
N6-methyladenosine reader YTHDF2 in cell state transition and antitumor immunity
- 1The Laboratory of Microbiome and Microecological Technology, Institute of Microbiology, Chinese Academy of Sciences, Beijing 100101, China
- 2Department of Radiation and Cellular Oncology, University of Chicago, Chicago, Illinois 60637, USA
- 3Ludwig Center for Metastasis Research, University of Chicago, Chicago, Illinois 60637, USA
- 4Department of Chemistry, The University of Chicago, Chicago, Illinois 60637, USA
- 5Howard Hughes Medical Institute, The University of Chicago, Chicago, Illinois 60637, USA
- 6Department of Biochemistry and Molecular Biology, Institute for Biophysical Dynamics, The University of Chicago, Chicago, Illinois 60637, USA
- Corresponding authors: wangll{at}im.ac.cn, chuanhe{at}uchicago.edu
Abstract
Recent studies have revealed that the YTHDF family proteins bind preferentially to the N6-methyladenosine (m6A)-modified mRNA and regulate the functions of these RNAs in different cell types. YTHDF2, the first identified m6A reader in mammals, has garnered significant attention because of its profound effect to regulate the m6A epitranscriptome in multiple biological processes. Here, we review current knowledge on the mechanisms by which YTHDF2 exerts its functions and discuss recent advances that underscore the multifaceted role of YTHDF2 in development, stem cell expansion, and immune evasion. We also highlight potential therapeutic interventions targeting the m6A/YTHDF2 axis to improve the response to current antitumor therapies.
INTRODUCTION
Among more than 170 different types of RNA modifications, N6-methyladenosine (m6A) is the most abundant and well-studied internal (non-cap) modification in eukaryotic mRNAs, present at a frequency of ∼0.15%–0.6% of all adenosines (He and He 2021). m6A is also present in ribosome RNA (rRNA), transfer RNA (tRNA), long noncoding RNA (lncRNA), small nuclear RNA (snRNA), and microRNA (miRNA). m6A methylation can be reversible with its deposition orchestrated by m6A-methyltransferases (writers) and m6A-demethylases (erasers) and recognized by m6A-binding proteins (readers). m6A is added by a complex consisting of METTL3 and many other accessory proteins, such as METTL14, METTL16, WTAP, VIRMA, RBM15/15B, ZC3H13, and HAKAI, forming a large methyltransferase holocomplex with a predicted size of 1000 kDa (Huang et al. 2021; Su et al. 2022; Han et al. 2023). Conversely, two demethylases, FTO and ALKBH5, can remove m6A marks from methylated RNA molecules (Jia et al. 2011; Zheng et al. 2013; Wei et al. 2018). Meanwhile, the readers preferentially recognize m6A-modified RNA to degrade or stabilize mRNA transcripts or impact translation (Roundtree et al. 2017a). They work jointly in a dynamic m6A modification process that affects RNA fate, including its stability, transport, processing, and translation (Frye et al. 2018; Shi et al. 2019; He and He 2021).
It has been reported that m6A is enriched within the METTL3/14 methyltransferase RRACH (where R = G/A and H = A/C/U) consensus in 3′ UTRs near stop codons (Liu et al. 2014). The outcome of RNA methylation is instructed, partially at least, by directly increasing the affinity of the binding site for YT521-B homology (YTH) domain-containing proteins, the so-called primary m6A readers. These proteins, including YTHDF1, YTHDF2, YTHDF3, YTHDC1, and YTHDC2, all contain an m6A-binding pocket in the YTH domain (Xu et al. 2015). YTHDF2 is the first identified m6A reader and promotes mRNA degradation by recognizing m6A and recruiting the mRNA decay machinery (Wang et al. 2014). YTHDF1 or YTHDF3 can facilitate the translation of its target methylated mRNAs (Wang et al. 2015; Shi et al. 2017). YTHDC1 affects RNA splicing and RNA degradation (Xiao et al. 2016; Roundtree et al. 2017b). YTHDC2 affects both mRNA decay and translation (Hsu et al. 2017). Recent studies also reveal the critical roles of YTHDC1 and YTHDC2 in chromatin regulation through m6A methylation of chromatin-associated RNA (Liu et al. 2020a, 2021; Chelmicki et al. 2021; Xu et al. 2021; Sun et al. 2023). Besides the YTH domain-containing readers, other readers, including IGF2BP, FMR1, and HNRNPA2B1, specifically recognize m6A-modified RNA to degrade or stabilize mRNA transcripts or impact translation. How these other proteins selectively bind m6A, however, has yet to be clearly elucidated (Han and Xu 2023).
In this review, we focus on the YTHDF2 protein. We summarize recent studies that elucidate the roles of YTHDF2 in modulating gene expression throughout development, stem cell differentiation, and immune effects. We also discuss known mechanisms regulating the expression of YTHDF2 in different cell types. Finally, we describe efforts to develop therapeutic strategies targeting YTHDF2 and the preclinical results reported with such agents to date.
m6A/YTHDF2 COMPLEXES REGULATE GENE EXPRESSION
YTHDF2 was initially found in pull-down experiments using m6A-containing RNA probes (Dominissini et al. 2012), and subsequently established as an m6A reader (Wang et al. 2014). YTHDF2 is composed of a proline/glutamine/asparagine (P/Q/N)-rich N terminus containing a tryptophan-xx-isoleucine (WxxI) motif interacting with ribosomal proteins and several low complexity regions enabling phase separation and partitioning into ribonucleoprotein (RNP) granules, and a C terminus containing a conserved YTH domain specifically binding to m6A-containing RNA (Stoilov et al. 2002; Zhang et al. 2010; Zou et al. 2023). In the N terminus, amino acids 1–100 have been reported to interact with HRSP12, a component of the RNase P/MRP complex involved in endoribonucleolytic RNA cleavage (Du et al. 2015; Zou and He 2024). In the C terminus, a lysine (K571), where an arginine is present in YTHDF1 and YTHDF3, creates a specific small ubiquitin-related modifier modification (SUMOylation) site (Hou et al. 2021). The unique sequence of YTHDF2, in particular in the low complexity regions, differentiates it from YTHDF1 and YTHDF3 and may explain its distinct functions.
Functionally, YTHDF2 preferentially binds m6A-methylated RNA probes over unmethylated ones assessed by gel-shift assays. It predominantly targets m6A sites around the stop codon and 3′ untranslated region (3′ UTR) as well as the coding region (CDS), and subsequently recruits the CCR4-NOT deadenylase complex to promote deadenylation (Wang et al. 2014) as well as the RNase P/MRP complex to promote endoribonucleolytic cleavage, both contributing to degrade m6A-marked transcripts (Du et al. 2016; Park et al. 2019). The YTHDF2-mediated mRNA decay occurs in the cytoplasm. YTHDF2 can bind to thousands of methylated mRNAs, including transcripts of many transcription factors, and shares common targets with YTHDF1 and YTHDF3 (Wang and He 2014; Shi et al. 2017). However, YTHDF2 cellular localization can change in response to stress. For instance, under heat shock, YTHDF2 enters the cell nucleus and competes with nuclear m6A eraser FTO to preserve 5′-UTR methylation of stress-induced transcripts thus promoting cap-independent translation initiation (Zhou et al. 2015; Zou and He 2024).
THE ROLE OF YTHDF2 IN DEVELOPMENT AND STEM CELL DIFFERENTIATION
m6A affects the translation and stability of the modified transcripts, thus providing a mechanism to coordinate the regulation of groups of transcripts during diverse eukaryotic biological processes (Frye et al. 2018). A wealth of recent studies highlighted a role of YTHDF2 in the regulation of transcriptome switching during development and stem cell differentiation.
An early clue that YTHDF2 is essential for normal development was the observation that the loss of Ythdf2 results in the failure to regulate transcription of a set of genes involved in oocyte maturation, enriched in the YTHDF2-binding consensus and modified with m6A (Ivanova et al. 2017). Besides the requirement of YTHDF2 for early zygotic development in mice, YTHDF2-mediated RNA decay also regulates various stages of zebrafish embryonic development. For example, YTHDF2 promotes the clearance of the maternal transcripts during maternal to zygotic transition (MZT), delaying MZT initiation and thus impeding zygotic genome activation (Zhao et al. 2017). In addition, Zhang et al. (2017) reported that YTHDF2 regulates Notch1a and Rhoca, which are required for endothelial cell fate in zebrafish and thereby blocks the endothelial-to-hematopoietic transition.
Similarly, YTHDF2 has been proved to be required for neural development. Loss of Ythdf2 in mice led to compromised neural development with impaired neural stem/progenitor cell self-renewal and spatiotemporal generation of neurons. Further studies revealed that Ythdf2 deletion resulted in increased transcription of Flrt2 and Ptprd, negative regulators of JAK-STAT cascade, which contributes to neuroprotection and neurite outgrowth (Li et al. 2018a). This phenomenon is aligned with the observation that genetic knockout of either Mettl3 or Mettl14 is developmentally lethal in mice, with embryos failing to differentiate (Batista et al. 2014; Geula et al. 2015). Mechanistically, YTHDF2 is overexpressed and destabilizes a group of m6A-modified mRNAs associated with neurodevelopment, thereby attenuating neural differentiation (Heck et al. 2020). Additionally, depletion of RNA m6A methylation in embryonic mouse brain prolongs the cell cycle of radial glia cells and extends cortical neurogenesis (Yoon et al. 2017), observations thought to be related to the YTHDF2-dependent transcriptome turnover during development. Altogether, these observations revealed that Ythdf2 knockout exhibits developmental defects in both mice and zebrafish because of the altered transcript turnover of its target genes, and this paradigm might be used to explain the effects of YTHDF2 on the differentiation of other cell types.
Much work has focused on investigating how the “m6A/YTHDF2” axis governs stem cell differentiation or expansion (Fig. 1). Linheng Li's group first reported that Ythdf2 knockout greatly increases the numbers of functional hematopoietic stem cells (HSCs) both in vitro and in vivo through stabilizing many key m6A-marked mRNAs, such as HOXB4 (Amsellem et al. 2003), RUNX1, and FOSB (Ebina and Rossi 2015), required for HSC self-renewal (Li et al. 2018b). Such a scenario is consistent with the observation that loss of Ythdf2 in adult HSCs of mouse bone marrow significantly increases the proportion and absolute number of long-term HSC, short-term HSC, and multipotent progenitors (Wang et al. 2018), highlighting the importance of YTHDF2 in HSC homeostasis and regeneration.
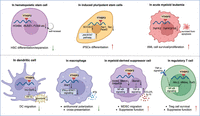
The role of YTHDF2 in HSCs, iPSCs, AML cells, and different immune cells. (Top) The m6A/YTHDF2 influences the differentiation or proliferation of HSCs, iPSCs, and AML cells. (Bottom) YTHDF2 affects DC migration and the cross presentation of antitumoral macrophages and maintains the suppressive function of MDSCs and Tregs. In general, inhibiting YTHDF2 triggers the antitumor immunity. Figure created with BioRender.
Later, researchers also found that YTHDF2 plays an essential role in induced pluripotent stem cells (iPSCs). For instance, YTHDF2 reprogrammed somatic cells into iPSCs by clearing somatic mRNAs, especially Yap1, Taz, and Tead2, through the m6A-dependent deadenylation pathway. YTHDF3 is also involved in this reprogramming process (Liu et al. 2020b). YTHDF2, orchestrated with YTHDF1, is involved in METTL3-m6A-mediated maintenance of the pluripotent state in porcine iPSCs by initiating the YTHDF1-mediated JAK2 translation and the YTHDF2-dependent SOCS3 (a negative regulator of JAK2-STAT3) mRNA decay, thereby provoking the activation of the STAT3/KLF4/SOX2 signal axis (Wu et al. 2019). The m6A- and YTHDF2-dependent transcriptome switch are critical for cell state transition.
In leukemia stem cell development and acute myeloid leukemia (AML) initiation, which is an aggressive clonal disorder of HSCs, loss of YTHDF2 impairs AML cell survival and engraftment, promotes stem or primitive progenitor cell expansion, and enhances the myeloid differentiation potentials (Wang et al. 2018; Paris et al. 2019). Mechanistic analysis revealed that YTHDF2 promotes the degradation of TNFR2 mRNA in an m6A-dependent manner, resulting in reduced sensitivity of AML cells to TNF stimulation. Consistently, TNF receptor superfamily member 1b (TNFRSF1b) has been shown to be another key YTHDF2 target mRNA in AML cells harboring the t(8;21) translocation and can be degraded by YTHDF2, thus protecting AML cells from TNFRSF1b-induced apoptosis (Chen et al. 2021). The notion that YTHDF2 is highly expressed in AML patients and associated with a higher risk of relapse makes YTHDF2 a potential therapeutic target for leukemia or other hematologic diseases.
THE SUPPRESSIVE EFFECTS OF YTHDF2 ON ANTITUMOR IMMUNITY
Given its important roles in the regulation of gene expression in mammals, it is unsurprising that YTHDF2 is closely associated with many cancers or cancer treatments (Chen et al. 2023a,b; Deng et al. 2023). For example, high levels of YTHDF2 expression clinically correlate with a poor prognosis in glioma patients, and YTHDF2 promotes glioblastoma cell proliferation, invasion, and tumorigenesis in preclinical models (Fang et al. 2021); YTHDF2 induces the epithelial-to-mesenchymal transition (EMT) by degrading transcripts encoding proteins in the MAPK/ERK pathway in MYC-driven triple-negative breast cancer (TNBC) cells (Einstein et al. 2021), or by attenuating YAP signaling in pancreatic cancer cells or non-small cell lung cancer cells (Chen et al. 2017; Jin et al. 2020). The role of YTHDF2 in different cancer types and its underlying target mRNAs in corresponding tumor cells have been well reviewed in 2023 (Deng et al. 2023), covering glioma (UBXN1, LXRA, HIVEP), lung cancer (AXIN1, DAPK2, BTG1, TSUC7), liver cancer (SLC7A11, IL11, SERPINE2), gynecological cancer (BMF, FENDRR), prostate cancer (LHPP, NKX3-1), colorectal cancer (GSK2B), and melanoma (PER1, TP53). Herein we summarize the recent discoveries focusing on the contribution of YTHDF2 in immune cells (Fig. 1).
A collection of results points to a critical role of YTHDF2 in reprogramming the tumor microenvironment. In DCs, silencing of Ythdf2 with siRNA up-regulated lnc-Dpf3 (a direct target of YTHDF2) accelerates C-C motif chemokine receptor (CCR7)-mediated DC migration (Liu et al. 2019). YTHDF2 also targets Stat1 mRNA to decrease interferon-γ-STAT1 signaling, thereby suppressing macrophage polarization and antigen cross-presentation capacity (Ma et al. 2023). In lymphocytes, YTHDF2 promotes B-cell development and interleukin-7-induced pro-B-cell proliferation (Zheng et al. 2020). While in regulatory T cells (Tregs), YTHDF2 degrades negative regulators of TNF signaling, leading to Treg activation and suppression of antitumor immunity (Zhang et al. 2023). Emerging evidence also suggests critical roles of YTHDF2 in natural killer cells (NKs) (Ma et al. 2021).
Here, we wish to emphasize the recent results regarding the role of YTHDF2 in myeloid-derived suppressor cells (MDSCs), which impair anti-cancer therapies. In two independent clinical trials of metastatic solid cancer patients receiving radiotherapy and immunotherapy, the investigators noted that the expression of YTHDF2 was markedly induced posttreatment in MDSCs but not in other tumor-infiltrating myeloid cell populations, representing a unique role of YTHDF2 in MDSCs and a promising target to alleviate extrinsic resistance to radio-immunotherapy (Wang et al. 2023a). The strong association between YTHDF2 and poor patients survival also occurred in murine cancer models, evidenced by the observations that LyzCre+;Ythdf2fl/fl (Ythdf2 deficiency in myeloid cells) conditional knockout mice exhibited delayed primary tumor growth in the context of radiation or anti-PD-L1 treatment. Interestingly, these knockout mice also displayed obvious reduction in radiation-induced lung metastasis size, suggesting that YTHDF2 suppression might not only enhance the local anti-tumor effects but also enhance the control of distant metastasis or metastatic disease.
Detailed mechanistic investigations showed that YTHDF2 promotes the migration and suppressive function of MDSCs in two independent ways: (1) YTHDF2 promotes m6A-modified RNA degradation of genes that encode negative regulators (Adrb2, Metrnl, or Smpdl3b) of NF-κB signaling to activate NF-κB signaling (Wang et al. 2023a); (2) YTHDF2 directly binds and degrades BAMBI mRNA, a TGF-β pseudoreceptor, in an m6A-dependent manner to enhance TGF-β signaling activation (Wang et al. 2023c). These two YTHDF2-regulated signaling pathways amplify the expression of multiple cytokines in MDSCs, which promote MDCS infiltration/differentiation and inhibit T-cell function, inducing tumor growth in multiple murine cancer models. Interestingly, Ythdf2 knockout accelerated MDSC differentiation toward M1-like macrophages, inhibited MDSC differentiation toward neutrophiles, and thereby reversed the radiation-mediated suppressive tumor immune microenvironment. Considering the fundamental roles of both NF-κB signaling and TGF-β signaling in influencing immune response, we believe that YTHDF2 may be important in immune-related diseases, including not only cancers but also inflammation and autoimmune diseases, and it may be a feasible target for developing effective therapeutic strategies against these diseases.
We are only now beginning to understand the YTHDF2-mediated molecular pathways that regulate MDSC differentiation, trafficking, and suppressive function in the context of radiotherapy (Wang et al. 2023b, 2024). How other signaling pathways are regulated by YTHDF2 in response to other antitumor therapies remains unknown. Based on results obtained so far of YTHDF2 in both tumor cells and immune cells, we can conclude that YTHDF2 acts as a key suppressor of antitumor immunity, contributing to immune evasion, and it is a highly attractive target for developing future new anticancer therapy agents in combination with radiotherapy and immunotherapies.
MECHANISMS REGULATING YTHDF2 EXPRESSION
While most of the attention has been focused on the downstream signaling of YTHDF2 and physiological importance, we would like to discuss the recently emerged mechanisms by which YTHDF2 expression is regulated. Early observations indicated that hypoxia induces down-regulation of YTHDF2 probably via both hypoxia-inducible factor-1α (HIF-1α) and HIF-2α in hepatocellular carcinoma (HCC) cells (Hou et al. 2019; Zhong et al. 2019). HIF-2α can directly bind to the promoter region of YTHDF2, evidenced by ChIP-qPCR assay (Hou et al. 2019), and likely recruits other elements/proteins to inhibit transcription of YTHDF2. However, conflicting observation also suggested that HIF1α is able to bind to the hypoxia-response elements at the YTHDF2 promoter and regulates YTHDF2 transcription in AML (Chen et al. 2021). EGFR signaling was also shown to be responsible for the up-regulation of YTHDF2 protein in glioblastoma cell lines (Fang et al. 2021). The expression of YTHDF2 in tumor-associated macrophages (TAMs) was regulated by IL-10/STAT3 signaling (Ma et al. 2023). Wang and colleagues identified RELA (P65, an NF-κB subunit) as a principal transcription factor in the induction of Ythdf2 in MDSCs, thus forming an NF-κB/YTHDF2/NF-κB circuit (positive feedback) (Wang et al. 2023a). Based on these observations, YTHDF2 could be regulated by different transcription factors in different cellular contexts. In addition to transcriptional regulation, specific posttranslational modifications (PTMs) such as SUMOylation, O-GlcNAcylation, and phosphorylation of YTHDF2 also occur (Hou et al. 2021; Chen et al. 2023a,b), suggesting networks of PTMs to modulate YTHDF2 activity in distinct cell types.
TARGETING YTHDF2 FOR STEM CELL EXPANSION AND CANCER TREATMENT
Given the important roles played by YTHDF2 in stem cells, cancer cells, and immune cells, this protein is a promising target for developing stem cell and anticancer therapies. YTHDF2 inhibition would lead to ex vivo expansion of stem cells, such as HSCs (Li et al. 2018b) but also other types of stem cell. These stem cells display increased stemness and could be ideal for stem cell therapies. YTHDF2 inhibition also dramatically elevates host antitumor immunity in several mouse models (Ma et al. 2023; Wang et al. 2023a; Zhang et al. 2023). A screening assay with an in-house compound library identified a YTHDF2 inhibitor-DC-Y13-27 (Wang et al. 2023a). DC-Y13-27 can inhibit YTHDF2 fourfold more selectively than YTHDF1 (IC50 = 165 μM for YTHDF1 and IC50 = 38 μM for YTHDF2). It induced effects consistent with Ythdf2 knockout both in vitro (inhibiting NF-κB activation) and in vivo (inhibiting MDSCs and enhancing antitumor immunity of radio-immunotherapy). DC-Y13-27 targets the YTH domain, resulting in the inhibition of YTHDF2 binding to m6A-containing RNA. Note that the YTH domains of YTHDF1/2/3 proteins are almost identical, which makes selective inhibition of the individual YTHDF2 challenging; further efforts are required to develop highly specific and potent small-molecule YTHDF2 inhibitors in order to explore its potentials in therapies.
In an alternative strategy targeting YTHDF2, YTHDF2 siRNA was conjugated to a toll-like receptor 9 (TLR9) agonist, which, when internalized by antigen-presenting cells, slowed tumor growth of both murine colon and melanoma tumors (Ma et al. 2023). Taken together, we propose that YTHDF2 inhibition, overcoming the suppressive milieu induced by MDSCs or TAMs, can improve the responses to current cancer therapies, including radiotherapy and immune checkpoint blockade (ICB).
COMPETING INTEREST STATEMENT
C.H. is a scientific founder, a member of the scientific advisory board and equity holder of Aferna Bio Inc. and Ellis Bio Inc., a scientific cofounder and equity holder of Accent Therapeutics Inc., and a member of the scientific advisory board of Rona Therapeutics and Element Biosciences. R.R.W. had stock and other ownership interests with Boost Therapeutics, Immvira LLC, Reflexion Pharmaceuticals, Coordination Pharmaceuticals Inc., Magi Therapeutics, Oncosenescence, Aqualung Therapeutics Corporation, Cyntegron, and PersonaDX. He has served in a consulting or advisory role for Aettis Inc., AstraZeneca, Coordination Pharmaceuticals, Genus, Merck Serono S.A., Nano Proteagen, NKGen Biotech, Shuttle Pharmaceuticals, Highlight Therapeutics, S.L., and Aqualung Therapeutics Corporation. He has research grants with Varian and Regeneron. He has a patent pending: “Methods and kits for diagnosis and triage of patients with colorectal liver metastases” (PCT/US2019/028071). He owns stock in and is a founder of PersonaDx. He has received compensation, including travel, accommodations, and expense reimbursement from AstraZeneca, Boehringer Ingelheim, and Merck Serono SA. C.H., R.R.W., and L.W. have a patent application pending (provisional application: ARCD.P0780US.P1/1001213214).
ACKNOWLEDGMENTS
C.H. is a Howard Hughes Medical Institute Investigator. This work was funded by National Institutes of Health (NIH) R01 grant no. R01CA262508 to R.R.W.
Footnotes
-
Article is online at http://www.rnajournal.org/cgi/doi/10.1261/rna.080259.124.
-
Freely available online through the RNA Open Access option.
This article, published in RNA, is available under a Creative Commons License (Attribution-NonCommercial 4.0 International), as described at http://creativecommons.org/licenses/by-nc/4.0/.








