERS
In addition to tagging RNAs with RNA aptamers, other methods of RNA purifications have previously been compared (Janvier et al. 2024). These techniques include the modification of RNA oligonucleotides with various chemical groups, which allows for these RNAs to be directly purified using the chemical group as an affinity tag (Moritz and Wahle 2014). Similar approaches can be used for RNA aptamer-based purifications, either by modifying the RNA or by using a tagged oligo that binds to the target RNA via base-pair interactions. While direct modification of the target RNA offers a variety of purification options, it is restricted to in vitro prepared or isolated RNAs. Meanwhile, the usage of tagged oligonucleotides that bind RNA via base-pair interactions are limited by the availability of the target sequence for base-pairing within the RNP complex and the strength of the base-pair interaction (Rogell et al. 2017).
Genetically encoded RNA aptamers are often of most benefit when the goal is to purify an in vivo assembled RNP. Using RNA aptamers to facilitate an RNP purification is a versatile method that works in a variety of circumstances and offers a modularity in the selection of aptamers and purification conditions (Fig. 2). However, due to the complexity of living systems and the variety of downstream applications, there is no “one size fits all” approach for the design of an RNP purification. The selection of an aptamer and purification system must consider the stability of the RNP complex and the requirements for the future analysis. Furthermore, as is the case with protein tags, careful selection of a location to introduce the RNA aptamer tag and thorough controls are necessary to ensure the RNA aptamer is not altering the expression or function of the target RNP. The major considerations for experimental design therefore depend both on the RNP and the downstream analysis.
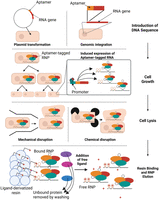
Workflow of a RNP purification using RNA aptamers as affinity tag. The RNA gene of interest can be tagged with an aptamer in the cell by introduction of a plasmid or by incorporation into the genome. During cell growth, expression of the aptamer may be induced chemically depending on the promoter used to generate cells containing the RNP. Cells are then lysed by one of several methods of chemical or mechanical lysis such as lysozyme digestion or high-pressure cell disruption, respectively. RNPs are subsequently isolated from the lysate by binding of the RNA aptamer to a ligand-derivatized resin. Following removal of unbound cell proteins and other RNAs in the lysate, the RNP is eluted by addition of excess ligand or through other methods based on the aptamer-ligand system used. Created in BioRender (Rocca and Kothe 2025, https://BioRender.com/u96m804).
It is a well-established method to purify an RNP based on the properties of the RNP other than the RNA component, that is, by size or by chromatography targeting protein components of the complex. While aptamer-based pulldowns offer specific targeting of RNA components of an RNP, purification of a complete RNP complex is most effective when two purification methods are combined. Size exclusion methods offer the advantage of separating complete complexes from free RNP components such as isolated aptamer-tagged RNAs, which bind a resin without the associated proteins. For example, size exclusion chromatography was used by the Unrau lab in combination with aptamer-based pulldown of a Mango tag in order to purify the RNP associated with the U1 snRNA (Panchapakesan et al. 2017).
Purification of RNPs based on the binding of protein components to a chromatography resin is often used in combination with RNA aptamer-based purifications. For instance, the purification of ribosome precursors often combines the purification of an aptamer-tagged rRNA precursor with a purification of a known protein component of the preribosome to ensure that intact ribosome precursors are isolated (Chaker-Margot et al. 2015; Zhang et al. 2016).
RNA APTAMER SELECTION AND PURIFICATION STRATEGIES
The most critical features in the selection of an RNA aptamer for RNP purifications are the size of the aptamer, its ligands, and its binding properties for each ligand (Table 1). Strong and specific binding of the target ligand is one of the defining characteristics of an RNA aptamer. However, strong binding can refer to affinities that range in dissociation constant from the micromolar range to subnanomolar (Patel et al. 1997). On the one hand, a higher affinity of the RNA aptamer for the ligand allows for improved binding of the RNP complex, especially at low concentrations. On the other hand, recovery of the RNP complex relies on an elution step that can effectively disrupt this interaction between the RNA aptamer and its ligand without affecting the stability of the RNP. Therefore, the binding kinetics of the RNA aptamer are also critical features although they are often unknown. As outlined above, an ideal aptamer binds with a fast on rate to its ligand during incubation of a lysate with a chromatography resin. However, an RNA aptamer should have a slow off rate preventing its dissociation from the ligand during washing steps but requiring a long elution time.
An important criterion for the selection of the RNA aptamer is the nature of the ligand, which determines how the aptamer interacts with the chromatography resin during purification (Fig. 3). Protein-binding aptamers are often selected for RNP purifications for various reasons. Protein ligands are easily customized and produced through protein overexpression and purification. As well, chromatography resins for affinity-based protein purifications tend to be inexpensive and robustly characterized. For instance, the MS2 coat protein-binding RNA aptamer has been used in conjunction with a multitude of fusion proteins combining the MS2 coat protein domain that binds the aptamer with known affinity tags such as the hexahistidine tag or tandem affinity purification (TAP) tags. For example, a fusion of the MS2 coat protein with the binding domain of maltose binding protein (MBP) allows for the purification of RNPs tagged with the MS2 coat protein-binding aptamer using maltose- or dextrose-derivatized chromatography resins (Jurica et al. 2002; Chaker-Margot et al. 2015; Zhang et al. 2016). The same MS2 coat protein-binding RNA aptamer has been used in conjunction with a hexahistidine-tagged MS2 coat protein for purification via Ni-NTA chromatography resins or a fusion to streptavidin binding protein for purification using streptavidin agarose (Slobodin and Gerst 2010). When using RNA aptamers binding to a protein ligand, it is common for fluorescent protein domains to be incorporated into protein ligands to detect the cellular localization of the tagged RNA or for fluorescence resonance energy transfer (FRET) analysis to analyze for colocalization with other factors (Slobodin and Gerst 2010; Laprade et al. 2017). Whereas the addition of a fluorescent protein to the protein ligand does not necessarily improve RNP purifications, the fluorescent proteins remain a useful tool for analysis in combination with the purification (Slobodin and Gerst 2010).
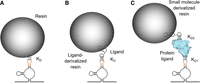
RNA aptamer interactions with chromatography resins. Noncovalent interactions are denoted as triple dotted lines and the respective dissociation constant (KD), and elution of the RNP occurs through disrupting these interactions, for example, by adding competing molecules. (A) Some RNA aptamers (orange) can interact directly with the chromatography resin. (B) Small ligand-binding RNA aptamers bind to chemical groups, which are covalently linked to the resin. (C) Protein-binding RNA aptamers interact with a protein (light blue, KD1) that itself interacts noncovalently with the resin (KD2), typically through a functional group linked to the resin.
However, the use of RNA aptamers binding to proteins can also introduce several disadvantages. Most notably, the protein ligand introduces a new large molecule to the RNP, which can sterically clash with the other RNP components. The presence of large quantities of a protein ligand can also complicate the downstream analysis of the protein components of the RNP. For example, when purifying RNPs containing the MS2 aptamer, gel-based analysis will show large bands corresponding to the MS2 protein ligand, and structures determined by cryo-EM or X-ray crystallography of the RNP will contain densities corresponding to the MS2 protein that is not a part of the native structure.
For RNA aptamer binding to a small ligand, a different set of advantages and concerns need to be considered for RNP purifications. The tobramycin-binding aptamer has been often chosen for these purifications due to its high affinity, small size, and the ready availability of the ligand. A particular advantage of this system is the coupling of the tobramycin ligand to an activated ester support resulting in a stable cross-link of tobramycin to the chromatography resin (Fig. 3; Hartmuth et al. 2002; Hogg and Collins 2007). In the past decade, there have also been large strides made toward the improvement and development of new fluorogenic RNA aptamers, whose ligands exhibit a strong increase in fluorescence when bound to the RNA aptamer. The Unrau lab has demonstrated that the fluorogenic aptamer Mango can be used for the RNP purifications. This was accomplished using desthiobiotin-derivatized versions of the ligand thiazole orange in combination with the Mango aptamer to purify the U1 snRNA (Panchapakesan et al. 2017).
For each RNA aptamer, there are often multiple options for the elution of the bound RNP from the chromatography resin. The simplest form of RNA aptamer elution is providing an increased concentration of free ligand in the elution buffer in order to outcompete the binding of the RNA aptamer to the immobilized ligand (Hartmuth et al. 2002; Hogg and Collins 2007). More complex RNA aptamer elution systems have also been developed offering several advantages. For example, when using the strong streptavidin–biotin interaction to immobilize a biotin-conjugated ligand, the initial capture of the RNA aptamer is performed using derivatives like desthiobiotin to allow for a weaker binding of the ligand to the chromatography resin. Subsequently, the aptamer is eluted by a relatively low concentration of biotin, which binds more tightly to the streptavidin chromatography resin than desthioiotin (Panchapakesan et al. 2017). The streptavidin binding aptamer is also eluted using biotin, as biotin outcompetes the aptamer for binding to the streptavidin resin (Lemay et al. 2011). Recently, the Unrau lab reported two other clever approaches to elute the Mango aptamer (Lu et al. 2024). While elution with competitive ligands typically must occur at higher temperature which may be detrimental to the RNP, they developed a photocleavable thiazole orange-biotin ligand that allows using UV light to cleave the RNA aptamer binding thiazole orange dye from the biotin ligand interacting with the streptavidin chromatography resin. Alternatively, elution of the Mango aptamer can also be achieved by photobleaching thiazole orange such that it dissociates from the aptamer. Both approaches yield higher RNP recovery than eluting with free biotin.
In protein-binding aptamers, the elution can occur either by disrupting the aptamer–protein interaction or the protein interaction with the chromatography resin (Fig. 3). Another possibility is the inclusion of a protease cleavage site in the protein such as the TEV or Factor Xa recognition sequences, which allows for the separation of the aptamer binding protein domain from the resin binding protein domain by the addition of the respective protease to the elution buffer (Hogg and Collins 2007; Falaleeva et al. 2016; Roake et al. 2019). Alternatively, the interaction of the protein with the chromatography resin can be directly disrupted similarly as in protein purifications by using a buffer containing a competitive elutant such as maltose for maltose binding protein fusions (Jurica et al. 2002; Said et al. 2009; Lalaouna and Masse 2015). In cases where the goal of the RNP purification is only the identification of associated proteins, elution can also be accomplished by degrading the RNA component of the RNP with the help of RNases (Leppek and Stoecklin 2014). A variant of this method uses a conditional form of the ribonuclease Csy4 as a ligand. This Csy4 variant, Csy4*, is inactive in the absence of imidazole. The elution of RNPs tagged with the Csy4 binding hairpin involves activating the nuclease activity of the protein by adding imidazole. This cleaves the hairpin from the tagged RNA and simultaneously elutes the RNP (Lee et al. 2013).
INSERTION OF THE APTAMER INTO THE RNA SEQUENCE
Due to limitations in the prediction of the tertiary structure of RNAs, it is difficult to ensure that the introduction of an aptamer to an RNA sequence will not disrupt the folding and subsequent function of the RNA of interest. The preservation of structure and function of the tagged RNA in the cell is often referred to as the “biological compatibility” of the aptamer sequence (Lu et al. 2023). To maintain the structure and function including protein binding of the target RNA upon insertion of an RNA aptamer, several features can be optimized. In particular, reducing the size and structure of the aptamer itself is a frequent strategy to preserve RNA fold and function, using one of the previously described minimal RNA aptamer binding structures. The size of the RNA aptamer can also be controlled during SELEX by limiting the maximum size of RNA used for each selection. In some cases, shorter RNA aptamers are also preferred to minimize the potential of steric clashes or to incorporate of multiple aptamers in the same transcript as seen in the RAT system (Hogg and Collins 2007). In other cases, it is possible to incorporate larger aptamers such as the S1m aptamer.
Another common strategy to insert an RNA aptamer is to combine the aptamer with an existing structural element of the RNA. This approach leverages the fact that most aptamers contain a terminal closing helix formed by their 5′ and 3′ ends that is essential structurally but that can be replaced with any other sequence that also adopts the helical structure (Fig. 1). Therefore, it is often possible to replace an existing stem–loop within the target RNA by the aptamer, which also minimizes number of nucleotides added to the RNA (Fig. 4A; Panchapakesan et al. 2017). Alternatively, it is also possible to incorporate the RNA aptamer by replacing a smaller section of a loop structure thereby converting a loop into a bulge in the target RNA (Fig. 4B). This strategy has been successfully used in a small nucleolar RNA by inserting the S1 aptamer into a nonessential region of a structured H/ACA snoRNA (Lemay et al. 2011). This approach has also been used for the incorporation of the Mango aptamer into the 6S RNA as well as spliceosomal and scaRNAs (Dolgosheina and Unrau 2016; Autour et al. 2018).
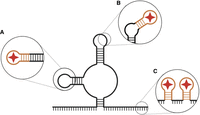
Common strategies for incorporating RNA aptamers into existing RNAs. (A) Aptamers can be inserted by integrating the terminal stem of the aptamer into an existing stem–loop region, replacing the existing loop with the aptamer. (B) The aptamer may be incorporated into a loop region, adding an additional stem to the structure. (C) A single aptamer may be added to an unstructured region, usually on the 5′ or 3′ ends of the RNA. Multiple copies of an aptamer, or multiple aptamers, may be added, separated by spacer regions. Created in BioRender (Rocca and Kothe 2025, https://BioRender.com/i49j579).
By far the most common method of inserting an RNA aptamer into an existing structure is the addition of the aptamer at either the 5′ or 3′ end of the target RNA with a linker sequence. This approach reduces the probability that the aptamer will disrupt the RNA by forming stems with nearby sequence elements in the RNA of interest (Fig. 2C). Moreover, this strategy is least likely to impair protein binding to the RNA of interest. Thereby, it is possible to incorporate single aptamers (Hartmuth et al. 2002; Lee et al. 2013; Flather et al. 2016) as well as repeats of multiple aptamers (Hogg and Collins 2007; Said et al. 2009; Leppek and Stoecklin 2014; Chaker-Margot et al. 2015; Zhang et al. 2016). However, tertiary interactions may still occur, affecting the structure of the target RNA. The inclusion of linker sequences also expands the number of additional nucleotides added to the target RNA. This raises the likelihood that the RNA aptamer and linker could cause steric disruption of the folded RNA within the context of the larger RNP, especially in cases where the RNA 5′ or 3′ ends are “buried” within the core of the RNP. Therefore, it is often necessary to experimentally test different locations in the target RNA for inserting an RNA aptamer.
When an aptamer is inserted into an RNA, it is important to verify that the structure and function of the target RNA is maintained, that is, the biological compatibility of the RNA aptamer must be verified. This can be accomplished in a variety of ways depending on the existing knowledge about the target RNA. As an initial step, the RNA aptamer and the target RNA sequences can be analyzed for complementary sequences that could form unintended helices disrupting the function of the RNA. More insight can be gained by secondary structure prediction algorithms such as RNAfold that predict base-pairing changes upon the addition of an aptamer sequence to the target RNA (Lorenz et al. 2011). In cases of RNAs with known loss-of-function phenotypes, a repression of wild-type RNA and complementation with the “tagged” variant, that is, the RNA containing the aptamer is useful in establishing that the tagged RNA remains functional in vivo (Lemay et al. 2011). While the goal of RNA aptamer purifications is generally to produce high quality pure RNPs following in vivo assembly, in vitro analysis of the RNA can increase confidence that the RNA continues to function as the wild type following the insertion of the aptamer (Hartmuth et al. 2002). Another technique for RNAs with some known interactions with proteins or small molecules are in vitro binding assays to ensure known interactions are maintained (Leppek and Stoecklin 2014).
EXPRESSION OF THE RNP
Expression of the tagged RNA for RNP purification varies based on the model organism (Fig. 2). Designing the expression of the tagged RNA must balance the high levels of expression that are beneficial to a high-yield RNP purification with the potential consequences that perturbations to the expression will have on the formation of the RNP. To achieve high expression of the tagged RNA, researchers frequently use promoters that have high basal expression (Slobodin and Gerst 2010) or inducible expression (Zhang et al. 2016) and that are appropriate for the cell and organism used. Usage of promoters with a high basal expression for the given organism such as the human cytomegalovirus promoter (hCMV) can be advantageous in instances where a high yield of RNPs per cell is desired (Slobodin and Gerst 2010).
In some cases, RNA processing steps are dependent on the context of the RNA transcription. For instance, the Ye lab noted small differences in the composition of ribosome precursors originating from tagged rRNAs expressed from a plasmid by RNA-polymerase II using the Gal7 inducible yeast promoter compared to ribosome precursors assembled from rRNAs transcribed by RNA-polymerase I using the endogenous promoter (Zhang et al. 2016). Therefore, the endogenous promoter for the RNA is often used to ensure expression levels and RNP assembly processes that are as close to native as possible. For example, the Unrau lab used the native promoter of the U1 snRNA to transcribe a U1 construct tagged with the Mango aptamer (Panchapakesan et al. 2017).
It is also important to consider the presence of other untagged copies of the target RNA within a cell. While engineering a system for the purification of RNPs, a choice must be made whether to allow the cell to assemble RNPs using an endogenous, untagged RNA in addition to the aptamer-tagged RNA. For example, Hogg and coworkers introduced several variants of an aptamer-tagged 7SK RNA and purified the associated RNP (Hogg and Collins 2007). This was accomplished without any perturbation of the endogenous 7SK RNA, which was present in a greater abundance than the tagged RNA. This example indicates that the depletion of untagged RNAs is not always necessary to ensure high yields in purification, which is especially convenient in cases where the target RNA is essential for growth. Alternatively, it is sometimes possible to generate a system where only the tagged RNA expressed without endogenous, untagged RNA. This is accomplished either through the deletion of the endogenous RNA, or by the direct insertion of the aptamer coding sequence into the RNA gene in the genome.
CELL TYPE, LYSIS, AND ORGANELLE ISOLATION
For RNP purifications, the cells must first be lysed, and special considerations must be taken when purifying RNPs from cellular organelles. While these concerns are common to many types of biomolecular purification, extra care must be taken to avoid a disruption of the noncovalent interactions between the subunits of the RNP. For instance, when purifying ribosomal, nuclear, and nucleolar RNAs, additional cell lysis steps are required in human cells to remove the dense heterochromatin layer in the human nucleus. For this purpose, Singh et al. use DNase I (Singh et al. 2021), whereas DNase I is not needed when purifying yeast preribosomes due to the lack of a robust heterochromatin layer in yeast (Chaker-Margot et al. 2015).
Efficient cell lysis can be challenging in cells with rigid cell walls such as Saccharomyces cerevisiae, which frequently require intense mechanical disruption in order to achieve high yield lysis. In yeast, several common lysis methods are detrimental to RNA integrity, especially for large RNAs such as rRNA (López de Heredia and Jansen 2004). In early comparisons of lysis methods, glass bead milling and high-pressure cell lysis via cell press both show reduced recovery of RNA compared to milling of deep frozen samples (López de Heredia and Jansen 2004). Since then, the usage of mechanical cryo-mills has emerged as a gold standard for cell lysis with minimal RNA degradation across a variety of cell types (Oeffinger 2012; Cheng et al. 2017; Desbois et al. 2023). As this approach requires the availability of expensive mechanical cryo-mills, high-pressure cell disruption, for instance, is still used and has been shown to often produce lysate of sufficient quality for purification of RNPs and generation of high resolution structures (Rigaut et al. 1999; Zhang et al. 2023).
When working with cell types lacking a rigid cell wall, such as human cells, cell lysis can often be achieved through usage of enzymes and freeze-thaw cycles without grinding of the cells (Hogg and Goff 2010; Nieto et al. 2021; Singh et al. 2021). Here, the isolation of organelles such as the nucleus, nucleolus, or mitochondria is highly feasible using differential centrifugation (Nieto et al. 2021; Singh et al. 2021) or affinity-based methods (Barrey et al. 2011) thereby reducing the potential for contaminants in the RNP purification originating from other cellular compartments.
BUFFER SYSTEMS AND PURIFICATION CONDITIONS
Selection of buffers, both for lysis and purification, is also important to consider. For the purification of an RNP, it is paramount that the RNP remains functional and intact without disrupting the noncovalent interactions between RNAs and proteins. Therefore, buffer selection involves the consideration of both RNA and protein stability. Many RNP purifications use a combination of additives to improve the stability of both RNA and protein by adding RNase and protease inhibitors as well as reducing agents such as dithiothreitol and β-mercapthoethanol. Additionally, RNP purifications are often performed at 4°C in order to reduce the degradation of RNAs and proteins, enzymatic or otherwise. While high pH buffers cannot be used to avoid alkaline lysis of the RNA components of the RNP, the unique composition of each RNP and its host cell often require adjustments of pH and ionic strength of the buffer to maximize the complex stability.
Additionally, it is important to consider the nature of downstream analysis when lysing cells and purifying the RNPs. Certain additives that maximize efficiency in lysis and improve protein stability can interfere with later analysis. For instance, the inclusion of glycerol in buffers is a common strategy for increasing the stability of proteins and RNPs (Castro et al. 2017). Glycerol can, however, be detrimental to future analysis including cryo-electron microscopy (Basanta et al. 2022) and can also destabilize RNA secondary structure (Lambert and Draper 2007). Additionally, the inclusion of certain detergents is incompatible with downstream applications such as LC-MS, and detergent removal steps may affect RNP integrity (Yeung et al. 2008). To obtain a comprehensive insight into suitable purification conditions for a particular RNA aptamer and RNP, the reader is encouraged to review the methods in the primary literature, for example, as listed in Table 1.
POTENTIAL OFF-TARGET INTERACTIONS OF THE RESIN
Not all RNA aptamers are designed with the final goal of RNP purifications in mind. As such, either the RNA aptamer or its ligand may have potential off-target binding activity, where a component of the cellular lysate or buffer system can compete for binding with either the aptamer or the ligand. Competitive binding of these other molecules can prevent a high yield of RNP purification. In the selection of an aptamer for purification, it is important to select a compatible ligand or implement strategies for mitigating their off-target binding. For instance, the S1 aptamer, which binds streptavidin, has been used for the purification of mRNPs, but it is necessary to deplete avidin-binding compounds such as biotin from the cell to prevent competition for binding with the streptavidin resin (Leppek and Stoecklin 2014). In cases of biotin-derivatized ligands, such as those used in combination with Mango RNA aptamers, streptavidin resins can be carefully saturated with the biotin-derivatized ligand prior to the introduction of the lysate to help prevent binding of cellular biotin to the resin (Panchapakesan et al. 2017). To further increase the stringency of an RNP purification using RNA aptamers, a frequent strategy is to couple the aptamer pulldown to a second purification step such as protein affinity purifications (Chaker-Margot et al. 2015; Zhang et al. 2016).
CONCLUSION
In the study of RNP complexes, it is often advantageous to purify in vivo assembled RNPs from the complex environment of cellular lysates rather than reconstituting RNP in vitro. RNA aptamers serve as a powerful and versatile tool for in vivo tagging of RNP complexes that are easily purified by well-established chromatography methods from a complex cellular mixture. This approach allows investigating the structure and composition of the RNP as well as creating opportunities to examine partially assembled complexes. Importantly, using RNA aptamers as a tool for the RNP pulldowns requires a careful consideration of the many factors that can affect the specificity and yield of the pulldown as well as the stability and composition of the RNP. As outlined here, a wide array of potential aptamers may be incorporated into a given RNA, each with varying size, structural characteristics, and binding affinity for a given ligand and in turn different RNP purification strategies.
An important advantage of RNA aptamers for RNP purification is the direct targeting of the RNA components of an RNP complex. The system allows for genetic encoding as opposed to chemical modification of the RNA facilitating the study of RNPs that expressed and processed similar to endogenous RNA. With appropriate considerations, RNA aptamer purification systems are portable to any cell types and can be combined easily with a variety of other purifications or analyses of the RNP.
ACKNOWLEDGMENTS
This research was supported by the Canadian Institutes for Health Research (CIHR, project grant 437623).
Footnotes
-
Article is online at http://www.rnajournal.org/cgi/doi/10.1261/rna.080460.125.
-
Freely available online through the RNA Open Access option.
This article, published in RNA, is available under a Creative Commons License (Attribution-NonCommercial 4.0 International), as described at http://creativecommons.org/licenses/by-nc/4.0/.








