
A general RNA-templated RNA extension activity of E. coli RNA polymerase
- 1Department of Genetics, Stanford University, Stanford, California 94305, USA
- 2Department of Pathology, Stanford University, Stanford, California 94305, USA
- 3Laboratory of Molecular Biophysics, The Rockefeller University, New York, New York 10065, USA
- Corresponding authors: dgalls{at}stanford.edu, afire{at}stanford.edu
-
↵4 These authors contributed equally to this work.
-
Handling editor: Erik Sontheimer
Abstract
Multisubunit “DNA-dependent” RNA polymerases (RNAPs) have noncanonical RNA-directed RNA synthesis activity; this allows the synthesis of complementary RNA from RNA templates. Such noncanonical RNAP activities are biologically significant, serving RNA pathogens such as hepatitis delta virus (HDV) and contributing to cellular gene regulation. Despite the broad biological implications of these processes, our understanding of the underlying RNAP mechanisms remains incomplete. Using Escherichia coli RNAP, a multisubunit RNAP, as a model, we describe here the general RNA-templated RNA extension activity of that enzyme. Our data argue that the 3′ end of an added RNA template can fold back and pair with upstream bases in the template, creating an intramolecular primer:template duplex as short as 1–2 base pairs. The RNAP then extends this intramolecular duplex, incorporating nucleotides complementary to the template. RNA-templated RNA extension occurred in minutes and did not appear to be suppressed by the presence of a promoter-containing DNA template. Excepting oligonucleotides implicitly designed to prevent any possibility of 3′ end self-priming, every RNA template we tested could be extended by the enzyme, highlighting the general nature of this reaction. These data define a general activity of a cellular RNAP. Unrestricted, this activity could contribute to the emergence and replication of RNA-based agents such as HDV and viroids; if highly regulated, the activity could limit these same elements.
Keywords
INTRODUCTION
In the central dogma, “DNA-dependent” RNA polymerases (RNAPs) transmit information from DNA to RNA. In addition to having this canonical function, some RNAPs can accept RNAs as templates to generate at least partially complementary RNAs (Biebricher and Orgel 1973; Mühlbach and Sänger 1979; Konarska and Sharp 1989; Glenn et al. 1990; Wassarman and Saecker 2006). These RNA-templated activities have implications in evolution, biology, and biotechnology. Regarding evolution, extant RNA-templated activities of RNAPs may be remnants of a primordial RNA–Protein World; these activities may offer insight into nearly extinct pathways along the origin of life (Wettich and Biebricher 2001). In terms of biology, RNA pathogens like hepatitis delta virus (HDV) and viroids infect humans and plants, respectively, and are replicated by endogenous RNAPs in the absence of any DNA intermediate (Glenn et al. 1990; MacNaughton et al. 1991). In addition, several examples illustrate that the RNA-templated activities of cellular RNAPs can be used by cells to modulate or amplify RNA signals and regulate gene expression (Wassarman and Saecker 2006; Haag et al. 2012; also see discussion in Volloch 1986). Lastly, in the context of biotechnology, such activities could be engineered so that endogenous RNAPs could amplify exogenously provided therapeutic RNAs (e.g., mRNAs, siRNAs) in a form of nongenomic but durable gene therapy (Hsieh and Taylor 1992).
Despite the evolutionary, biological, and biotechnological significance of RNAP activities on RNA, our understanding of such activities remains incomplete. We focus herein on noncanonical activities of cellular “DNA-dependent” RNAPs on RNA. While various noncanonical RNA-templated activities of the RNAP from bacteriophage T7 (T7 RNAP) have been characterized in vitro (Konarska and Sharp 1989; Cazenave and Uhlenbeck 1994; Arnaud-Barbe et al. 1998; Gholamalipour et al. 2018; Jain et al. 2020), the utility of T7 RNAP as a model for RNA-templated RNAP activities in cells is limited. T7 RNAP is a single-subunit RNAP that is distantly related to viral RNA-dependent RNA polymerases (Koonin et al. 2020). In contrast, chromosomal transcription in all known cells is catalyzed by multisubunit RNAPs distinct from single-subunit RNAPs (Cramer 2002).
Historically, RNA-templated activities of multisubunit RNAPs have been studied through a lens of activities on specific, “special” RNA templates (e.g., HDV, 6S RNA), which can be grouped into several categories. First, pathogens like HDV and viroids have RNA genomes but do not encode their own polymerases (Gross et al. 1978; Wang et al. 1986; Hernández and Flores 1992). Multiple lines of evidence, including the reconstitution of a potential replication cycle in vitro, suggest that these RNA genomes are replicated by host-encoded RNAPs, such as eukaryotic RNAP II (Rackwitz et al. 1981; MacNaughton et al. 1991; Filipovska and Konarska 2000; Pelchat et al. 2001; Lehmann et al. 2007). Second, RNAPs have activity on regulatory noncoding RNAs. In bacteria, 6S RNA binds to RNAP to repress DNA-templated transcription; RNA-templated transcription on RNAP-bound 6S RNA causes dissociation of the 6S RNA:RNAP complex, derepressing transcription (Wassarman and Storz 2000; Wassarman and Saecker 2006). In eukaryotes, RNAP II can extend constructs derived from the transcriptional repressors B2 RNA and FC RNA in vitro; this extension has been suggested to derepress transcription in a manner analogous to that of 6S RNA (Espinoza et al. 2004; Lehmann et al. 2007; Wagner et al. 2013). Third, various RNAPs have been observed to have activity on several not-obviously-physiological RNA templates. Escherichia coli RNAP replicated several AU-rich, repetitive RNA species in vitro (Biebricher and Orgel 1973; Wettich and Biebricher 2001). Yeast RNAP II (Johnson and Chamberlin 1994) and E. coli RNAP (Altmann et al. 1994) added nucleotides to the 3′ end of diverse RNA templates. In these studies, this activity was interpreted as a nonspecific, nontemplated tailing activity; however, a templating event was not excluded. Finally, E. coli RNAP appeared to add C to the 3′ end of an RNA helix that contained two G bases on its 5′ end (Kashlev and Komissarova 2002). Outside of the context of these “special” RNAs discussed in this paragraph, there is little knowledge of RNA-templated activities of multisubunit RNAPs.
We chose to use E. coli RNAP as a tractable model for characterizing RNA-templated activities of multisubunit RNAPs in a template-agnostic manner. Key advantages of E. coli RNAP as a model system include (i) its relative simplicity; (ii) its homology with eukaryotic RNAPs, including human RNAP II; (iii) the known RNA-templated activities of E. coli RNAP on certain “special” RNA templates (e.g., 6S RNA and AU-rich repetitive RNAs) (Biebricher and Orgel 1973; Wettich and Biebricher 2001; Wassarman and Saecker 2006); and (iv) the potential relevance of bacterial RNAPs to the replication of putative HDV- and viroid-like RNA-based agents thought to replicate in bacteria (Weinberg et al. 2021; Edgar et al. 2022; Forgia et al. 2023; Lee et al. 2023; Zheludev et al. 2024).
In the course of these studies, we have characterized a general RNA-templated RNA extension activity of E. coli RNAP. We propose a model in which the 3′ end of an RNA template molecule can fold back and anneal upstream in the RNA template, creating an intramolecular primer for RNA extension. Excepting oligonucleotides designed to be nonextendable, every template we tested could be extended by the enzyme, supporting the generality of this reaction. We discuss the implications of these data for RNAP biology and for the emergence and replication of RNA-based agents.
RESULTS
An RNA extension activity of E. coli RNAP
To characterize the potential activities of a multisubunit RNAP on RNA templates, we set up reactions containing E. coli RNAP σ70 holoenzyme and all four natural nucleotide triphosphates (NTPs). Instead of a classic promoter-containing DNA template, we provided a 68 nt RNA oligonucleotide (LR-3) containing a fragment of the coding region of GFP. This oligonucleotide was produced for another experiment and was chosen arbitrarily based on its availability in the laboratory. Gel electrophoresis of the resulting reaction products yielded a slower-migrating band in addition to the RNA template band, despite rigorous post-reaction protein removal (Fig. 1A; Supplemental Fig. S1A). The upshifted, slower-migrating band also appeared when we used a 5′ endlabeled fluorescent template and visualized the fluorophore (Fig. 1A), demonstrating that the product retained the template strand. Bands of intermediate mobility (in between that of the template and the final product) were detectable within minutes of adding the template to the reaction; the intensity of these additional bands first increased and then decreased over several hours, thus appearing to chase into the largest product (Fig. 1B). This reaction required the addition of RNAP, RNA template, NTPs, and MgCl2 (Supplemental Fig. S1B,C). We hypothesized that E. coli RNAP extended the LR-3 RNA oligonucleotide at its 3′ end, and we preliminarily termed this activity “RNA extension” (Fig. 1C).
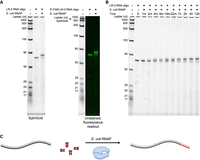
E. coli RNAP-catalyzed RNA extension. (A) Extension of the unlabeled (left) or 5′ endlabeled (right) RNA oligonucleotide LR-3. The right panel is pseudocolored to reflect the 5′ 6-FAM endlabel. Oligonucleotide sequence is an arbitrary segment of 68 nt from the coding region of GFP (sequence in Supplemental Table S1). In the left panel, the gel was stained with SybrGold and imaged. In the right panel, the fluorophore was imaged (to visualize the 5′ 6-FAM); the gel was then stained with SybrGold and reimaged (to visualize the ladder). Extension was overnight (∼12 h). (B) Time course of RNA extension with the unlabeled LR-3 RNA oligonucleotide. (C) Schematic of proposed model for RNA extension. The LR-3 RNA oligonucleotide is in black, the RNAP in blue, and the additional nucleotides in red. Note that the diagram is not to scale.
To characterize this reaction, we next performed a series of additional assays. First, to verify the canonical DNA-dependent RNAP activity of our enzyme preparation, we transcribed a defined promoter-containing DNA template with E. coli RNAP; the enzyme was active in this assay (Supplemental Fig. S1D,E). Both the DNA-templated transcription reaction and the RNA extension activity showed sensitivity to rifampicin (Supplemental Fig. S1D,F). Rifampicin is a small molecule inhibitor of E. coli RNAP that sterically hinders initiation after the nascent transcript is 2–3 nt in length (Campbell et al. 2001); it is in clinical use as an antibiotic (Goldstein 2014).
Second, to determine the role of the σ factor (which adds specificity for promoter sequences during DNA-templated transcription) in RNA extension, we compared the RNA extension activities of σ70 holoenzyme (our default preparation, which we used for all experiments unless otherwise specified) and core enzyme (no σ factor) preparations. Indeed, holoenzyme and core enzyme preparations displayed similar RNA extension activities (Supplemental Fig. S2A). Our primary preparations of E. coli RNAP holoenzyme and core enzyme were obtained commercially from New England Biolabs (NEB). Upon analysis by electrophoresis, these preparations yielded major bands consistent with the expected composition of the enzyme preparations (Supplemental Fig. S2B). Additionally, a separate preparation of the core enzyme (Chen et al. 2019) showed an equivalent extension activity (Supplemental Fig. S2A).
Third, to determine whether RNA extension was multiturnover, we varied RNAP concentration. Although most of our experiments were performed with approximately equimolar RNAP and RNA template, robust RNA extension also occurred when the template was in ∼10-fold excess (Supplemental Fig. S2D). The observed near-complete extension under conditions of substrate excess is indicative of a multiturnover reaction (i.e., one in which each enzyme molecule extends multiple RNA templates).
Extension of diverse RNA templates
Next, to characterize the specificity of this reaction for different RNA templates, we assayed RNAP activity on a variety of other RNA oligonucleotide templates. These templates varied in sequence, structure, and length (Fig. 2). As with LR-3 (used above), most templates were RNAs that had been synthesized for unrelated experiments (see Supplemental Table S1 for details; note that oligonucleotide names [e.g., LR-3] are arbitrary laboratory inventory labels). With several RNA templates generally unrelated in sequence, E. coli RNAP yielded additional, slower-migrating bands, analogous to the reaction with the LR-3 RNA template above (Fig. 2; Supplemental Table S1; e.g., R-12, DG-141, DG-36, DG-37, DG-144, and DG-145). This result suggests that E. coli RNAP can extend diverse RNA templates. In other cases, the RNAP appeared to yield a smear of products of higher molecular weight (e.g., LR-4, DG-48, and DG-143). These RNAs could be heterogenous extension products, in which the extension reaction may not begin or terminate at a defined position. For some templates, extension appeared to be stochiometric (as judged by a disappearance of species of the length of the template; e.g., LR-3, DG-48), whereas for other templates only a fraction of molecules appeared to be extended (e.g., R-12). In some cases, we observed additional faster-migrating material (e.g., DG-36); one of several possibilities is that this could stem from nuclease activity in our enzyme preparation (Altmann et al. 1994). We did not observe E. coli RNAP to have any activity on one oligonucleotide, JTG-20, which contains a 28 nt predicted hairpin flanked by CC dinucleotides (Supplemental Table S1) in which the 3′ CC end sits unpaired at the end of a duplex. Across all of our experiments, JTG-20 and two other oligonucleotides designed to be nonextendable (see below) were the only oligonucleotides on which we did not observe E. coli RNAP to have extension activity.

RNA extension on diverse RNA templates. RNA extension reactions were templated with a variety of RNA oligonucleotides (sequences in Supplemental Table S1) and analyzed on 10% (left) or 15% (center and right) gels.
Sequence analysis of RNA extensions
To characterize the sequences of the RNA extensions catalyzed by E. coli RNAP, we used RNA sequencing (RNA-seq). We customized a published dual-end capture RNA-seq protocol in which adaptors are directly ligated to the 3′ and 5′ ends of the RNA targets (Kim et al. 2019). A major advantage of this approach is that for RNAs under ∼135 nt, we can capture the exact end-to-end sequence of the RNA molecule on a single Illumina 150 cycle run. We note that RNA-seq has many potential sources of bias. Thus, we focused on semi-quantitative comparisons between RNA-seq libraries. Several caveats for the specific interpretation of read numbers and sequencing completeness are presented in Materials and Methods.
We sequenced RNA extension reactions in the absence and presence of E. coli RNAP (i.e., templates and extension products). We present RNA-seq data for all oligonucleotides described in Figure 2 except R-12 (which was prepared using T7 RNAP rather than by chemical synthesis so does not have a homogenous 3′ end [Cazenave and Uhlenbeck 1994; Gholamalipour et al. 2018]) and DG-36, DG-37, and DG-48 (which contain Illumina sequencing adaptors, meaning building an Illumina RNA-seq library from these templates would have been challenging). Before the RNAP reaction, almost all template-containing reads terminated exactly at the predicted last base in the template (see Materials and Methods). Typically, fewer than 1% of such reads had extensions (i.e., any additional 3′ bases). In contrast, for post-reaction RNA, most template-containing reads contained 3′ extensions of various lengths (Fig. 3A). Notably, this enzyme-dependent increase in the number of extended reads provides strong orthogonal support for the electrophoresis results described above. We also observed by sequencing that JTG-20 was not extended (Fig. 3A), consistent with our results obtained by gel electrophoresis (Fig. 2).
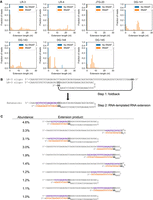
RNA extensions are templated. (A) RNA-seq analysis of the length distribution of RNA extensions. (B) An example extension product of the LR-3 oligonucleotide and a model for its formation. The LR-3 oligonucleotide sequence is in black, the extension sequence is in orange, the region complementary to the extension sequence is in purple, and priming bases are bolded. (C) For the LR-3 oligonucleotide, a table of the 10 most abundant extension products of at least 5 nt. Colors are as in B. Note that the percentages reflect the abundance of a specific extension product relative to all extension products and not just extension products of at least 5 nt. See Supplemental Figure S3B for the five most abundant extension products of less than 5 nt and Supplemental Table S2 for all extension products regardless of length and abundance.
Evidence for RNA-templated RNA extension
We next characterized the sequences of these RNA extensions. Extension sequences were highly correlated between independent experiments (Supplemental Fig. S3A; see Materials and Methods). We found that the vast majority of extended reads were comprised of sequences that were complementary to the template RNA (Fig. 3B,C; Supplemental Figs. S3B,C and S4A; Supplemental Table S2). For example, consider the LR-3 oligonucleotide template derived from GFP. Of the 10 most abundant extension sequences of at least 5 nt, all were complementary to the LR-3 template (Fig. 3B,C). In addition, extensions typically began at a point where the 3′ end of the input oligonucleotide would have been capable of self-priming (what we term “priming bases”; Fig. 3B,C; Supplemental Figs. S3B,C and S4A; Supplemental Table S2). For example, the LR-3 oligonucleotide terminus (ending in GG) chooses priming sites where the complementary sequence is CC or C (Fig. 3B,C). Since the extension products we detected via RNA-seq were diverse in length (Fig. 3A), the abundance of any given extension product was low. Thus, we grouped extension sequences by their first 5 nt (“initial extension sequences”; for example, the extensions GUAUCU and GUAUCUC both would have the same 5 nt initial extension sequence GUAUC). Of the five most common 5 nt initial extension sequences (GUAUC, GCAUG, CACUC, GGCAU, and CAUGG; representing >80% of all initial 5-mers), all were exactly complementary to the LR-3 template and contained priming bases (Supplemental Fig. S3C).
Similar results were obtained for five other RNA templates, all of which were described in Figure 2 (LR-4, DG-141, DG-143, DG-144, and DG-145; Supplemental Fig. S4A; Supplemental Table S2). In light of these data, we suggest that the templates fold back and sample duplex conformations (intermolecularly or intramolecularly). This creates a “primer” for what we term “RNA-templated RNA extension”; some of these primers form with only 1–2 nt of apparent complementarity (Fig. 3B,C; Supplemental Figs. S3B,C and S4A; Supplemental Table S2).
Priming of RNA-templated RNA extension provides a model for the absence of extension activity on oligonucleotide JTG-20. JTG-20 is predicted to fold into an almost perfect duplex, so its 3′ end cannot easily sample an intramolecular (or intermolecular) base-pairing interaction (Supplemental Fig. S4B). To further explore the importance of priming to RNA-templated RNA extension, we designed an oligonucleotide (DG-253) that cannot prime via canonical Watson–Crick–Franklin base-pairing: DG-253 ends in CC and does not contain G. A parallel oligonucleotide (DG-254) was designed with an additional 2 nt (GG) that allows priming. A striking increase in extension activity on DG-254 (with priming bases) compared to that on DG-253 (no priming bases) is consistent with priming bases playing an important role in RNA-templated RNA extension (Supplemental Fig. S5A).
To further explore the range of RNA-templated RNA extension, we designed an additional RNA oligonucleotide (DG-225) in which the 3′ end consists of a 4 nt loop bracketed by a 4 nt stem (Supplemental Fig. S5B). The resulting molecule would present four priming bases on which straightforward extension of up to 14 nt should be possible based on the length to the 5′ end of the potential template RNA. DG-225 was readily extended by E. coli RNAP (Supplemental Fig. S5B). We synthesized a marker oligonucleotide with the electrophoretic mobility expected for the full extension (an oligonucleotide with the same initial sequence as DG-225 and with the complete 14 nt extension) (Supplemental Fig. S5B). The electrophoretic mobilities of the extended DG-225 and of the synthetic completely extended marker RNA were indistinguishable (Supplemental Fig. S5B), consistent with efficient extension to or near the 5′ end of the primer/template.
Beyond the simple extension products predicted and described in this section, we observed a minority of longer and more complex products both on gels and in sequencing data. Upon examination of products from the sequencing data, we observed a fraction of hyper-extended reads containing evidence for multiround, discontinuous RNA-templated RNA extension (Supplemental Fig. S5C). We propose a mechanism for the synthesis of these products in which the RNAP extends the template oligonucleotide by several nucleotides; the RNA:RNA duplex unfolds; the 3′ end anneals elsewhere in the template oligonucleotide; and the RNAP extends the oligonucleotide further (Supplemental Fig. S5C). Such products raise the possibility that a similar multistep mechanism could underlie the synthesis of large RNAs by RNA-templated RNA extension.
Evidence for cis (self-primed) RNA-templated RNA extension
To assess whether RNA-templated RNA extension occurs in cis (i.e., foldback of the template onto itself) or in trans (annealing of the 3′ end of one template molecule to a different template molecule), we performed a mixing experiment. We designed a variant of LR-3 with a single base difference near its 5′ end (LR-3′; A-to-U substitution not overlapping with any common initial extension sequence or its priming bases; Supplemental Fig. S6A). We first extended LR-3 and LR-3′ individually and sequenced the products. We found that LR-3 and LR-3′ had similar but nonidentical extension profiles (Supplemental Fig. S6A–C; Supplemental Table S2); in particular, an extension beginning with GUAUC was common on the LR-3 template but rare on the LR-3′ template. As expected, at the region of the template with the single base difference, we found that extensions of the LR-3 template were complementary to LR-3 (and not LR-3′) and extensions of the LR-3′ template were complementary to LR-3′ (and not LR-3) (Fig. 4A,B; Supplemental Fig. S6A). We then mixed LR-3 and LR-3′ in the same RNA extension reaction and sequenced the products. We reasoned that if extension occurs primarily in cis, LR-3 templates should have extensions complementary to LR-3, and LR-3′ templates should have extensions complementary to LR-3′. In contrast, if extension occurs primarily in trans, both LR-3 and LR-3′ templates should have equal proportions of extensions complementary to LR-3 and LR-3′.
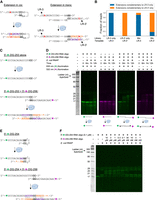
RNA-templated RNA extension in cis and in trans. (A) Schematic of experiment to test cis versus trans nature of the extension of LR-3. The single base difference between LR-3 and LR-3′ is bolded in red, and RNAP is in blue; otherwise, colors for the extension sequences are as in Figure 3B. (B) Oligonucleotides LR-3, LR-3′, and an equimolar mixture of LR-3 and LR-3′ were extended with E. coli RNAP, and the extension products were sequenced. For each library (LR-3, LR-3′, and mixture) and for each template (LR-3 and LR-3′), the fraction of extensions complementary to LR-3 alone (blue) or LR-3′ alone (orange) was counted. Extensions complementary to both LR-3 and LR-3′ were not counted here. (C) Schematic of experiment to test the possibility of trans RNA-templated RNA extension. (D) Trans extension of two RNA oligonucleotide templates. White space indicates gel splicing, and RNAP is in blue. (E) Schematic of experiment to attempt to assess cis and trans RNA-templated RNA extension in the same reaction. (F) Analysis of cis and trans RNA-templated RNA extension in the same reaction. Note that since DG-254 concentration was reduced from our default of 1 to 0.1 μM, RNAP concentration was reduced from 0.85 μM to 85 nM. Extension was for 1 h. In C and E, colors for the extension sequences are as in Figure 3B, and RNAP is in blue. In D and F, RNAP reactions were analyzed crude on 15% gels with gel protocol 2 (see Materials and Methods). Panels pseudocolored green reflect 635 nm illumination and visualization of TYE 655 (green star), and panels pseudocolored red reflect 532 nm illumination and visualization of Cy3 (red star). In all cases, all fluorophores were imaged first; the gel was then stained with SybrGold and reimaged (to visualize the ladder). The diffuse stain at ∼30 nt with 635 nm illumination is xylene cyanol.
In the mixing experiment containing both templates, we found that 95% of extensions of LR-3 templates were complementary to LR-3 only, and the remaining 5% of such extensions were complementary to LR-3′ only (Fig. 4A,B). Similarly, 90% of extensions of LR-3′ templates were complementary to LR-3′ only, and the remaining 10% of such extensions were complementary to LR-3 only (Fig. 4A,B). Extensions complementary to both LR-3 and LR-3′ (i.e., extensions that did not overlap the A-to-U mutation) were not considered here. This result indicates that RNA-templated RNA extension of LR-3 primarily occurs in cis (i.e., intramolecularly) under the conditions used in these experiments. The ∼5%–10% of LR-3 reads with LR-3′ extensions (and vice versa) could reflect trans (intermolecular) RNA-templated RNA extension or template switching during PCR; we suspect substantial contributions from template switching (see Materials and Methods). Overall, these data support a model in which the primary mode of extension of LR-3 under the conditions used relies on intramolecular self-priming followed by templated synthesis.
Analysis of trans RNA-templated RNA extension
We also wished to test if RNA-templated RNA extension could occur in trans. We used two differently labeled oligonucleotides for this experiment: DG-253 ends in CC, does not contain G (see above), and has a TYE 655 fluorescent label (spectrally similar to Cy5) at its 5′ end; DG-256 ends in GG, does not contain CC dinucleotides, and has a Cy3 label at its 5′ end (Fig. 4C). Neither DG-253 nor DG-256 can easily prime in cis, but each can prime the other in trans (GG to CC) (Fig. 4C). We observed a substantial disparity in extension activity between the individual and mixed extension reactions with each oligonucleotide alone generally recalcitrant to extension, while an equimolar mixture of both oligonucleotides was readily extendable (Fig. 4D). These results suggest that RNA-templated RNA extension can occur in trans.
To attempt to assess cis and trans extension in the same reaction, we used the cis priming-capable DG-254 at a fixed concentration, while adding increasing concentrations of a potential trans priming partner (DG-256) (Fig. 4E). When the concentration of DG-256 is low, we expect the extension of DG-254 to be similar to that with DG-254 alone, consistent with cis extension (Fig. 4E). When the concentration of DG-256 is high, we expect the extension of DG-254 in trans (Fig. 4E). These predictions were fulfilled in that extensions of DG-254 with low concentrations of DG-256 were indistinguishable from the extension of DG-254 alone, while increasing concentrations of DG-256 produced a distinct pattern consistent with trans extension (Fig. 4F). With DG-254 and DG-256 both at 0.1 μM, the extension pattern of DG-254 was consistent with a mixture of both reaction types. Taken together, our data support a model in which RNA-templated RNA extension can occur both in cis and in trans depending on the substrate(s) and reaction conditions.
Evidence for RNA-templated RNA extension in the presence of a DNA template
To determine whether RNA-templated RNA extension might occur during DNA-templated transcription, we performed a canonical DNA-templated transcription reaction and sequenced the RNA products (Fig. 5A). We quantified the number and sequence of any nucleotides 3′ of the predicted run-off transcript sequence. We observed that 88% of reads beginning with the predicted sequence of the full-length transcript also contained 3′ extensions 13–15 nt in length (Fig. 5B). The five most abundant extension products were all complementary to the template-encoded transcript sequence and contained at least 1 nt of priming as defined above (Fig. 5C,D). The presence of extended reads in a DNA-templated transcription reaction is consistent with RNA-templated RNA extension occurring on the transcript made in the presence of a DNA template.
We note that this transcript, predicted to be 130 nt in length without any RNA extension, migrated at ∼150 nt on a gel (Supplemental Fig. S1D,E). Abundant RNA-templated RNA extension of 13–15 nt would result in an RNA molecule 143–145 nt in length, consistent with the observed ∼150 nt gel mobility.
DISCUSSION
In this work, we have described a noncanonical activity of an essential and universal enzyme: a general RNA-templated RNA extension activity of E. coli RNAP. In E. coli RNAP reactions with an arbitrary RNA template (LR-3) derived from the coding region of GFP and without DNA, we observed that E. coli RNAP extended the GFP RNA template in minutes. Strikingly, we found that E. coli RNAP had a similar extension activity on a wide variety of RNA templates, with the only exceptions being a set of templates designed to prevent the possibility of internal self-priming by the free 3′ end. Sequence analysis of the extension products indicated that RNA extension was templated. Priming by the 3′ end of an RNA was critical for the reaction, and we have found that such priming can occur both in cis and in trans.
The RNAs we observed to be recalcitrant to extension (oligonucleotides JTG-20, DG-253, and DG-256) lack the ability to effectively self-prime. JTG-20 is an almost perfect duplex, and its 3′ end cannot access any straightforward base-pairing interaction (Supplemental Fig. S4B). Oligonucleotides DG-253 and DG-256 lack the complementarity necessary for base-pairing: DG-253 ends in CC and does not contain G, and DG-256 ends in GG and does not contain a CC dinucleotide (Fig. 4C). From these templates and from the sequence analysis above, we infer that the 3′ terminus base-pairing upstream in the template RNA segment is critical to the extension reaction.
Although RNA-templated RNA extension was general in that almost every template we tested could be extended by E. coli RNAP, the enzyme appeared to have some sequence-specific preferences for extension. The LR-3′ oligonucleotide, which differs from LR-3 by just a single base outside of any common initial extension region or its priming bases, had an extension profile that appeared to differ slightly from that of LR-3 (for instance, initial extension GUAUC was common for LR-3 but rare for LR-3′). The details of the RNA substrate specificity of E. coli RNAP RNA-templated RNA extension activity remain an open question. Such specificity could provide cells with a capability to control the populations of RNA subject to copying and potential replication or to impact RNA stability by reducing degradation by 3′ exonucleases.
Canonical DNA-templated transcription has usefully been considered as a three-phase process (initiation, elongation, and termination) (von Hippel et al. 1996). From our observations here, RNA-templated RNA extension appears to have characteristics of both transcriptional initiation and elongation. DNA-templated transcription becomes resistant to rifampicin during the transition from initiation to elongation; once the nascent transcript is 2–3 nt in length, it occupies the rifampicin binding site (McClure and Cech 1978; Campbell et al. 2001). The sensitivity of RNA extension to rifampicin suggests that RNA extension may have characteristics of transcription initiation from DNA promoters. We do note some residual rifampicin-resistant RNA extension activity. Formally, this could arise because of rifampicin-resistant E. coli RNAP activity or the activity of a separate, copurifying enzyme. Because elongation is known to be resistant to rifampicin, rifampicin-resistant RNA extension activity could stem from an elongation-like activity. Indeed, the fact that core RNAP (no σ factor) could specifically extend RNA is suggestive of a mechanism akin to the canonical elongation reaction. The structure of the RNA template required for extension or the RNA:RNA duplex region could conceivably bypass and/or lead to the displacement of bound rifampicin to a minor degree, and, therefore, allow residual extension activity to occur. In this regard, our findings are not inconsistent with genetic interactions between 6S RNA and rifampicin sensitivity (Esberard et al. 2022). While the modest rifampicin-resistant RNA extension activity provides for intriguing speculation, the fact that the reaction is at least partially rifampicin-sensitive confirms our overall conclusion of RNA extension by E. coli RNAP.
The generality of the E. coli RNAP-catalyzed RNA-templated RNA extension activity we observed adds to a literature of noncanonical multisubunit RNAP activities on constructs derived from specific, “special” RNAs, such as pathogens like HDV and viroids (Rackwitz et al. 1981; Filipovska and Konarska 2000; Pelchat et al. 2001; Lehmann et al. 2007), regulatory noncoding RNAs like 6S RNA, B2 RNA, and FC RNA (Wassarman and Storz 2000; Espinoza et al. 2004; Wassarman and Saecker 2006; Lehmann et al. 2007; Wagner et al. 2013), and AU-rich replicons (Biebricher and Orgel 1973; Wettich and Biebricher 2001). A previous report on RNA extension by E. coli RNAP (Altmann et al. 1994) (and a similar report on yeast RNAP II [Johnson and Chamberlin 1994]) interpreted RNA extension as a nonspecific, terminal transferase-like activity. If this terminal transferase-like activity were the only DNA-independent polymerase activity of these RNAPs, then these RNAPs would lack the ability to transmit genetic information in an RNA-templated manner. Here we show a general RNA-templated RNA extension activity for the well-studied E. coli RNAP.
We further note that a similar RNA-templated RNA extension activity has been characterized for T7 RNAP (Cazenave and Uhlenbeck 1994; Zaher and Unrau 2004; Gholamalipour et al. 2018), which is a single-subunit RNAP with no evident phylogenetic relationship to multisubunit RNAPs like E. coli RNAP. RNA-templated RNA extension and other noncanonical enzymatic activities can create substantial challenges for the production of mRNA therapeutics (Karikó et al. 2005; Mu et al. 2018; Baiersdörfer et al. 2019; Dousis et al. 2023). The general RNA-templated RNA extension activity of E. coli RNAP—and that this activity appeared to occur during DNA-templated transcription—suggests that efforts to use E. coli RNAP to produce mRNA therapeutics may face similar challenges to those with T7 RNAP.
The general RNA-templated RNA extension activity of a multisubunit, cellular RNAP like E. coli RNAP has biological and evolutionary implications.
First, while various RNA-templated RNAP activities have been observed both in vitro and in vivo (e.g., RNA-templated transcription on 6S RNA [Wassarman and Storz 2000; Wassarman and Saecker 2006]), RNA-templated RNA extension has not, to our knowledge, been described in the literature on 3′ ends in vivo (Dar et al. 2016; Maes et al. 2017; Herzel et al. 2022). Our own preliminary analyses suggest that RNA-templated RNA extension is rare in wild-type E. coli at steady state (see Materials and Methods). Together, these data imply that RNA-templated RNA extension may be suppressed inside cells. The simplest model is that this suppression stems from differences between our in vitro reaction conditions and the cellular environment. Other models are possible, including that RNA-templated RNA extension could be actively suppressed by a cellular factor in vivo. We note that eukaryotic transcription elongation factor TFIIS, which stimulates transcript cleavage in stalled elongation complexes, has been shown to suppress longer-than-expected transcripts during RNAP II-catalyzed, DNA-templated run-off transcription in vitro (Izban et al. 1998). Although a multitude of other models are plausible, one potential model is that these longer-than-expected transcripts were generated by RNA-templated RNA extension, and this activity was suppressed by TFIIS. Bacterial Gre factors, which have a similar role in promoting transcript cleavage in prokaryotes as TFIIS does in eukaryotes (Sekine et al. 2012), could be candidate suppressors of RNA-templated RNA extension in E. coli and other bacteria.
Second, RNA-templated RNA extension by E. coli RNAP has relevance for the life cycle of RNA agents that replicate via “DNA-dependent” RNAPs (e.g., Biebricher and Orgel 1973; Rackwitz et al. 1981; Pelchat et al. 2001; Lehmann et al. 2007). The two classic examples of such agents are HDV and viroids, which are hosted by humans and plants, respectively, and replicated by host RNAPs. However, recent metagenomic and metatranscriptomic studies (Weinberg et al. 2021; Edgar et al. 2022; Forgia et al. 2023; Lee et al. 2023; Zheludev et al. 2024) have characterized a diversity of RNAs that do not appear to be encoded in DNA, that do not encode polymerases, and that have been likened to HDV- or viroid-like RNA agents in bacterial hosts. The RNA-templated RNA extension activity of E. coli RNAP illustrates that bacterial RNAPs have polymerase activity on RNA templates, supporting a model in which bacterial “DNA-dependent” RNAPs might replicate these RNA agents. The general nature of RNA-templated RNA extension is consistent with the substantial sequence diversity of these RNA agents.
Third, RNA-templated RNA extension provides a plausible origin narrative for such RNA agents. Viroids and HDV have extensive self-complementarity in their primary sequences, a feature shared by many of the polymerase-nonencoding RNA agents found in metatranscriptomes (Weinberg et al. 2021; Edgar et al. 2022; Forgia et al. 2023; Lee et al. 2023; Zheludev et al. 2024). It is possible that this self-complementarity initially arose by templated extension of cellular RNAs (Diener 1995; Brazas and Ganem 1996). The general RNA-templated RNA extension activity of E. coli RNAP suggests both that bacterial RNAPs could have such an activity and that diverse RNA sequences could be extended. Of course, such an extension event would only be the first step on the path to a replicable RNA agent.
On a broader level, the remarkable flexibility shown by E. coli RNAP in extending RNA templates raises some interesting points about the potential for emergence of replicable RNAs from standard RNA transcripts. We speculate that the path from a standard cellular RNA to an RNA replicable via an RNAP could be short indeed if the RNA used a form of genetic amplification known as rolling hairpin replication (RHR). In the simplest form of RHR, a nucleic acid template with terminal hairpins is extended at its 3′ end by a processive polymerase with strand displacement activity (Tattersall and Ward 1976). Once the polymerase has reached the end of the template, conformational isomerization of the terminal hairpin frees a new 3′ end for another extension reaction, enabling the next round of genome amplification (Tattersall and Ward 1976). Parvoviruses, a family of ssDNA viruses that can cause disease in humans, use RHR to solve the end replication problem (Tattersall and Ward 1976; Young and Brown 2004), and RHR has been reconstituted in vitro with synthetic DNA templates and recombinant polymerases (Kato et al. 2012; Jung and Ellington 2016). An advantage of RHR is that no new nucleic acid chains must be initiated. To our knowledge, RHR of RNA templates in vivo or in vitro has not yet been described, although there do not appear to be obvious reasons why such a process should not occur. It is also possible that RHR could serve as a precursor to rolling circle replication (HDV and viroids are thought to be replicated via rolling circle replication [Glenn et al. 1990; Diener 1995]). We note that general RNA-templated RNA extension by E. coli RNAP suggests that it—and possibly other cellular RNAPs—could conceivably perform RHR. RNA-templated transcription on 6S RNA mutants is consistent with E. coli RNAP having strand displacement activity on RNA templates (Panchapakesan and Unrau 2012; Oviedo Ovando et al. 2014). In addition, given that RNA-templated RNA extension requires an intramolecular duplex of just 1–2 nt at the 3′ end of the template, the extension of almost any RNA with a 5′ terminal hairpin could result in a substrate for RHR.
MATERIALS AND METHODS
General
Synthetic oligonucleotides were synthesized by Integrated DNA Technologies (IDT) or GenScript (Supplemental Table S1). For RNA oligonucleotides, secondary structures were predicted with RNAfold (Lorenz et al. 2011) using the default parameters (Supplemental Table S1). Oligonucleotide R-12 was a gift of I.N. Zheludev. All commercial kits were used as directed unless otherwise specified. For all Zymo Research nucleic acid purification kits, the spin column was spun empty prior to elution to reduce carryover of the wash buffer. For the Oligo Clean & Concentrator-5 kit (Zymo Research), two wash steps, instead of one, were performed. pH values are given for room temperature solutions unless otherwise specified.
E. coli RNAP
Our primary preparations of E. coli RNAP core enzyme and σ70 holoenzyme were purchased from NEB as custom high-concentration orders of commercially available enzymes (catalog numbers M0550 and M0551, respectively). We also prepared an additional aliquot of E. coli RNAP core enzyme as previously described (Chen et al. 2019), except for removal of the C-terminal His10-tag from the β′ subunit. Specifically, His-tagged 3C protease was added at a 1:40 molar ratio to the sample pool eluted from the IMAC column. After dialysis against 20 mM Tris-HCl (pH 8.0 at 4°C), 1 M NaCl, 5% glycerol, 0.1 mM EDTA, 1 mM β-mercaptoethanol, and 0.5 mM DTT overnight at 4°C in a 12–14 kDa membrane (SpectraPor), the sample was passed over the IMAC column to remove the protease and remaining tagged protein, followed by dialysis of the flowthrough overnight at 4°C in a 12–14 kDa membrane against 10 mM Tris-HCl (pH 7.8), 0.1 M NaCl, 0.1 mM EDTA, 5% glycerol, and 5 mM DTT. The dialyzed sample was further purified by chromatography over Biorex resin and gel filtration as described previously (Chen et al. 2019).
RNAP reactions
RNAP reactions contained 40 mM Tris-HCl (pH 8.0), 80 mg/ml PEG-8000, 20 mM MgCl2, 5 mM DTT, 1 mM spermidine, 0.01% Triton X-100, 75 μg/mL kanamycin, 4 mM NTPs (“KS buffer”), rifampicin if necessary, 0.85 μM RNAP, and 1 μM RNA oligonucleotide template unless otherwise specified. In general, for RNAP reactions, 4 μL buffer (containing everything except enzyme and oligonucleotide template) was slowly added to 0.5 μL enzyme (σ70 holoenzyme from NEB unless otherwise specified). For mock (no enzyme) reactions, buffer was added to 0.5 μL RNAP storage buffer (20 mM Tris-HCl, 0.1 M NaCl, 0.1 mM EDTA, 1 mM DTT, and 50% glycerol; final pH 7.5) (the reactions were subsequently processed identically). RNA oligonucleotide was then added to the RNAP:buffer mixture (5 pmol of oligonucleotide usually in 0.5 μL). For experiments with multiple templates (e.g., the mixing experiment with DG-253 and DG-256), the total amount of oligonucleotide in the reaction was 5 pmol unless otherwise specified. Reactions were incubated at 37°C overnight (∼12 h) unless otherwise specified. Reactions were quenched with an equal volume of 0.1 M EDTA. Unless otherwise specified, reactions were purified via an Oligo Clean & Concentrator-5 kit and eluted in 10 μL (usually IDTE [pH 7.5; IDT]). Depending on the sample, 5%–40% of the eluted reaction was analyzed via gel electrophoresis. Although we used KS buffer for most of our experiments, RNA extension also occurred in “MD buffer” (see DNA-templated transcription; Supplemental Fig. S2C).
Phenol-chloroform chloroform extraction
For experiments in which protein was to be rigorously removed from reactions, unquenched reactions were brought to ∼1% SDS and ∼2 mg/mL Proteinase K in 10 μL total volume (1 μL of 10% SDS and 1 μL of 20 mg/mL Proteinase K), incubated at 65°C for 30 min, and brought to 50 μL total volume with water. Samples were then extracted with one volume of phenol:chloroform (1:1 mixture of chloroform and phenol equilibrated with 10 mM Tris-HCl [pH 8.0] and 1 mM EDTA) and reextracted with one volume of chloroform. The aqueous phase was purified via an Oligo Clean & Concentrator-5 kit and eluted in 10 μL IDTE (pH 7.5).
Analysis of RNA by gel electrophoresis
Samples (including the RNA ladder, 0.25 μL Low Range ssRNA Ladder [NEB] optionally mixed with 1 μL ZR small-RNA Ladder [Zymo Research]) were diluted to 5 μL with water and combined with an equal volume of Gel Loading Buffer II (GLBII; 95% formamide, 18 mM EDTA, 0.025% SDS, trace bromophenol blue [some samples], and trace xylene cyanol [some samples]). We used two protocols for electrophoresis. In the first (“protocol 1”; our default protocol unless otherwise specified), samples were heated to 95°C for 5 min, snap-cooled on ice, loaded on a Tris-borate-EDTA- (TBE)-urea polyacrylamide gel (Bio-Rad Laboratories), pre-run in TBE running buffer (0.1 M Tris, 0.1 M boric acid, and 2 mM EDTA), and run at 200 V for ∼1 h. In the second (“protocol 2”; specified in the figure legend), TBE running buffer was heated to ∼70°C and added to the gel tank. Samples were heated to 95°C for 2–5 min and loaded directly on a TBE-urea polyacrylamide gel without cooling; the gel was run at 200 V for ∼20 min. Gels were 10% unless otherwise specified. Gels were stained with SybrGold/Helixyte Gold (AAT Bioquest; Helixyte Gold is chemically equivalent to SybrGold) diluted ∼1:10,000 from the provided “10,000× stock” in TBE for ∼10–30 min at room temperature with gentle agitation. Gels were imaged with a Typhoon Phosphorimager with the default EtBr settings. For the experiments with 5′ endlabeled oligonucleotide templates, the gel was imaged with the Phosphorimager (the default FAM, Cy5, or Cy3 settings) to visualize the endlabel prior to SybrGold staining. The gel was then stained with SybrGold and imaged with the usual protocol (to visualize the ladder).
Transcription from a DNA template
To create a DNA template for transcription, we prepared PCR reactions containing 1 pM pAR1707 (Levin et al. 1987), 0.5 μM each primer (DG-234 and DG-235), and 1× Taq 2× Master Mix (NEB) in a total volume of 200 μL. Thirty cycles of PCR were performed (95°C for 30 sec, 48°C for 30 sec, and 72°C for 1 min; initial denaturation at 95°C for 30 sec; final extension at 72°C for 10 min). The PCR product was purified with a DNA Clean & Concentrator-5 kit (Zymo Research) and eluted/diluted so that the final concentration was ∼0.5 μM. Transcriptions were performed as previously described (Saecker et al. 2024). Specifically, reactions contained 40 mM Tris-HCl (pH 8.0), 0.12 M KCl, 10 mM MgCl2, 1 mM DTT, 20 μg/mL BSA, optional 0.01% methanol (as a control for experiments with rifampicin, which was stored in methanol), 1 mM NTPs, 10 nM DNA template, and 85 nM E. coli RNAP. Enzyme and template were added to 1× buffer in a total volume of 8 μL, and the reactions were incubated at 37°C for 10 min. NTPs in 1× buffer equilibrated to 37°C were added to bring the total volume to 10 μL, and the reactions were incubated for 15 min at 37°C. 1/10 volume Turbo DNase (Thermo Fisher Scientific) was added, and the reactions were incubated for 10 min at 37°C. Reactions were quenched with an equal volume of GLBII and analyzed via electrophoresis as above without further purification.
We considered whether the observed RNA-templated RNA extension of this transcript (Fig. 5) could occur during transcription, DNase treatment, or both. Electrophoresis of a non-DNase treated transcript revealed a minor band migrating slightly faster than the 150 nt marker, consistent with a nonextended transcript (in addition to the primary band at ∼150 nt) (Supplemental Fig. S1E). This data supports a model in which RNA-templated RNA extension can occur during transcription in the presence of a DNA template.
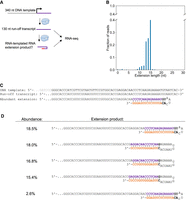
RNA-templated RNA extension during DNA-templated transcription. (A) Schematic of experiment assessing RNA-templated RNA extension during DNA-templated transcription. The nontranscribed portion of the DNA template is in gray, the transcribed portion of the DNA template and the RNA transcript are in purple, and the RNA extension is in red. (B) RNA-seq analysis of the length distribution of RNA extensions during DNA-templated transcription. (C) An example extension product of the run-off transcript and a model for its formation. (D) Table of the five most abundant extension products. In C and D, colors for the extension sequences are as in Figure 3B. Reaction conditions were as described in Materials and Methods.
Analysis of protein via gel electrophoresis
Ten picomoles of RNAP was diluted in water to a final volume of 10 μL and combined with 2 μL 6× Laemmli Sample Buffer (Bio-Rad Laboratories). Samples were run on a 4%–15% Tris-glycine gel (Bio-Rad Laboratories) at 150 V for ∼45 min. Gels were stained with SYPRO Ruby (Thermo Fisher Scientific) as recommended by the manufacturer and imaged with a Typhoon Phosphorimager with the default SYPRO Ruby settings.
RNA-seq
This customized RNA-seq protocol was derived from the TruSeq Small RNA Library Preparation Kit (Illumina) and AQ-seq (Kim et al. 2019). Key changes from the commercial TruSeq kit include (i) buffer composition of the ligation reaction; (ii) randomization of an adaptor sequence to add unique molecular identifiers (UMIs); and (iii) reverse transcription (RT) and PCR enzymes. These changes reduce per-reaction cost by ∼10-fold and ligation bias (data not shown) without substantially impacting hands-on time.
The 3′ adaptor (DG-35) was enzymatically adenylated with the 5′ DNA adenylation kit (NEB). The adenylation reaction was purified with the Oligo Clean & Concentrator-5 kit, eluted in 15 μL IDTE (pH 8.0; IDT), and diluted to 6 μM.
RNAP reactions for sequencing were assembled as above except with ∼2 μM (instead of 1 μM) 5′ phosphorylated templates (instead of 5′ hydroxyl; the 5′ phosphate allows for direct adaptor ligation). For RNA-seq, we exclusively used chemically synthesized oligonucleotides, given the known 3′ end heterogeneity of RNAs synthesized with T7 RNAP (Gholamalipour et al. 2018). Two hundred picomoles of oligonucleotide was phosphorylated with T4 polynucleotide kinase (NEB; as directed except without heat inactivation). The reaction was purified with the Oligo Clean & Concentrator-5 kit and eluted in 10 μL IDTE (pH 7.5). In the RNAP reaction, the template was ∼2 μM (1/20 of the eluate; ∼10 pmol) instead of 1 μM. For the mixing experiments with LR-3 and LR-3′, the total amount of oligonucleotide in the reaction was ∼10 pmol (∼5 pmol each). For RNA-seq of the DNA-templated transcription reaction, a transcription reaction was purified via the Oligo Clean & Concentrator-5 kit and eluted in 10 μL IDTE (pH 7.5). 5′ triphosphate groups were removed with RppH (NEB) as directed by the manufacturer except enzyme concentration was reduced twofold; the reaction was purified with a second Oligo Clean & Concentrator-5 kit and eluted in 6 μL IDTE (pH 7.5).
The 3′ adaptor ligation reaction contained 0.6 μM adenylated adaptor (3 pmol), 20,000 U/mL T4 RNA Ligase 2, truncated KQ (NEB; 100 U), 20% PEG-8000, and 1× T4 RNA Ligase Buffer (NEB) in a total volume of 5 μL. A total of 1.5 μL RNA (eluate from the purification of RNAP extension reactions [∼1 μM RNA oligonucleotide template] or from the DNA-templated transcription reaction) was combined with the adaptor in a total volume of 2 μL. The RNA:adaptor mix was heated at 72°C for 2 min and cooled on ice. Then, 3 μL of solution containing enzyme, PEG-8000, and buffer was added. The reaction was incubated at 25°C for 1 h. Then, STP oligonucleotide (DG-37) was added to a final concentration of 1.8 μM in a volume of 0.5 μL (10 pmol), and the reaction was incubated for a further 15 min at 25°C. The 5′ adaptor ligation reaction contained 1.4 μM 5′ adaptor (DG-48; 10 pmol), 714 U/mL T4 RNA ligase 1 (NEB; 5 U), 1 mM ATP, 16.3% PEG-8000, and 1× T4 RNA Ligase Buffer (NEB) in a total volume of 7 μL. The 5′ adaptor was heated at 72°C for 2 min and cooled on ice. Then, 1.5 μL of solution containing adaptor, enzyme, ATP, PEG-8000, and buffer was added to the 5.5 μL 3′ ligation reaction:STP oligonucleotide mixture; the resulting 5′ ligation mixture was incubated for 1 h at 25°C.
The RT reaction contained 14,814 U/mL Protoscript II (NEB; 200 U), 460 μM dNTPs, 7.41 mM DTT, 0.74× Protoscript II buffer, and the entire volume of the ligation reaction in a total volume of 13.5 μL. To the 7 μL of the unpurified ligation reaction, RT primer (DG-3) was added to 0.5 μM in a volume of 1 μL (4 pmol). The RNA:primer mixture was heated at 72°C for 2 min and cooled on ice. Then, 5.5 μL of solution containing enzyme, dNTPs, DTT, and buffer was added. The reaction was incubated at 42°C for 1 h.
The PCR reaction contained 0.24 μM universal primer (DG-4; 12 pmol), 0.8 μM indexing primer (varies; 40 pmol), 1× NEBNext High-Fidelity 2× PCR Master Mix (NEB), and the entire volume of the RT reaction in a total volume of 50 μL. For some experiments, we used indexing primers from the TruSeq Small RNA Library Preparation Kit (2 μL of primer). The source of the indexing primer did not substantially impact sequencing results (data not shown). Twenty-five microliters of PCR reaction was amplified with 12 cycles of PCR (98°C for 10 sec, 60°C for 30 sec, and 72°C for 30 sec; initial denaturation at 98°C for 30 sec; final extension at 72°C for 15 min). The libraries were loaded on a 2% agarose gel and run for ∼2 h at 5 V/cm. DNA in the range ∼125–300 nt (corresponding to RNA fragments ∼0–175 nt) was excised with a razor blade. For the library prepared from the DNA-templated transcription reaction, DNA in the range ∼400–500 nt was excised. Libraries were purified with the Zymoclean Gel DNA Recovery Kit (Zymo Research); elution was in 6 μL IDTE (pH 8.0). Libraries were quantified via the Qubit dsDNA High Sensitivity Assay (Thermo Fisher Scientific). Libraries were sequenced on a MiSeq (Illumina) in various configurations.
In general, we note that RNA-seq has substantial biases depending on RNA sequence, structure, and length. In our libraries, sequences of extensions are likely to substantially alter the structure of the RNA, which along with the increased length of extended molecules is likely to impact the efficiency of capture for RNA-seq. In addition, we note that possible nuclease activity in our enzyme preparation (see above) and possible other noncanonical enzyme activities likely substantially increased the complexity of our RNA-seq data. We emphasize the qualitative, rather than quantitative, nature of RNA-seq, and we focus on comparisons between RNA-seq libraries. Additionally, regarding the mixing experiment with LR-3 and LR-3′, we suspect that the ∼5%–10% of LR-3 reads with LR-3′ extensions (and vice versa) were primarily due to artifacts during library preparation (e.g., template switching during PCR) and not bona fide transRNA-templated RNA extension. When we reacted LR-3 and LR-3′ individually with E. coli RNAP, purified the reactions, and mixed the samples prior to library preparation, we observed LR-3 reads with LR-3′ extensions (and vice versa), even though we know when sequenced individually LR-3 had only LR-3 extensions (Supplemental Fig. S6D).
Bioinformatic analysis of RNA extension
All code necessary to create the figures has been deposited to the Stanford Digital Repository (see Data Deposition). RNA-seq reads were trimmed and, if necessary for paired-end libraries, merged with the scripts TrimSE.py and MergePE_v2.py. We note that we did not use the 5′ UMIs associated with the randomized 5′ adaptor because our RNA-seq libraries were nondiverse (e.g., template oligonucleotides), and collapsing reads based on UMIs would overrepresent the abundance of rare species. Using functions described in the script ExtensionPaperFunctions241010.py (and scripts for individual figures), we selected reads beginning with exact matches to the template (“template-containing reads”) and analyzed the sequences (if any) 3′ of the match (i.e., extension sequences). This approach focuses our analysis on reads for which we can offer a strong interpretation. We note that this approach ignores reads with oligonucleotide synthesis or sequencing errors and may not capture certain complexities of the RNA-seq library. We generated Supplemental Table S2 with the script ParseRabbit_ag00_110524_DGedits.py. For Figures 3 and 5, Supplemental Figures S3B, S4A, S6C, and Supplemental Table S2, we counted the abundance of each extension. For Supplemental Figure S3A and S3C, we subset for reads extended at least 5 nt and counted the abundance of 5 nt initial extension sequences (e.g., the extensions GUAUCU and GUAUCUC would both have the same 5 nt initial extension sequence GUAUC). In Supplemental Figure S3A, for each replicate, we divided the abundance of each 5 nt initial extension sequence by the total number of reads extended at least 5 nt to obtain the relative abundance of each 5 nt initial extension sequence (this normalizes across RNA-seq libraries with nonequal read counts). For Figure 4B and Supplemental Figure S6D, we counted the number of extensions complementary to LR-3 only and LR-3′ only; extensions complementary to both templates (i.e., of ambiguous origin) were not counted. We used ChatGPT (OpenAI) and Copilot (GitHub) to assist during routine coding tasks (e.g., plotting).
Preliminary analysis of RNA-templated RNA extension in vivo
We downloaded total and 3′ end RNA-seq data from the Sequence Read Archive (SRA) (PRJNA640168). Read 2s (R2s) were reverse complemented with seqkit (command seq -r -p). R2s were filtered for quality, and adaptors were trimmed with fastp (options –qualified_quality_phred 25 –unqualified_percent_limit 10 –length_required 20). A Bowtie2 index was built from the NCBI MG1655 reference (NC_000913.3). Reads were mapped with Bowtie2 against the MG1655 index (option –very-sensitive-local). A python script was used to parse the output SAM file to count reads that had at least 6 nt of 3′ soft-clipping but otherwise alignment to the genome and in which the reverse complement of the last two aligned bases and the first six soft-clipped bases was found in the aligned portion of the read (“cis extended reads”). Cis extended reads were rare (∼1 in 105) in both total RNA-seq and 3′ end RNA-seq data (see Discussion).
DATA DEPOSITION
All RNA-seq data have been deposited to the SRA (PRJNA1143695). All code has been deposited to the Stanford Digital Repository (https://purl.stanford.edu/vz333ss5687). We are happy to provide additional information about any data, code, or methods upon request.
SUPPLEMENTAL MATERIAL
Supplemental material is available for this article.
ACKNOWLEDGMENTS
We thank I.N. Zheludev for oligonucleotide R-12, D. Yue for custom aliquots of E. coli RNAP, A.N. Pogson and A. Brunet for SYPRO Ruby, and R. Grosely for advice on polyacrylamide gel electrophoresis. We thank C.R. Altmann, S.A. Darst, D. Herschlag, W.J. Greenleaf, K. Kirkegaard, R.E. Kingston, J.B. Li, J.D. Puglisi, J.H. Shin, D.E. Solow-Cordero, and current and former members of the Fire Laboratory, especially K.L. Artiles, N. Jain, and I.N. Zheludev, for helpful comments and feedback. Some graphics were made with BioRender. D.G. was supported in part by the National Institutes of Health- (NIH-) T32HG000044-26. A.U.M. is an Agouron Institute Awardee of the Life Sciences Research Foundation. E.G. was supported by the Division of Graduate Education-1656518, NIH-R01MH125244, and NIH-R01AG066490. This work was funded by NIH-R35GM130366 and NIH-R35GM118130.
Footnotes
-
Article is online at http://www.rnajournal.org/cgi/doi/10.1261/rna.080238.124.
- Received August 24, 2024.
- Accepted January 13, 2025.
This article is distributed exclusively by the RNA Society for the first 12 months after the full-issue publication date (see http://rnajournal.cshlp.org/site/misc/terms.xhtml). After 12 months, it is available under a Creative Commons License (Attribution-NonCommercial 4.0 International), as described at http://creativecommons.org/licenses/by-nc/4.0/.








