
Positioning of sperm tail longitudinal columns depends on NSUN7, an RNA-binding protein destabilizing elongated spermatid transcripts
- Ekaterina A. Guseva1,2,3,
- Olga A. Averina2,3,
- Sergey V. Isaev4,
- Philipp I. Pletnev3,
- Elizaveta E. Bragina2,5,
- Oleg A. Permyakov2,3,
- Vitaly S. Buev3,6,
- Anastasia V. Priymak2,
- Mariia A. Emelianova1,2,
- Lizaveta Pshanichnaya3,
- Evgeny A. Romanov7,
- Svetlana E. Novikova8,
- Kirill S. Petriukov3,
- Anna Ya. Golovina2,
- Olga O. Grigorieva2,3,
- Vasily N. Manskikh2,
- Diana S. Korshunova9,
- Yuliya Yu. Silaeva9,
- Alexey V. Deykin10,
- Maria P. Rubtsova3,
- Victor G. Zgoda8,
- Alexander M. Mazur11,
- Egor B. Prokhortchouk11,
- Olga A. Dontsova1,2,3,12 and
- Petr V. Sergiev1,2,3
- 1Center of Molecular and Cellular Biology, Skolkovo Institute of Science and Technology, 143025 Skolkovo, Russia
- 2Belozersky Institute of Physico-Chemical Biology, Lomonosov Moscow State University, 119991 Moscow, Russia
- 3Faculty of Chemistry, Lomonosov Moscow State University, 119991 Moscow, Russia
- 4Center for Brain Research, Medical University Vienna, 1090 Vienna, Austria
- 5Research Centre for Medical Genetics, Moscow 115522, Russia
- 6Faculty of Bioengineering and Bioinformatics, Lomonosov Moscow State University, 119991 Moscow, Russia
- 7National Medical Research Center for Obstetrics, Gynecology and Perinatology named after Academician V.I. Kulakov, 117198 Moscow, Russia
- 8Institute of Biomedical Chemistry, 119121 Moscow, Russia
- 9Institute of Gene Biology, 119334 Moscow, Russia
- 10Belgorod State University, 308015 Belgorod, Russia
- 11Institute of Bioengineering, Research Center of Biotechnology of the Russian Academy of Sciences, 119071 Moscow, Russia
- 12Shemyakin–Ovchinnikov Institute of Bioorganic Chemistry, 117997 Moscow, Russia
- Corresponding author: petya{at}genebee.msu.ru
-
Handling editor: Maria Carmo-Fonseca
Abstract
Spermatozoid's flagella assemble in transcriptionally silent spermatids and thus depend on posttranscriptional regulation of gene expression. Mutations in Nsun7 gene are known to cause male infertility in human and mice. We identified m5C-specific NSUN7 RNA methyltransferase as a protein present in elongated spermatids and interacting with RNAs specific for this type of spermatozoid's precursor cells. Inactivation of the Nsun7 gene in mice leads to upregulation of its RNA interactors, thus indicating that NSUN7 downregulates a set of RNAs in the elongated spermatids. A physiologic consequence of Nsun7 gene knockout is male infertility, which is mechanistically explained by the observed mispositioning of longitudinal columns relative to the axonemal microtubular doublets leading to a motility defect.
Keywords
INTRODUCTION
Spermatogenesis is the process by which male germ cells develop into mature spermatozoa (Griswold 2016). Maturation of spermatogonia, resident at the periphery of the seminiferous epithelium, proceeds via formation of spermatocytes and spermatids, ultimately leading to the functional sperm (Fig. 1A). Central to the sperm tail structure is a microtubule-based axoneme essential for sperm motility (Inaba 2011). Mutations affecting axonemal proteins can lead to asthenozoospermia and male infertility (Linck et al. 2016). Besides motility, the axoneme acts as the basis for the assembly of periaxonemal accessory structures and as a railway for anterograde and retrograde intraflagellar transport (IFT) (San Agustin et al. 2015). Flagellum of mammalian spermatozoa consists of three main parts: the midpiece, the principal piece, and the endpiece (Fig. 1B). The midpiece is characterized by the mitochondrial sheath, which tightly surrounds the axoneme and the nine outer dense fibers (ODFs). The mitochondrial sheath is essential for providing the energy required for sperm motility through oxidative phosphorylation. The principal piece is separated from the midpiece by an annulus, a cytoskeleton structure formed by septins. The fibrous sheath covers the principal piece of the sperm tail, providing mechanical support that modulates flagellar bending and defines the shape of the flagellar beat (Eddy et al. 2003). The fibrous sheath is the site of localization of glycolytic enzymes, which contribute to the energy supply of the sperm tail (Eddy et al. 2003). Major periaxonemal structures of the principal piece are circumferential ribs of the fibrous sheath surrounding the dense fibers and two longitudinal columns (LC) of the fibrous sheath replacing ODFs precisely at the microtubule doublets 3 and 8.
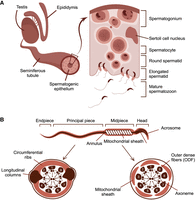
Structure of male reproductive system and spermatozoa. (A) Structure of male reproductive organs and schematic representation of the cross section of the spermatogenic epithelium. (B) Structure of spermatozoa and cross section of midpiece and principal piece.
NSUN7 was identified as a testes-enriched protein required for male fertility in mice almost two decades ago (Harris et al. 2007). Later it was shown that C26232T-transition and T26248G-transversion mutations in exon 7 of the NSUN7 gene (Khosronezhad et al. 2015a), as well as A11337-deletion (Khosronezhad et al. 2015b), were associated with asthenozoospermia in humans. However, the molecular function of NSUN7 was never studied in vivo and most of the studies were conducted on cancer-derived cell lines. In mouse hepatocellular carcinoma, NSUN7 has been reported to methylate specific enhancer RNAs, such as Pfk1, Sirt5, Idh3b, and Hmox2, in association with the transcriptional coactivator PGC-1α (Aguilo et al. 2016). Epigenetic silencing of NSUN7 has been linked to liver cancer, impacting the NSUN7-dependent methylation of CCDC9B mRNA (Ortiz-Barahona et al. 2023).
NSUN7 is annotated as a member of the NOP2/Sun m5C RNA methyltransferase family specific for higher eukaryotes (Supplemental Appendix, Supplemental Fig. S1A). Other members of this family, NSUN1–NSUN6, are responsible for the posttranscriptional addition of the methyl group to the C5 atom of cytosine in tRNAs and rRNAs (Kuznetsova et al. 2019). Moreover, most of the family members were previously reported to modify mRNA (Guarnacci and Preiss 2024). Spermatogenesis uses a network of RNA-binding proteins to orchestrate the timing and localization of protein synthesis (Kotaja et al. 2006). Previously, it was shown that another member of the NOP2/Sun family, NSUN2, is essential for testicular function (Hussain et al. 2013).
Here we demonstrate that RNA methyltransferase NSUN7 interacts with a multitude of mRNAs, specific for the elongated spermatids, in a process crucial for sperm tail assembly. Inactivation of mouse Nsun7 leads to an increase in the abundance of its RNA targets, leading to male subfertility characterized by abnormal positioning of sperm LC and, consequently, sperm locomotion defect.
RESULTS
NSUN7 is localized to the forming tails of elongating spermatids and is absent in mature spermatozoa
While NSUN7 function has been previously addressed on a model of hepatocellular carcinoma (Aguilo et al. 2016; Ortiz-Barahona et al. 2023), according to gene expression databases (Uhlén et al. 2015; Cardoso-Moreira et al. 2019), Nsun7 expression occurs at a high level only in testes, while other tissues characterized by the presence of ciliated cells, such as lung and ependymal epithelium, demonstrate moderate expression. Using reverse transcription polymerase chain reaction (RT-PCR), we verified that Nsun7 is specifically expressed in testes (Fig. 2A), while the expression level in other tissues was below the detection limit. Immunohistochemical (IHC) and immunoblotting analysis of NSUN7 with commercially available anti-NSUN7 antibodies, as well as antibodies generated in-house, yielded no positive results. Targeted mass-spectrometry analysis of whole testes lysates revealed peptides corresponding to the NSUN7 at a concentration of 72 ± 34 attomole/μg total peptides after trypsin digestion (Supplemental Appendix, Supplemental Fig. S3C,D).
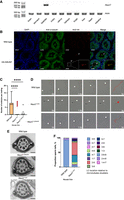
Nsun7 is expressed in testes. (A) RT-PCR analysis of Nsun7 gene expression in mouse organs. ActB gene is used as a positive control. (B) Immunostaining of testes cross sections of the wild-type and HA-Nsun7 mice. White bars = 100 μm. Arrows indicate HA-NSUN7 localization in flagella of elongated spermatids. The colocalization of HA-NSUN7 and α-tubulin is shown in the frames on a higher magnification. (C) Decrease of fertility of Nsun7i2/i2 (n = 21) and Nsun7Δ38/Δ38 (n = 18) mouse lines relative to the WT (n = 47). For each mouse line, the number of embryos was analyzed 10 dpc. ANOVA test result: (****) P < 0.0001. (D) Motion tracks of spermatozoa of the WT (top panel), Nsun7i2/i2 (middle panel), and Nsun7Δ38/Δ38 (bottom panel) mice. Red lines show the motion tracks of spermatozoa, white arrowheads indicate head position, black arrowheads—cytoplasmic bugling position. (E) Electron microscopy of spermatozoid tails of the WT (top panel), Nsun7i2/i2 (middle panel), and Nsun7Δ38/Δ38 (bottom panel) mice. White arrows indicate LC. Black bars = 100 nm. (F) Distribution of LC position in the WT (n = 3 mice) and Nsun7i2/i2 (n = 3 mice).
To overcome the lack of assay sensitivity, we generated a transgenic mouse line expressing an N-terminally HA-tagged allele of Nsun7 by sgRNA/Cas9 cleavage and single-stranded oligonucleotide-guided homologous recombination in mice zygotes (Supplemental Appendix, Supplemental Fig. S1B). Insertion of the tag does not affect mice fertility (Supplemental Appendix, Supplemental Fig. S1C). Specific staining of HA-NSUN7 within the testes was observed in the tails of elongated spermatids where it is colocalized with α-tubulin (Fig. 2B). No specific HA-NSUN7 staining was observed for lungs or brain which are known to possess ciliated cells (Supplemental Appendix, Supplemental Fig. S2), in agreement with the results of RT-PCR. We were also unable to detect HA-NSUN7 in mature spermatozoids residing in the epididymis (Supplemental Appendix, Supplemental Fig. S2A).
Inactivation of Nsun7 causes male subfertility in mice and sperm locomotion defect due to mispositioning of longitudinal columns
To study the functional role of NSUN7 in vivo, we generated two mouse lines carrying inactivating i2 and Δ38 frameshifting alleles of Nsun7 using sgRNA/Cas9 mediated Nsun7 gene exon 8 cleavage in mice zygotes (Supplemental Appendix, Supplemental Fig. 3A–D). Homozygous Nsun7i2/i2 and Nsun7Δ38/Δ38 mice were viable and have no apparent abnormalities except male infertility (see below). Mating of both Nsun7i2/i2 and Nsun7Δ38/Δ38 males with WT females demonstrated a marked reduction in fertility for both alleles (Fig. 2C), but no pathological changes of the testes or other tissues containing ciliated cells (Supplemental Appendix, Supplemental Fig. S4A–C,E–J). Yet, we observed a significant decrease in the sperm count of knockout mice (Supplemental Appendix, Supplemental Fig. S4D). The majority of Nsun7i2/i2 and Nsun7Δ38/Δ38 mice spermatozoa were either motionless or unable to perform the progressive movement (Fig. 2D; Supplemental Appendix, Supplemental Fig. S4K,L). The majority of motile knockout sperm exhibited a trajectory of movement deviating significantly from the norm, such as rotational movements around a single point (Fig. 2D). We also noted a higher incidence of residual body retaining and spermatozoa folding in the annulus region (Supplemental Appendix, Supplemental Fig. S4L,M). The fertilization capability of knockout sperm was also reduced as measured by in vitro fertilization assays. Even under experimental conditions that are extremely favorable for fertilization of the egg, knockout sperm showed significantly reduced levels of fertilization (Supplemental Appendix, Supplemental Fig. S4N).
To investigate whether the abnormal motility of Nsun7−/− sperm is caused by structural defects in the flagella, we analyzed axonemes using transmission electron microscopy. Upon closer examination of spermatozoa structure, we observed mispositioning of LC within the fibrous sheath of both Nsun7i2/i2 and Nsun7Δ38/Δ38 mice (Fig. 2E). In wild-type mice, LC are attached to the outer microtubule doublets 3 and 8, while upon Nsun7 gene inactivation, only 6% of LC exhibit normal attachment (Fig. 2F). Most spermatozoa had columns connected to doublets 2 and 8 (42%) or 4 and 7 (24%) (Fig. 2F). We also observed infrequent cases of tails exhibiting proximal localization of LCs, as well as additional LCs. Interestingly, doublets 5 and 6 were underrepresented in final statistics. Those doublets are characterized by bridge-like connecting structures that could potentially sterically limit the attachment of LCs (Chen et al. 2023). LCs offer structural support, harbor crucial proteins involved in signaling pathways, and restrict spermatozoa tail motility along one of three dimensions, which is essential for specific patterns of sperm movement. It is likely that male subfertility and sperm locomotion defect are a result of LC mispositioning.
Nsun7 knockout affects testes transcriptome and proteome
To gain a better understanding of the molecular pathways associated with NSUN7, we performed a comparative analysis of proteomes and transcriptomes of the entire testes from wild-type and Nsun7i2/i2 mice. RNA-seq of testicular samples obtained from three individual mice revealed 34 genes that were upregulated and 32 genes that were downregulated in the Nsun7i2/i2 mice (Fig. 3A). A Gene Ontology (GO) term enrichment analysis revealed that the downregulated genes are associated with FGF signaling and lipid metabolism (Supplemental Appendix, Supplemental Fig. S5A). The upregulated genes are associated with the synthesis and metabolism of steroid hormones (Supplemental Appendix, Supplemental Fig. S5A). The upregulated genes also included four members of the serine protease kallikrein protein family (Lundwall et al. 2006). They are exclusively expressed in Leydig cells and play a role in paracrine regulation of the seminiferous epithelium (Monsees et al. 1997).

Comparison of proteomes and transcriptome of wild-type mice and Nsun7 knockouts. (A) Volcano plot showing genes of testes transcriptome differentially expressed in the WT (n = 3) and Nsun7i2/i2 (n = 3) mice. (B) Volcano plot showing proteins whose abundance is different in the testes of the WT (n = 3) and Nsun7i2/i2 (n = 3) mice.
To investigate the influence of NSUN7 on testes proteome, we performed panoramic LC–MS/MS analysis. We revealed that 14 proteins were downregulated and 25 were upregulated in Nsun7i2/i2 mice (Fig. 3B; Supplemental Appendix, Supplemental Fig. S5B). As expected, the statistically significant results of proteome analysis correlated with that of the transcriptome (Supplemental Appendix, Supplemental Fig. S5C). We applied western blotting to verify a difference in the abundance of several proteins, such as RNF151, Septin10, and KIFAP3, in testes of the wild-type and Nsun7i2/i2 mice (Supplemental Appendix, Supplemental Fig. S5D). While we have not observed any changes in the abundance of KIFAP3, the downregulation of RNF151 and Septin10 was confirmed.
NSUN7 binds and destabilizes multiple RNA targets in the elongated spermatids
While the inactivation of the Nsun7 gene affects the expression of multiple genes in the testes, the observed effects might be indirect. Spermatogenesis is orchestrated by a multitude of cell–cell interactions and hypothetically, Nsun7 inactivation might even affect cell populations where this very gene is not expressed. To identify RNAs that interact with NSUN7 directly, ex vivo cross-linking of the testes cell suspension with UV light and immunoprecipitation, known as the CLIP, was employed (Supplemental Appendix, Supplemental Fig. S6A). Next-generation sequencing of the cross-linked RNAs, mapping, and peak calling procedure allowed us to determine 100 binding sites of NSUN7 in 71 transcripts. The vast majority of the identified binding sites are located in the coding sequences (CDS) of protein-coding genes (Fig. 4A,B). We identified a consensus sequence of the NSUN7 binding site, which comprises two highly conserved cytosines in the U-rich region (Fig. 4C).
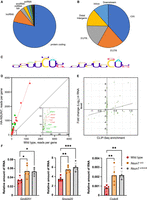
NSUN7 binds elongated spermatid RNAs. (A) Distribution of NSUN7 cross-link sites (FC > 2, P < 0.05) between transcript types. (B) Distribution of NSUN7 cross-link sites (FC > 2, P < 0.05) between mRNA parts. (C) Logo of consensus binding motif of NSUN7. (D) Coverage scatter plot of cross-linked and precipitated RNAs for the HA-NSUN7 and the wild-type control mice. Red dots correspond to the transcripts characteristic to elongated spermatids according to the scRNA-seq data, green dots correspond to other transcripts. Only RNAs that passed a statistical significance threshold (P < 0.05) are shown. (E) Correlation between the efficiency of RNA cross-linking to the NSUN7 as determined by CLIP-seq with the difference RNA abundance in wild-type and Nsun7i2/i2 mice as determined by RNA-seq. (F) Comparison of the mRNA levels of NSUN7 potential binding targets in wild-type and Nsun7i2/i2 mice. T-test was used, stars indicate P-value: (*) P < 0.05, (***) P < 0.001.
We used available single-cell transcriptome data of mice testes (Jung et al. 2019) to determine cell-type specificity of mRNAs cross-linked to NSUN7. GSEA analysis revealed that one-third (22) of the NSUN7 cross-linked and coprecipitated RNAs were elongated spermatid-specific RNAs, while only one-tenth (eight) were round spermatid-specific RNAs (Fig. 4D; Supplemental Appendix, Supplemental Fig. S6B). Among the genes whose transcription products were significantly cross-linked and coprecipitated with HA-NSUN7, there are numerous genes associated with flagellar motility and sperm abnormalities. For example, loss of function of Tcte1 (Castaneda et al. 2017), Adam3 (Yamaguchi et al. 2009), Adam32 (Italiya et al. 2023), Tppp2 (Zhu et al. 2019), and Crisp2 (Lim et al. 2019) leads to a decrease and/or abnormalities in sperm motility, while Stat1 (Bastián et al. 2007) and Crisp2 (Lim et al. 2019) affect the ability of sperm to undergo the acrosome reaction. Additionally, among mRNAs that coprecipitate with HA-NSUN7 are Gstm5, which is responsible for binding hexokinase1-s and phosphofructokinase to the fibrous sheath of sperm flagella (Nakamura et al. 2010), and Gsg1. The expression of the latter and other important structural proteins of the fibrous sheath, such as ROPN1B, AKAP3, and AKAP4, was found to be associated with oligozoospermic hypogonadism (Grande et al. 2022). This finding corresponds well to the observed NSUN7 localization and sperm abnormalities in Nsun7−/− mice described above.
We have demonstrated that NSUN7 is an RNA-binding protein specific for elongated spermatids at late stage of development. Since NSUN7 belongs to the family of Sun-domain-containing RNA methyltransferases, we set up to determine its methylation targets. To this end, we performed bisulfite sequencing of RNA extracted from elongated spermatids sorted from nine wild-type and nine Nsun7i2/i2 mice.
The BS-RNA-seq analysis (Supplemental Appendix, Supplemental Fig. S6C) revealed 3373 methylation sites where the level of methylation in the wild-type exceeded that for the Nsun7i2/i2 mice. Among them, 250 sites were 10%–100% methylated in the wild-type mice, while being completely unmethylated in the mice devoid of Nsun7. However, differential methylation sites demonstrated little correlation with NSUN7 binding sites revealed by CLIP-seq and have little statistical support due to the low coverage of sites demonstrating differences in methylation between the WT and Nsun7i2/i2 mice. Since that, none of the differential methylation sites has sufficient statistical support after correction for multiple testing. We do not exclude a possibility that BS-RNA-seq has not revealed a primary RNA target for NSUN7 methylation due to the ambiguity of deaminated reads mapping or low abundance of target RNA. Thus, the following correlation analysis is based on the data generated by CLIP-seq.
What might be a function of RNA methylation by NSUN7 RNA methyltransferase? RNA modifications, such as m6A, might trigger RNA degradation (Wang et al. 2014). To check for the possibility that m5C modification by NSUN7 RNA methyltransferase might affect the stability of its targets, we compared the results of transcriptome analysis of the wild-type and Nsun7i2/i2 mice testes with the results of CLIP-seq (Fig. 4E). We found that RNAs cross-linked to NSUN7 at P < 0.01 are on average stabilized by Nsun7 inactivation, suggesting that NSUN7 destabilizes its targets.
To test the hypothesis that NSUN7 binding affects RNA levels, we conducted RT-qPCR analysis of 10 RNA-binding targets of NSUN7. The analysis of the testes from wild-type and Nsun7i2/i2 mice revealed that the presence of functional NSUN7 in cells resulted in a reduction in the levels of RNAs interacting with NSUN7. For three genes, reliable changes were observed between the mouse lines, while for the remaining genes, a decreasing trend was noted (Fig. 4F; Supplemental Appendix, Supplemental Fig. S6D). An intron of the Tcp1—snoRNA Snora20—is of particular interest. According to predictions, it is a small nucleolar H/ACA box snoRNA. There is accumulating evidence that snoRNAs in male germ cells can regulate gene expression (García-López et al. 2015).
DISCUSSION
Assembly of spermatozoid's flagella is a complex process coordinated by a multitude of protein–protein interactions, precise localization and transport of the components as well as an intricate orchestration of gene expression. The latter is especially problematic, since transcription is ceased at the meiotic stage, before the assembly of flagella (Monesi 1964). Due to this fact, posttranscriptional control of gene expression is of primary importance for altering stages of spermatogenesis (Morgan et al. 2021).
We have shown that inactivation of m5C RNA methyltransferase Nsun7 in mice results in a decline in sperm count and motility, accompanied by an increase in spermatozoa morphological abnormalities. These findings are consistent with the abnormalities described in humans with NSUN7 gene mutation (Khosronezhad et al. 2015a). Moreover, we have demonstrated that knockout of Nsun7 leads to the mispositioning of LC to noncanonical axonemal doublets. The mechanisms underlying the proper positioning of LC within the axoneme remain largely elusive. Historically, axoneme, the core structure of flagella, has long been regarded as a highly symmetrical and regular structure. With the uprising of advanced transmission electron microscopy and lately, cryo-electron tomography, the uneven distribution of axonemal components has become increasingly evident (Chen et al. 2023). This was further corroborated by studies involving knockout mice, showing a differential distribution of certain protein complexes and posttranslational modifications within the axoneme. For example, inactivation of Ttll5 and Ttll9 encoding enzymes responsible for polyglutamylation of tubulin, led to the complete elimination of microtubule doublets 4 and 7, respectively. Moreover, posttranslational glutamylation and glycylation were reported to be unevenly distributed among microtubule doublets in sperm tails (Lee et al. 2013; Konno et al. 2016). These findings suggest that the inequality of axonemal doublets appears to be a fundamental characteristic of the structure influencing the assembly and function of flagella. Our observations, along with those from other studies, allow us to suggest that proper orientation of LC might be mediated by a machinery that is capable of recognition of some specific features of doublets 3 and 8. Previous studies in Trypanosoma brucei showed that localization of both retrograde and anterograde intraflagellar trafficking (IFT) complexes is mostly restricted to two sets of doublets (3–4 and 7–8) (Bertiaux et al. 2018). If this type of IFT localization is conserved among species, we may assume that the assembly and positioning of LC could be mediated by the on-site availability of fibrous sheath components. As was described by Fawcett in his foundational research (Fawcett 1970), mammalian fibrous sheath grows around LC precursors which may represent a perfect docking site for the assembly of periaxonemal structures in the spermatozoa.
Inactivation of the Nsun7 gene is not the only one leading to LC misposition. According to previously published data, the inactivation of several genes, such as Ube2b, Dnali, and Fused (Escalier 2003; Nozawa et al. 2014; Wu et al. 2023; Guseva et al. 2024), leads to mispositioning of LCs at noncanonical microtubule doublets and even to the appearance of additional LC-like structures. However, the precise contribution of each of these genes in this process remains to be clarified. Dnali and Fused are involved in sperm-specific transport systems associated with dynein and kinesin, respectively (Nozawa et al. 2014; Wu et al. 2023), and the proper functioning of these systems is important for the assembly of LC structural proteins AKAP3 and AKAP4 (Wu et al. 2023). Furthermore, loss-of-function mutations in a testes-specific E2-ubiquitin ligase Ube2b (Escalier 2003) also led to the mispositioning of LCs, suggesting the involvement of ubiquitin-conjugating machinery in the assembly of the fibrous sheath. In the same vein, RNF151, a testes-specific E3-ubiquitin ligase (Nozawa et al. 2022) which, among other testes-specific E3-ubiquitin ligases RNF133 and RNF148, functions at the spermatid stage of spermatogenesis and affects sperm motility. In our study, we observed a significant downshift of RNF151 protein abundance upon Nsun7 inactivation, which may partially explain the observed phenotype. Curiously, an RNA bound by NSUN7 and upregulated upon Nsun7 gene inactivation, Gm8251 also codes for a putative ubiquitin ligase, E1 class.
The CLIP-seq experiment performed on testes extract allowed us to identify multiple NSUN7 binding RNAs. The majority of those belong to mRNAs, although several noncoding RNAs have also been identified. In line with the NSUN7 localization in the elongated spermatids, the mRNA set interacting with NSUN7 was also found to be enriched for the elongated spermatid-specific RNAs. This result fits well the hypothesis that NSUN7 is a posttranscriptional regulator of RNAs at postmeiotic spermatogenesis stages. Posttranscriptional control of gene expression in spermatogenesis is known to be mediated by RNA-binding proteins (Morgan et al. 2021), specialized ribosomes carrying testes-specific ribosomal proteins (Zou et al. 2021; Li et al. 2022), and mRNA methylation (Hussain et al. 2013; Lin et al. 2017) and demethylation (Zheng et al. 2013; Tang et al. 2018). Deficiency in testes-specific ribosomal protein Rpl39L (Zou et al. 2021; Li et al. 2022) and RNA demethylase Alkbh5 (Zheng et al. 2013; Tang et al. 2018) resulted in flagellar misassembly and motility defects.
We observed that the lack of a functional Nsun7 gene resulted in upregulation of its target RNAs. A similar effect of methylation has been demonstrated for NSUN2; however, the precise mechanism of m5C methylated RNA degradation remains to be clarified (Mei et al. 2020). The other methylation types, for example, m6A catalyzed by protein METTL3 (Shulman and Stern-Ginossar 2020), promote deadenylation of the target RNA, which results in facilitating RNA degradation (Du et al. 2016). Altogether, the obtained data speak in favor of the model where NSUN7 functions to destabilize a set of transcripts at the late stages of spermatogenesis for the sake of proper flagella assembly.
MATERIALS AND METHODS
Mice housing, breeding and genome editing
The experiments were conducted in strict accordance with the relevant national and international guidelines for the Care and Use of Laboratory Animals. The animal study was carried out following the ARRIVE guidelines. The work with animals was approved by the local bioethics committee “Institute of Mitoengineering MSU” LLC, protocol #79, dated April 28, 2015. The animals were housed in individually ventilated cages (IVC system, TECNIPLAST S.p.A.) with free access to food and water purified by reverse osmosis. The environment was free of specific pathogens and provided with light. The animals were housed in a 12/12 light cycle (light on at 09:00), in rooms with an air exchange rate exceeding 15 r/h, at 20°C–24°C, and 30%–70% humidity. Wood chips with minimal dust formation were utilized as bedding material. Shelters and building materials for nests were constructed from natural materials to enhance the environmental enrichment. All materials provided to the animals were sterilized by autoclaving.
Inactivation of Nsun7 gene was done by CRISPR/Cas9 system. sgRNA was designed by https://chopchop.cbu.uib.no/. sgRNA with the guiding sequence ACACCGAGGCTGGAACAGCG was obtained by T7 transcription in vitro (MEGAscript T7 Transcription Kit, Thermo Fisher) from PCR amplified templates, obtained by PCR amplification of the plasmid pX458 (Ran et al. 2013) with the forward primer TGTAATACGACTCACTATAGGACACCGAGGCTGGAACAGCGGTTTTAGAGCTAGAAATAGCAAG and the reverse primer AAAAGCACCGACTCGGTGC. Obtained sgRNA was mixed with GeneArt CRISPR Nuclease mRNA (Thermo Fisher: A29378) and diluted in filtered microinjection buffer (10 mM Tris, 0.1 mM EDTA, pH 8) to final concentrations of 25 ng/mL sgRNA and 50 ng/mL Cas9 mRNA.
Insertion of HA tag to the N-end of Nsun7 was done similarly, using sgRNA GACATCAGGGGCAAACATGC guide sequence. Single-stranded DNA oligonucleotide, 200 nt length, containing HA sequence inserted after AUG codon was used as a homology recombination template; 10 ng/mL f.c. added to 25 ng/mL sgRNA and 50 ng/mL Cas9 mRNA for microinjection.
Mice inbred strains C57Bl/6J and CBA (Federal Research Center Institute of Cytology and Genetics, Siberian Branch, Russian Academy of Sciences [ICG SB RAS] Novosibirsk, Russia), SPF status, were mated to obtain F1 hybrids C57Bl/6J × CBA. Zygotes were obtained by mating of the hybrid superovulated female mice with males using standard procedure (Averina et al. 2020). Zygotes were microinjected into the pronucleus and transferred into the oviducts of pseudo-pregnant outbred CD1 female mice. For further experiments, we selected the mutant mice line with the insertion of 2 nt and deletion of 38 nt, leading to a frameshift as Nsun7 knockout and mice with precise in-frame insertion of HA-coding sequence between the first AUG and second codons of the gene as Nsun7 was tagged. To avoid any potential influence of unlikely secondary mutations, descendants of the founder were back-crossed to inbred strain C57BL/6J. The wild-type and homozygous strains were obtained by crossing heterozygous mice Nsun7+/−. Wild-type mice were used to establish a reference control group.
Animal's DNA samples from the tiny pieces of the distal tail (taking the tail biopsy was carried out in accordance with the FELASA July 2013 decision on genotyping of transgenic rodents) were extracted with QuickExtract DNA Extraction Solution (Lucigen: QE0905T) according to manufacturer's instructions and analyzed by PCR and Sanger sequencing of the amplicons. Founder pups, their descendants, and wild-type and homozygous mice were genotyped by genomic DNA amplification with the primers TCAGATTTTCCATATTTAACACGAGTG and ATTTAGAAATCAAAACAAGTACCTTCG (knockouts for Nsun7), CGATGCTTAAGACTACAAGGATAG and GTAATTTATTATGACTTTGTCTGGCGACC (HA-Nsun7), followed by Sanger sequencing (Center of Collective Use «Genome» at Engelhard Institute of Molecular Biology, Moscow, Russia). Experiments were performed on 2–6-month-old male mice. To control the specificity of introduced mutations during knockout generation, we amplified three regions of genome using the following primers: GCGCTTACCGCCTAGATGAA and GCTACCTGGAAGTCAGTCAGG (chr2:–168001826), TTCAGGTATGTGATTTGGTGATTTG and TGGGGGACTTTTGGTATAGCAT (chrX:+56651059), TTTGGAGACAGGGTTTCCTGG and GTTGGCTACCAGGGCACTTA (chr5:+34535215), followed by Sanger sequencing.
Measurement of sperm motility
Sperm was collected from the epididymis of wild-type (n=3), Nsun7i2/i2 (n=3) and Nsun7 Δ38/Δ38 (n=3) mice. After extraction, sperm was incubated in PBS at 37°C for 30 min, diluted to 10 ml and analyzed in a Makler chamber. Spermatozoa were counted three times in 10 fields of view. The final values represent the average of all calculations. For analysis of spermatozoa motion tracks, Fiji ImageJ (version 1.54f) (Schindelin et al. 2012) was used.
Fertility tests
The process of in vitro fertilization (IVF) in mice was initiated with the hormonal stimulation of female mice aged 4–5 weeks, belonging to the F1 C57Bl/6/CBA strain. The initial procedure involved the administration of an intraperitoneal injection of anti-inhibin serum in conjunction with pregnant mare serum gonadotropin (PMSG) to the mice. Subsequently, the mice were administered human chorionic gonadotropin (hCG) to induce ovulation. Approximately 13–14 hours after the hCG injection, the oocytes and sperm were collected and incubated separately in HTF medium at 37°C with 5% CO2 for a period of time. A 60–90 minute incubation period was required to enhance sperm fertilizing capacity and to remove cumulus cells from the oocytes. Subsequently, the prepared oocytes were transferred to Petri dishes containing sperm at an adequate concentration, where they were co-incubated for 4–6 hours. Following fertilization, the zygotes were carefully washed and transferred to fresh HTF medium. The efficacy of the IVF procedure was assessed by examining the formation of two-cell embryos after 16–18 hours of incubation.
Histology
Male testes, epididymis, brains and lungs were used for histopathological examination. The specimens were fixed with 10% buffered formalin solution (pH 7.4), trimmed, dehydrated with 99.7% isopropanol, and paraffin-embedded. Microtome sections (3 µm) were deparaffinized, hydrated, and stained with hematoxylin and eosin. Pathologies were diagnosed and classified according to published recommendations (Maronpot et al. 1999).
Immunohistochemistry
Paraffin sections were washed with xylen three times, then dehydrated in two changes of 100% pure ethanol, followed by overnight rehydration in TBS (10 mM Tris-HCl pH 7.5, 150 mM NaCl). For permeabilization, tissue sections were incubated in a citric buffer (10 mM citric acid, 0.05% Tween-20) for 12 min at 96°C in a stirred water bath WB-4MS (Biosan). Blocking was conducted in TBS supplied with 2% bovine serum albumin (BSA, Proliant Biologicals). The following primary antibodies were used in the corresponding dilutions: anti-HA (1:80, 20970300, Roche Diagnostics GmbH), anti-α-tubulin Alexa 488 (1:100, RH233965, Invitrogen), anti-acetylated α-tubulin Alexa 488 (1:100, K0821, Santa Cruz Biotechnology). For detection, the following secondary antibodies were used: Alexa 647 anti-rat (1:500, A-21247, Thermo Fisher). Tissue sections were covered with Mowiol supplied with DAPI (Invitrogen).
Immunoblotting
Immunoblotting of tissue lysates was done as previously described (Averina et al. 2023). For immunoblot analysis, samples of testes tissue were lysed in lysis buffer (5% SDS, 0.1% SDC, 100 mM TEAB) with protease inhibitor cocktail (Thermo Fisher). The transfer ready PVDF membrane (Thermo Fisher) was blocked for 1 h in TBST (10 mM Tris-HCl pH 7.5, 150 mM NaCl, 0.1% Tween-20) containing 5% bovine serum albumin (BSA, Proliant Biologicals). Primary antibodies anti-HA tag were diluted 1:2500, others 1:1000 in TBST containing 5% BSA. The following primary antibodies were used: anti-HA HRP-conjugated (1:4000, 54193500, Roche Diagnostics GmbH), anti-RNF151 (1:1000, 24708-I-AP, Proteintech), anti-KIFAP3 (1:1000, XF3612423, Invitrogen), anti-SEPTIN10 (1:1000, 12420-I-AP, Proteintech). The secondary HRP-conjugated anti-rabbit (1706515, Bio-Rad) antibodies were used at a 1:5000 dilution. GAPDH (ab8245, Abcam) was used as a loading control.
Electron microscopy
Three testes samples from wild-type, Nsun7Δ38/Δ38 and Nsun7i2/i2 mice lines were fixed with 2.5% glutaraldehyde in 0.1 M cacodylate buffer (pH 7.2-7.4), postfixed in 1% osmic acid and embedded in epoxy resin. Ultrathin sections were prepared on a Reichert Jung Ultramicrotome, Ultracut E and contrasted with 1% aqueous uranyl acetate and lead citrate solution. The preparations were examined at 80 kV using a JEM-1011 transmission electron microscope (JEOL) equipped with an Orius SC1000 W camera (Gatan Inc.). In each sample was determined the position of LC of fibrous sheath in the principal piece of spermatozoa tail.
Proteome analysis
Testes of wild-type (n=3) and Nsun7i2/i2 (n=3) mice lice were lysed in lysis buffer (5% SDS, 0.1% SDC, 100 mM TEAB) with protease inhibitor cocktail (Thermo Fisher) and ultrasonicated (three cycles for 10 sec of ultrasonication on Ultrasonic professor, Cole-Parmer). The protein concentration was measured with DC Protein Assay Kit II (Bio-Rad). 100 mkg of proteins were taken for further analysis. Reduction, alkylation and trypsin (Promega) digestion were conducted under manufacturer recommendations of S-Trap Micro MS Sample Prep Kit (ProtiFi Innovations omics solutions). Obtained peptides were collected under centrifugation.
The sample peptides in a volume of 2 µl were loaded onto the Acclaim µ-Precolumn (0.5 mm × 3 mm, 5 µm particle size, Thermo Fisher) at a flow rate of 15 µL/min for 4 min in an isocratic mode of Mobile Phase C (2% acetonitrile, 0.1% formic acid). Then the peptides were separated with high-performance liquid chromatography (HPLC, Ultimate 3000 Nano LC System, Thermo Fisher) in a 25 cm long C18 column (Peaky C18 column, inner diameter of 100 μm, Molecta). The peptides were eluted with a gradient of buffer B (80% acetonitrile, 0.1% formic acid) at a flow rate of 0.4 μL/min. Total run time was 90 min, which included an initial 4 min of column equilibration to buffer A (0.1% formic acid), then gradient from 5% to 35% of buffer B over 65 min, then 6 min to reach 99% of buffer B, flushing 10 min with 99% of buffer B and 5 min re-equilibration to buffer A.
MS analysis was performed at least in triplicate with a Q Exactive HF-X mass spectrometer (Q Exactive HF-X Hybrid Quadrupole-Orbitrap Mass spectrometer, Thermo Fisher). The temperature of the capillary was 250°C, and the voltage at the emitter was 2.1 kV. Mass spectra were acquired at a resolution of 60,000 (MS) in a range of 400−1500 m/z. Tandem mass spectra of fragments were acquired at a resolution of 15,000 (MS/MS) in the range from 140 m/z to m/z value determined by a charge state of the precursor. The maximum integration time was 50 ms and 30 ms for precursor and fragment ions, respectively. The AGC targets for precursor and fragment ions were set to 1×106 and 1×105, respectively. An isolation intensity threshold of 100,000 counts was determined for the precursor's selection, and up to the top 20 precursors were chosen for fragmentation with high-energy collisional dissociation (HCD) at 29 NCE. Precursors with a charge state of +1 and more than +5 were rejected, and all measured precursors were dynamically excluded from triggering of a subsequent MS/MS for 50 sec.
The obtained raw data files were processed with MaxQuant software (versions 2.1.0.0) (Cox and Mann 2008) using the internal search engine Andromeda (Cox et al. 2011), and searched against the UniProtKB database restricted to Mus musculus (51,444 entries). For the quantification, the same parameters as in the previous study (Fang et al. 2021) were used. Briefly, for the peptide, the following parameters were set: trypsin digestion only with maximum of two missed cleavages, minimum length,- six amino acids. N-terminal acetylation and methionine oxidations were set as variable modifications, and cysteine carbamidomethylation as a fixed. The fragment mass tolerance was 0.05 Da and the precursor mass tolerance was 20 ppm. The Match between run function was enabled. Label-free quantification (LFQ) was estimated with the MaxLFQ algorithm (Cox et al. 2014), and a minimum ratio count of 1 was set. The FDR for protein identification was set at 0.01, and at least two unique peptides were required.
Bioinformatic analysis of the identified and quantified data was performed with package DEP (version 1.23.0) (Zhang et al. 2018) on R (version 4.1.2). Statistical significance was determined using the protein-wise linear models and empirical Bayes statistics. For filtration of differentially expressed proteins, the following thresholds were set: an adjusted P-value <0.05 and a fold change >1.5.
UV CLIP assay
Testes were obtained from 30 mice of wild-type and 30 HA-Nsun7 mice. Testes were decapsulated in PBS and irradiated twice with 400 mJ/sm2 UV (GS Gene Linker UV Chamber, Bio-Rad). After that, seminiferous tubules were lysed in PXL buffer (0.1% SDS, 0.5% deoxycholate, 0.5% NP-40 in PBS, without Ca and Mg) and exposed to five cycles of 8 sec ultrasonication with 70% amplitude. Then samples were incubated with Rnase T1 0.01 u/μl (Thermo Fisher) for 10 min at 37°C, with shaking at 1000 rpm. For immunoprecipitation, Pierce Anti-HA Magnetic Beads (Thermo Fisher) were used. Before elution, RNA-protein conjugates, which were bound to magnetic beads, were exposed to dephosphorylation with СIAP (0.5 U/μl) (2386334, Invitrogen) for 15 min at 37°C, with shaking at 1000 rpm, which was followed by phosphorylation with T4 PNK (Thermo Fisher) for 30 min at 37°C, with shaking at 1000 rpm. RNA was eluted from the magnetic beads with proteinase K (Thermo Fisher) for 20 min at 37°C, with shaking at 1000 rpm.
RNA libraries were prepared with the NEBNext Small RNA Library Prep Set for Illumina (Multiplex Compatible, E7330S), in accordance with the manufacturer's recommendations, omitting the size-select step at the final stage of the protocol. The quality and concentration of the libraries were determined using the automated electrophoresis system Agilent 4200 Bioanalyzer with High Sensitivity ScreenTape Kits (Agilent) and a Qubit 4 fluorimeter (Thermo Fisher), respectively. Sequencing was conducted on Illumina sequencing system HiSeq 1500 (Illumina), with a minimum of 5 million reads generated for each sample.
Radioactive labeling and PNK assay
Testes were obtained from five mice of wild-type and five HA-Nsun7 mice. The sample preparation procedure was the same as in UV CLIP assay followed by sequencing; however, in the reaction with T4 PNK (Thermo Fisher), we added 1 μCi γ-[32P] ATP. RNA-protein conjugates were eluted from the magnetic beads with 0.1% SDS 5 min at 95oC with shaking at 1000 rpm. The obtained samples were applied to the 8% PAAG gel and then transferred on nitrocellulose membrane 0.2 μm (Bio-Rad). The membrane was exposed for 9 days with the Storage Phosphor Screen. Visualization of the results was conducted using Typhoon FLA 9500 (GE Healthcare Bio-Sciences AB).
Analysis of RNA methylation in elongated spermatids
One biological replicate consisted of testes obtained from three mice from wild-type and Nsun7i2/i2 mice lines. Cell sorting was performed according to the previously published procedure (Simard et al. 2015). Briefly, testes were decapsulated and incubated with collagenase type IV (2 mg/ml, Gibco) at 37°C for 20 min. After that, seminiferous tubules were applied to 5% Percoll solution and incubated on ice for 15 min. Seminiferous tubules, which had drowned, were homogenized with Dounce homogenizer type A. The obtained cell suspension was twice filtered through a 40 mkm cell strainer (Greiner Bio-One). After that, cells were centrifuged (5 min, 800 g), dissolved in 3 ml of PBS and fixed on ice for 15 min with 3 volumes of ethanol 100% supplied with 0.8 mM EDTA. Then cells were stained with SYTO16 (Invitrogen) on ice for 30 min.
Cell sorting was performed on a 4-Laser (405 nm: violet, 488 nm: blue, 561 nm: yellow-green, 633 nm: red) 20-parameter BD FACSAria III, and BD FACSDiva 6.1.3 software was used to visualize and analyze the data. Laser delay was set to 0 for the blue, –77.39 for the red, 37.33 for the violet and –39.85 for the yellow-green lasers. Area scaling was set to 1.14 for the blue, 1.0 for the red, 0.75 for the violet and 0.96 for the yellow-green lasers. Window extension was set to 2.00 µs and the FSC area scaling to 1.00. A sample filter line of 50 µm was placed at the end of the sample line to avoid clumping.
Cells on 10–16 steps of spermatogenesis (elongated spermatids) were collected in falcons covered with FBS. RNA was purified from fractions of elongated spermatids. rRNA depletion (RiboMinu Eukaryote Kit v2, Invitrogen) and bisulfite conversions (Abcam) were conducted under manufacturer's recommendations with an additional thermal cycle of bisulfite conversion. The RNA library preparation protocol and sequencing parameters were the same as in the UV CLIP assay.
Whole transcriptome assay
For the whole transcriptome assay, RNA was purified from testis tissue of wild-type (n = 3) and Nsun7 knockout (n = 3) mice. The isolated RNA was used for rRNA and globin depletion using Illumina Ribo-Zero Plus rRNA Depletion (Illumina). Libraries were then prepared using the NEBNext Ultra II RNA Library Prep Kit for Illumina (NEB) with dual indexing (NEB E6445, Set 3, NEBNext Dual). The resulting libraries were sequenced using NovaSeq6000 in paired-end read mode with a read length of 61 bp. The sequencing was carried out using resources of the Skoltech Genomics and Biovisualization Core Facility.
Analysis of RNA sequencing data
Analysis of BS-RNA-seq was performed with methylseq (v2.4.0) nf-core pipeline (Ewels et al. 2020) using mm39 version of the mouse genome. Briefly, in this pipeline, Bismark (Krueger and Andrews 2011) was used to prepare the reference with C-to-T substitutions in both orientations of DNA strands, and alignment itself was done via splicing-robust method HISAT2 (Kim et al. 2019). Reads with the same start and end were removed from the alignment, and methylation in each position was calculated as a fraction of nonconverted cytosines. Only positions that are covered with at least 10 reads in total in each group of comparisons (mutants and wild-types) were kept for further analysis. Statistical significance of differences between methylation levels in wild-type mice and mutants was calculated via a likelihood-ratio test of beta-binomial regression as described in Dolzhenko and Smith (2014), with Benjamini-Hochberg FDR multiple testing correction.
CLIP-seq reads were first trimmed with the Trim Galore tool and afterward aligned onto the mm39 mouse genome with STAR (Dobin et al. 2013), prohibiting novel splice junctions within the alignment. Resulting bam files were deduplicated and concatenated together to perform peak calling with Piranha (Uren et al. 2012). Coordinates of each peak were extracted and used to calculate the coverage of each peak in each sample with featureCounts (Liao et al. 2014). Differential testing of each peak coverage between wild-type mice and mutants was performed via the DESeq2 (Love et al. 2014) R package. Binding site identification was performed on a set of sequences of upregulated HA-NSUN7 peaks with the tool ChIP-Munk (Kulakovskiy et al. 2010). Benjamini-Hochberg procedure was used to correct P-values for multiple testing. A cutoff of FDR < 0.05 and FoldChange > 2 was used to determine NSUN7-associated peaks.
Bulk RNA-seq quanitification was performed based on raw reads with kallisto (v. 0.50.1) (Bray et al. 2016). Counts of each isoform of the gene were summarized and rounded (similarly to tximport [Soneson et al. 2015] pipeline) to work on a gene level instead of transcript level. Differential expression analysis between Nsun7 knockouts and wild-type mice was performed with DESeq2 package (v. 1.34.0) (Love et al. 2014) with Benjamini-Hochberg FDR correction of Wald test P-values. A cutoff of FDR < 0.05 and Log2FoldChange > 1.5 was used to determine differentially expressed genes.
Functional enrichment analysis was performed with GSEA (Subramanian et al. 2005). The transcriptome of individual mouse testicular cells, containing 32 sets of marker genes and allowing to distinguish 12 types of testicular cells, was used as a reference for comparison (Jung et al. 2019).
qPCR
For the measurement of Nsun7 levels, RNA was extracted from 5–15 mg of mice tissue. For the measurement of NSUN7 target RNA levels, mice testes were decapsulated and exposed to collagenase type IV (2 mg/ml, Gibco) treatment at 37°C for 20 min. The obtained tissue and cell suspensions were used for total RNA purification with RNA extract reagent (Evrogen), followed by cDNA synthesis with a Maxima First Strand cDNA Synthesis Kit for RT-qPCR (Thermo Fisher) with a random hexamer primer. Quantitative PCR was performed by a Maxima Hot Start DNA polymerase (Evrogen) in the presence of SYBR green.
The following primers were used for amplification of indicated mRNAs: Nsun7 (TACACAGTTGCCCATATGTC and ACCTTCACATTCTGTAACCG), ActB(TGCGTGACATCAAAGAGAAG and CGGATGTCAACGYCACACTT), Pi4k2a(TAGCCAGAAGAGCAAGGTTA and TCATGTGTGCTAGAGATGGA), Spata31d1d(CGAAGCTTTTGTGCTTATTCC and AGGGATAGCTAAAGGAGCAA), Ccdc6(TTTGGCCTTTCTAAAATGAAGC and AGCTCCCTGACACAAGTTA), Gm8251(CTTTTCCTTCTACGGATTCCC and AAACCCAGCCTTATGTCAAC), Gsg1 (CCCCAAGCTATTCACCAAAG and GTGCTCGTTGAGTAACAGAA), Abtb3(TCATTGTCAAGGGCAGAATG and CCATCTCTGGAAGATCCTGT), Pwp1(CATGCTGAGGAAGGTCTATTC and TCATGAGGAAGGAGAGATGC), LOC102635990(GGTTTTGAAAGAGAGTGGCTA and AGCTATACCACTTCTGGACC), 1700027A15Rik(AAGTCCTAGTGTTTGACTGTTC and ACCGAGCTCCTCACAAG), 4930505K14Rik(GATCCATCATCAACACAGCA and AAAAGCCTTGATCCATTCCC), Pabpc1(TGATGTCCCAAGCTATTCCA and GTTTTATAGACCCTGGGAAAAGA), Tcte1(CTCCAGAGTGGTAGCTCAG and CTCTTGTTCCTCTGTGTCCT), Gstm5(GAGAGAGGAGGAGACAGTTC and CCCAGAACCATAGACTTGGA), Snora20(TCACCAAGGAGAGAATTCAGA and TTTAAAACTCTCCTAACAGCCAT), Tcp1(TAGGCATGCCCAAGAGAATA and TCCAATTTCTCAGGGTCTGT), Hk1(AACAGACTTCGACAAAGTGG and AAACTTGGTCTCGAAGATGC), Prm1 (GCCGCTCATACACCATAAG and TTAGCAGGCTCCTGTTTTTC).
Targeted mass-spectrometry analysis of NSUN7
Solid-phase synthesis of stable isotope-labeled standard (SIS) ALEYQSSGVK and TVSQAGTSSQVR was carried out on an Overture automated peptide synthesizer (Protein Technologies). In the synthesis of SIS peptides, isotope-labeled amino acids Fmoc-Lys-OH-13C6,15N2 or Fmoc-Arg-OH-13C6,15N2 (Cambridge Isotope Laboratories) were incorporated in peptide sequences instead of the native counterparts. Experimental samples were spiked with SIS with a content of 10 fmole per μg of total peptides.
Targeted mass spectrometric analysis in the selected reaction monitoring (SRM) mode was carried out according to the following protocol. Each experimental sample was analyzed in three technical replicates. Chromatographic separation was carried out using an Agilent 1200 series system (Agilent Technologies) coupled to a TSQ Quantiva triple quadrupole mass analyzer (Thermo Fisher). A 3.5 μl sample containing 7 μg of native peptides and SIS standards was separated using a ZORBAX SB-C18 analytical column (150 × 0.5 mm, 5 μm particle diameter, Agilent Technologies) in an acetonitrile gradient at a flow rate of 20 μl/min. The column was initially equilibrated with 5% (v/v) solution B (80% [v/v] acetonitrile in 0.1% [v/v] formic acid) and 95% (v/v) solution A (0.1% [v/v] formic acid) for 5 min; then the concentration of solution B was linearly increased to 60% (v/v) over 30 min, after which the concentration of solution B was increased to 99% (v/v) over 1 min and the column was washed with 99% (v/v) solution B for 5 min; then the concentration was returned to the initial conditions over 1 min, in which the column was equilibrated for 9 min. Mass spectrometric analysis was performed in the dynamic selected reaction monitoring (dSRM) mode using the following MS detector settings: capillary voltage was 4000 V, drying gas flow rate (nitrogen) was 7 L/min, axillary gas flow rate (nitrogen) was 5 L/min, capillary temperature was 350°C, isolation window for the first and third quadrupoles was 0.7 Da, scan cycle time was 1.2 sec, gas pressure (argon) in the collision cell was 1.5 mTorr. All transitions from which the signal was recorded were used for quantification in Skyline software (version 4.1.0).
DATA DEPOSITION
The results of the transcriptome sequencing data are available in GEO via accession numbers GSE281899, GSE281866, and GSE281586. The mass-spectrometry proteomics data have been deposited in the ProteomeXchange Consortium via the PRIDE partner repository with the data set identifier PXD057007.
SUPPLEMENTAL MATERIAL
Supplemental material is available for this article.
ACKNOWLEDGMENTS
We are grateful to the Moscow State University Development Program for providing access to the CelenaX High Content Imaging System. The whole transcriptome sequencing of testicles was carried out using resources of the Skoltech Genomics and Biovisualization Core Facility. The study was conducted under the state assignment of Lomonosov Moscow State University. Experimental work was supported by Russian Science Foundation grant 24-14-00048 (P.V.S.). The animal study protocol was approved by the Institutional Ethics Committee of “The Institute of Mitoengineering of Moscow State University” LLC (Moscow, Russia), protocol code no. 79 dated April 28, 2015.
Author contributions: E.A.G., O.A.A., P.I.P., O.A.D., and P.V.S. designed the research; E.A.G., O.A.A., P.I.P., E.E.B., O.A.P., V.S.B., A.V.P., M.A.E., L.P., E.A.R., S.E.N., K.S.P., A.Y.G., O.O.G., V.N.M., D.S.K., Y.Y.S., A.V.D., M.P.R., V.G.Z., A.M.M., and P.V.S. performed the research; A.M.M. contributed analytic tools; S.V.I. analyzed the data; and E.A.G., P.I.P., and P.V.S. wrote the paper.
Footnotes
-
Article is online at http://www.rnajournal.org/cgi/doi/10.1261/rna.080320.124.
- Received November 10, 2024.
- Accepted February 16, 2025.
This article is distributed exclusively by the RNA Society for the first 12 months after the full-issue publication date (see http://rnajournal.cshlp.org/site/misc/terms.xhtml). After 12 months, it is available under a Creative Commons License (Attribution-NonCommercial 4.0 International), as described at http://creativecommons.org/licenses/by-nc/4.0/.
REFERENCES
MEET THE FIRST AUTHOR

Meet the First Author(s) is an editorial feature within RNA, in which the first author(s) of research-based papers in each issue have the opportunity to introduce themselves and their work to readers of RNA and the RNA research community. Ekaterina A. Guseva is the first author of this paper, “Positioning of sperm tail longitudinal columns depends on NSUN7, an RNA-binding protein destabilizing elongated spermatid transcripts.” Ekaterina is a PhD student in the laboratory of Professor Olga Dontsova at the Skolkovo University of Science and Technology and a junior scientist in the laboratory of Professor Petr Sergiev at the Lomonosov Moscow State University. Her research focuses on the study of RNA methyltransferases and RNA-binding ubiquitin ligases, mutations which are associated with human reproductive disorders.
What are the major results described in your paper and how do they impact this branch of the field?
In this paper, we investigated the role of RNA methyltransferase NSUN7 in spermatogenesis in vivo. We studied the phenotype of Nsun7 knockout mice, which appeared to be infertile, reproducing the phenotype of humans. The detailed characterization of the obtained mutants revealed structural abnormalities of longitudinal columns positioning in the sperm tail, which dramatically affected the beating ability of the sperm tail. Very little is known about the formation and positioning regulation of these structures. We established that Nsun7 is expressed during spermatogenesis and binds to a set of spermatid-specific RNAs, leading to a decrease in their levels. On a protein level, we observed the association of NSUN7 with components of the ubiquitin ligase system, which is also intriguing, as the same phenotype was described for mutants in the E2-ubiquitin ligase Ube2Bb. Collectively, these observations suggest that structural rearrangements during spermatogenesis may be orchestrated by RNA-binding methyltransferases; however, the involvement of methylation in this process remains to be investigated.
What led you to study RNA or this aspect of RNA science?
RNA-binding proteins are known to perform a wide range of functions, with many of them having significant impacts on human health. I've always been fascinated by the molecular mechanisms of human diseases that rely on RNA and RNA-associated proteins, so the opportunity to contribute to this field of research is particularly exciting for me.
During the course of these experiments, were there any surprising results or particular difficulties that altered your thinking and subsequent focus?
We started to study the RNA methyltransferase NSUN7 in cell lines, but when we characterized the effects of mutations in this gene, we decided to continue our studies exclusively in animals. This was because spermatogenesis is a very specific process, and the natural binding partners of NSUN7 and accessory structures such as the axoneme would probably be absent in most cell lines. And this has paid off, because many of the RNA-binding targets of NSUN7 that we have discovered are indeed specific for spermatogenic cells.
What are some of the landmark moments that provoked your interest in science or your development as a scientist?
I have always been interested in science, especially biology. At school, I attended class with the Advanced Biology program, and one of the key moments in our education process was participating in a real study at a scientific laboratory. In this project, I used PCR to screen for allelic variants of Alu repeats in my genome and in the genomes of two sets of twins. This simple school project showed me in practice that we all have diverse genomes. And from then on, I was inspired to study the functions of genes and the effects of mutations in them.
If you were able to give one piece of advice to your younger self, what would that be?
Don't be afraid to make mistakes—this experience is necessary and inevitable when working on something that is truly at the cutting edge of science.








