
RNA gain-of-function mechanisms in short tandem repeat diseases
- Department of Molecular Genetics and Microbiology, Center for NeuroGenetics and the Genetics Institute, University of Florida, Gainesville, Florida 32610, USA
- Corresponding author: mswanson{at}ufl.edu
Abstract
As adaptors, catalysts, guides, messengers, scaffolds, and structural components, RNAs perform an impressive array of cellular regulatory functions often by recruiting RNA-binding proteins (RBPs) to form ribonucleoprotein complexes (RNPs). While this RNA–RBP interaction network allows precise RNP assembly and the subsequent structural dynamics required for normal functions, RNA motif mutations may trigger the formation of aberrant RNP structures that lead to cell dysfunction and disease. Here, we provide our perspective on one type of RNA motif mutation, RNA gain-of-function mutations associated with the abnormal expansion of short tandem repeats (STRs) that underlie multiple developmental and degenerative diseases. We first discuss our current understanding of normal polymorphic STR functions in RNA processing and localization followed by an assessment of the pathogenic roles of STR expansions in the neuromuscular disease myotonic dystrophy. We also highlight ongoing questions and controversies focused on STR-based insights into the regulation of nuclear RNA processing and export as well as the relevance of the RNA gain-of-function pathomechanism for other STR expansion disorders in both coding and noncoding genes.
Keywords
INTRODUCTION
Dynamic interactions between RNA-binding proteins (RBPs) and their target RNA transcripts are required for the myriad roles required for cell/tissue development and maintenance. Specific RBP–RNA-binding events are generally driven by either single or multiple RNA-binding domains on each RBP that preferentially recognize relatively short and degenerate RNA motifs that are either regionally clustered or dispersed on the transcript (Ray et al. 2013; Corley et al. 2020). Importantly, the affinity and stability of these interactions are influenced by RNA structural dynamics, RBP–RBP interactions, and binding site availability as well as site occupancy competition between RBPs and RNPs (Ule and Blencowe 2019).
A particularly striking class of RNA motifs are short tandem repeats (STRs, generally 1–6 nt) since they are highly unstable genomic sequence elements characterized by frequent repeat contractions and expansions (Sun et al. 2012; Fotsing et al. 2019; Shi et al. 2023; Lamkin and Gymrek 2024). From the RNA perspective, STRs provide high-affinity RBP-binding sites depending on tandem repeat number that can influence gene expression at both the transcriptional and co/posttranscriptional levels (Miller et al. 2000; Batra et al. 2014; Li et al. 2022; Hamanaka et al. 2023). While STRs are polymorphic, a particularly striking feature of human STRs is the high level of instability leading to large expansions of repeat tracts that cause a wide range of hereditary neurological and neuromuscular diseases (Depienne and Mandel 2021; Malik et al. 2021; Rajan-Babu et al. 2024).
For this RNA Perspective, we first address potential nuclear and cytoplasmic functions of STRs in the general population, followed by an examination of the impacts of STR expansion mutations on disease using the RNA gain-of-function model developed through studies on the most common adult-onset muscular dystrophy, myotonic dystrophy (dystrophia myotonica, DM). We also discuss current ideas and questions focused on how STR pathomechanistic studies might provide further insights into RNA-associated pathways.
WHY ARE HUMAN TANDEM REPEATS SO PREVALENT AND WHAT ARE THE RNA IMPACTS?
Prior to the development of early DNA sequencing technologies, experiments analyzing DNA reassociation kinetics following heat denaturation provided clear evidence that vertebrate DNAs contained a large proportion of repetitive sequences (Britten and Kohne 1968). The recent telomere-to-telomere (T2T) gapless sequencing of the human genome that encompasses both euchromatic and heterochromatic regions reinforced the prominence of repetitive sequences (Nurk et al. 2022). Among repetitive DNAs, tandem repeats are divided by motif length and chromosomal location: (1) STRs (1–9 bp although 1–6 bp are linked to disease) that comprise ∼3% of the human genome; (2) variable number tandem repeats (VNTRs) (≥10 bp); (3) tandem segmental duplications (>1 kbp); (4) satellites (alpha centromeric, HSat1A, 1B, 2-4, beta and gamma) (5 bp to ∼2.4 kbp) (Gemayel et al. 2010; Chaisson et al. 2023). Of these repeat types, STR expansions in gene coding and noncoding regions have played an outsized role in disease pathogenesis (Sulovari et al. 2019; Mirceta et al. 2022).
Why are STRs so prevalent in the human genome? One possibility is that these repetitive elements have contributed to the evolution of complex tissues, such as the central nervous system (CNS), because of their high repeat instability with mutation rates that are 10 to 105 times those observed in other genomic regions (Chakraborty et al. 1997; Gemayel et al. 2010). This genomic STR instability results from intra-/inter-repeat recombination, strand slippage during DNA replication, and compromised DNA repair (Khristich and Mirkin 2020; Deshmukh et al. 2021; Kim et al. 2024).
How might STR polymorphisms in the general population impact RNA-mediated regulatory events in cells? For alternative splicing, STRs can function as intronic splicing enhancers and silencers (ISEs, ISSs) due to repeat tract length-dependent splicing factor (SF) recruitment (Hui et al. 2003; Gates et al. 2011; Ling et al. 2015; Donde et al. 2019) while an increase in CAG intronic repeats can generate alternative 3′ splice sites (Fig. 1; Anderson et al. 2024). Interestingly, some TDP-43 regulated nonconserved cryptic exons are constitutively silenced by STRs although future studies may reveal specific cell types or developmental stages where these splicing events occur (Ling et al. 2015). The polymorphic nature of STRs allows these regulatory modules to increase or decrease SF recruitment on a sliding scale allowing each splicing event to be fine-tuned and calibrated depending on additional exon and intron splicing regulatory elements. Besides an important role in pre-mRNA processing, splicing may also be impacted by the expression of lncRNA STRs. An example is the lncRNA PNCTR, which contains numerous (UC)n and UCUYY/YYUCUY repeats that sequester the SF PTBP1 in the perinucleolar compartment (PNC) to regulate splicing and cancer cell survival (Yap et al. 2018). Although less studied, STRs also have an impact on 3′-end processing and polyadenylation (Fig. 1). In Huntington's disease (HD), the expansion of CAG repeats in HTT coding exon 1 induces utilization of a cryptic poly(A) site in intron 1 that yields a highly toxic C-terminal truncated polyglutamine protein (Hoschek et al. 2024). In addition to these effects on RNA alternative processing, the length of an STR tract may alter RBP site occupancy with potential impacts on RNA–RNA, RNA–RBP, and RBP–RBP multivalent interactions and RNP condensate dynamics (Nedelsky and Taylor 2022).
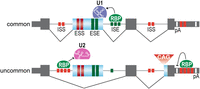
STR functions in pre-mRNA processing. STRs are distributed throughout genic coding and noncoding regions (gray boxes, constitutive exons; blue, alternative exon or 3′ss) and function as copy number dependent alternative processing signals based on RBP recruitment (red boxes, ISS and ESS; green, ISE and ESE). For this example, the top illustration depicts a hypothetical common pre-mRNA processing pattern in the general population. For splicing, exon 2 inclusion is driven by an intron 2 ISE composed of three tandem repeats with coordinate RBP-binding promoting U1 snRNP recruitment. For 3′-end cleavage and polyadenylation, the first polyadenylation site (pA) is selected since there are only two tandem repeats in the downstream region. The uncommon (bottom) splicing pattern shows that exon 2 is skipped due to an intron 2 ISE with only one repeat, while an intron 1 ISS contains four repeats that facilitate RBP recruitment resulting in a U2 snRNP block. This uncommon example also shows an increase in CAG repeats in intron 3 that promotes an alternative exon 4 3′ss together with 3′ UTR repeat number variations that increase RBP recruitment downstream from the proximal pA site leading to selection of the distal site.
HOW DO STR EXPANSIONS CAUSE DISEASE?
Disease-associated STR expansions occur in a wide array of genes and adversely impact multiple cellular pathways (Fig. 2). There are several remarkable characteristics associated with STR expansion disorders. First, some neurodegenerative disorders (e.g., HD and myotonic dystrophy type 2, DM2) show characteristic cell-specific and regional degeneration profiles even though the host gene containing the STR expansion is broadly expressed. Second, STR expansions in the pathological range may be inherited resulting in congenital disease, whereas somatic expansions play a more prominent role in late-onset neurodegenerative diseases. Somatic expansions occur at a higher frequency in postmitotic cells, such as neurons due to their long-term dependence on DNA repair pathways, particularly mismatch repair (MMR) (Schmidt and Pearson 2016; Miller and Usdin 2022). Another active area of research, which is focused on individuals that present with extremes of clinical phenotypes, is to understand the contributions of genetic modifiers to disease manifestations with an early emphasis on HD (McAllister et al. 2022). Third, there is limited length (3 bp) and sequence diversity (CNG, GCN, GAC) of repeats in genic CDS and UTR regions, while intronic repeats are considerably more divergent (3–6 bp in length, N1-5, C/G/T). Although coding limitations account for the usual CDS 3-mer length restriction, STR length diversity in 5′ and 3′ UTRs may also be restricted due to the presence of upstream and downstream open reading frames (uORFs, dORFs) (Dever et al. 2020; Wu et al. 2020).
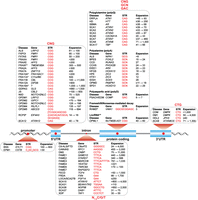
STR-mediated diseases. Shown are STRs (red font) in the promoter, 5′ untranslated (5' UTR), intron, coding, and 3′ untranslated (3' UTR) regions together with the disease acronym, gene name, and pathogenic repeat range (black font). Listed diseases are: amyotrophic lateral sclerosis (ALS); blepharophimosis, ptosis, and epicanthus inversus syndrome (BPES); Baratela–Scott syndrome (BSS); C9orf72-linked ALS and frontotemporal dementia (C9-ALS/FTD); cerebellar ataxia, neuropathy, and vestibular areflexia syndrome (CANVAS); cleidocranial dysplasia (CCD); congenital central hypoventilation syndrome (CCHS); congenital myotonic dystrophy (CDM); myotonic dystrophy type 1 (DM1) and type 2 (DM2); dentatorubral-pallidoluysian atrophy (DRPLA); early infantile epileptic encephalopathy 1 (EIEE1); progressive myoclonus epilepsy (EPM1); familial adult myoclonic epilepsy types 1–4,6,7 (FAME1-4,6,7); Fuchs endothelial corneal dystrophy (FECD); intellectual disability-associated fragile sites 2A, 7A, 10A*, 11A*, 11B, 12A, 16A* (FRA2A, FRA7A, FRA10A*, FRA11A*, FRA11B, FRA12A, FRA16A*) and fragile X syndromes A,E,F (FRAXA, FRAXE, FRAXF*), (*) indicates the disease link is questionable, and for FRA16A* a gene link has not been documented; fragile X-associated primary ovarian insufficiency (FXPOI); fragile X-associated tremor/ataxia syndrome (FXTAS); Friedreich's ataxia (FRDA); global developmental delay, progressive ataxia, elevated glutamine (GDPAG), Huntington's disease (HD); Huntington's disease-like 2 (HDL2); hand-foot genital syndrome (HFGS); holoprosencephaly (HPE); X-linked heterotaxy VACTERLX syndrome (HTX1); intellectual disability with growth hormone deficiency (IDGH); Jacobsen syndrome (JS); late-onset cerebellar ataxia/spinocerebellar ataxia 27B (LOCA/SCA27B); neuronal intranuclear inclusion disease (NIID); oculopharyngodistal myopathy types 1–5 (OPDM); oculopharyngeal muscular dystrophy (OPMD); oculopharyngeal myopathy with leukoencephalopathy (OPML1); pseudoachondroplasia/multiple epiphyseal dysplasia-1 (PSACH/EDM); Richieri-Costa-Pereira syndrome (not the common 5′-UTR pattern so noted as RCPS#); recessive hereditary motor neuropathy (not the common CDS pattern so noted as RHMD#); spinal-bulbar muscular atrophy (SBMA); spinocerebellar ataxia (SCA) types 1–3, 6–8, 10, 12, 17, 31, 36, 37 (SCA1–3, 6–8, 10, 12, 17, 31, 36, 37); synpolydactyly (SPD1); X-linked dystonia-parkinsonism (XDP). See Depienne and Mandel (2021), Malik et al. (2021), Mirceta et al. (2022) for references except ALS LRP12 (Kume et al. 2023), ALS NIPA1 (Tazelaar et al. 2019), FRA10A (Sarafidou et al. 2004), FRA11A (Debacker et al. 2007), FRA11B (Jones et al. 1995), FRAXF (Shaw et al. 2002), HTX1 (Wessels et al. 2010), LOCA/SCA27B (Pellerin et al. 2023; Rafehi et al. 2023), OPDM3 (Yu et al. 2021), OPDM4 (Yu et al. 2022; Zeng et al. 2022), OPDM5 (Cortese et al. 2024), RCPS (Favaro et al. 2014), and RHMD (Pagnamenta et al. 2021).
How do these expansions result in either neurodevelopmental defects or later-onset neurodegeneration? Current pathomechanistic models that are not mutually exclusive include: (1) recessive loss of function of STRexp host gene expression due to repeat-induced gene methylation or R-loop formation; (2) RNA gain of function due to sequestration of trans-acting factors such as RBPs; (3) protein gain of function resulting from canonical translation of coding region repeats (e.g., polyGln in HD) or noncanonical repeat-associated non-AUG (RAN) translation of coding and noncoding repeats (Malik et al. 2021). For this Perspective, we will focus on the RNA gain-of-function model.
MYOTONIC DYSTROPHY IS A DEVELOPMENTAL RNA-PROCESSING DISORDER
A pathomechanistic model for how STR length dynamics influence nuclear RNA processing and localization resulting in disease manifestations is the neuromuscular disease myotonic dystrophy (DM). DM is caused by the expansion of similar STRs in the noncoding regions of two different genes (Fig. 2). DM1 is associated with DMPK 3′ UTR CTG trinucleotide expansions (unaffected ≤37 repeats, disease range 50 to >4500) while a CCTG repeat expansion (≥75 to >11,000 repeats) in intron 1 of CNBP underlies DM2 (Brook et al. 1992; Liquori et al. 2001). Both of these repeats contain a YGCY motif that is the preferred RNA-binding site for the MBNL proteins, which are CCCH-type zinc-finger proteins encoded by the MBNL1, MBNL2, and MBNL3 genes (Miller et al. 2000; Kanadia et al. 2003; Du et al. 2010; Goers et al. 2010). MBNL proteins bind CUG repeats with high affinity (Kd = 1–15 nM; higher affinities for CCUG) with binding proportional to repeat length. As CUG repeat tract length increases, MBNL proteins bind directly to nuclear CUG expansion (CUGexp) RNAs, resulting in the formation of MBNL-CUGexp foci (Fig. 3; Goodwin et al. 2015). In DM1, cell/tissue-specific increases in CTG repeat lengths intergenerationally or somatically lead to progressive loss of MBNL activity thus providing a molecular explanation for genetic anticipation, a non-Mendelian inheritance pattern associated with intergenerational increases in disease severity coinciding with decreases in age-of-onset (Miller et al. 2000; Friedman 2011). Intriguingly, anticipation has not been observed in DM2. Additional proteins, including several hnRNPs and DEAD-box helicases (e.g., DDX5, DDX17, DHX9), have also been reported to colocalize in nuclear MBNL-CUGexp foci in DM1 although it is likely that MBNL proteins will remain the predominant RBP in these condensates since they bind proportional to repeat length (Miller et al. 2000; Paul et al. 2011). An intriguing question is whether future smRNA FISH and other experimental strategies will identify additional foci-localized RNAs or nuclease-generated fragments. In contrast to DM1, RBFOX proteins colocalize with MBNL-CCUGexp foci in DM2 cells and it is controversial whether DM2 nuclear RNP foci contain CCUGexp repeats with/without flanking sequences (Margolis et al. 2006; Sellier et al. 2018; Sznajder et al. 2018).
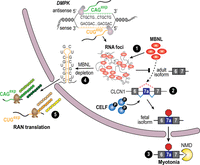
RNA gain-of-function pathomechanism in DM1. Bidirectional transcription of DM1 DMPK produces sense CUGexp RNAs that bind MBNL proteins (red ovals), which undergo RNA–RNA, RNA–protein, and protein–protein multivalent interactions to form RNA foci or condensates (1). In contrast to sense transcription, antisense transcription yields very low-abundance RNAs with/without CAGexp repeats (Gudde et al. 2017). MBNL sequestration in these foci together with phosphorylated CELF (blue sphere with black P) overexpression promotes fetal splicing patterns for target RNAs in adult tissues, and in DM1 skeletal muscle, exon 7A inclusion in CLCN1 mRNA (fetal isoform) (2) results in nonsense-mediated decay (NMD), CLCN1 loss, and myotonia (3). Somatic CTG expansions result in extremely long CUGexp tracts that deplete MBNL (4) from the dilute nucleoplasmic pool, leading to release of intact or fragmented mutant DMPK mRNAs into the cytoplasm where they undergo RAN translation, giving rise to toxic RAN proteins (green and brown/yellow beads) (5). Mutant DMPK antisense transcripts may be exported into the cytoplasm at a low level, resulting in RAN translation of polyGln (Zu et al. 2011), and possibly polySer and polyAla, proteins.
What is the connection between MBNL sequestration and DM disease onset and progression? MBNL1 and MBNL2 are pre-mRNA alternate processing factors that regulate postnatal switches in nuclear pre-mRNA alternative splicing and polyadenylation, as well as mRNA cytoplasmic localization, from fetal to adult patterns, while MBNL3 regulates similar functions during embryogenesis and adult tissue regeneration (Charizanis et al. 2012; Wang et al. 2012; Batra et al. 2014; Thomas et al. 2017; Nutter et al. 2023). Thus, DM1 and DM2 are caused by RNA gain-of-function mutations and expression of CUG or CCUG repeat expansion RNAs are toxic because they sequester RBPs in RNP foci resulting in the expression of developmentally inappropriate fetal isoforms incompatible with adult tissue functions (Fig. 3).
Another issue is that toxic peptides are also generated in DM1 by RAN translation (polyGln, antisense RNA; potential but undocumented polyLeu, polyCys, polyAla, sense; polySer, polyAla, antisense) and DM2 (poly LeuProAlaCys, sense; polyGlnAlaGlyArg, antisense) (Zu et al. 2011, 2017). For DM2, a “sequestration failure” model has been proposed whereby MBNL proteins retain C(C)UGexp RNAs in nuclear RNA foci but as somatic expansion events increase the length of the repeat tract, the sequestration capacity of the MBNL is exhausted and free RNAs are released into the cytoplasm leading to toxic RAN peptide production (Fig. 3; Zu et al. 2017). How DM1 and DM2 onset, progression and acute phases are impacted by RNA versus RAN protein gain-of-function mechanisms is currently unclear but could be resolved by the development of DM1 and DM2 animal models that replicate the corresponding multisystemic disease phenotypes as well as the endogenous levels of toxic RNAs and RAN proteins. However, the generation of mouse models may prove quite challenging for many STR diseases due to the high number of repeats required for disease-specific pathological phenotypes (Nutter et al. 2019).
Current questions, and some answers, designed to provide a mechanistic understanding of RAN translation include: (1) what are the roles of different STR sequences and structures; (2) does initiation occur at noncanonical codons upstream of the repeat tract or at random sites; (3) does RAN translation occur due to impaired ribosomal scanning over long repeat tracts leading to compromised start codon fidelity; (4) what are the functional relationships between IRES, RAN and canonical translation; (5) does frameshifting occur across these repeats and/or does alternative splicing introduce canonical start codons upstream or within repeat tracts (Nguyen et al. 2019; Malik et al. 2021; Anderson et al. 2024)?
MBNL-C(C)UGexp RNP FOCI: PATHOGENIC OR PROTECTIVE CONDENSATES?
The formation of RNP foci in DM and other STR expansion diseases elicits several provocative questions about their formation, stability, possible functions, and whether they provide any insights into the generation, processing, and nuclear export of nonexpansion RNPs. First, what cellular events facilitate the formation of MBNL-C(C)UGexp RNP foci? For RNP granules, formation is driven by RNA–RNA, RNA–protein, and protein–protein multivalent interactions, but RNP granules are generally defined as functional compartments composed of multiple types of RNAs and proteins, such as nuclear Cajal bodies and cytoplasmic stress granules (Ripin and Parker 2023). Another classification, “incidental” condensates, might be a bit more accurate for RNP foci. These condensates do not add functionality to the corresponding soluble RNP complexes but result from phase separation above a critical concentration and are characterized by viscoelastic properties appearing both liquid-like and solid-like with a defined boundary between condensed and dilute phases (Putnam et al. 2023). MBNL-CUGexp RNP foci possess these condensate properties with droplet-like behaviors, and both RNA and MBNL proteins are required for foci formation with MBNL undergoing rapid exchanges with the dilute nucleoplasmic pool as assessed by FRAP (Querido et al. 2011; Sznajder et al. 2016). Additionally, RNA foci possess a clear boundary that is particularly striking with repeat expansions in the severe pathogenic range with smaller and more intense foci as assessed by RNA-FISH or MBNL immunofluorescence suggesting repeat length-dependent focal compression. While CUG RNAs expressed at relatively high concentrations also undergo dynamic phase transitions in the nucleus (Jain and Vale 2017), the characteristic condensed morphology and maintenance of DM1 RNP foci requires MBNL protein interactions (Dansithong et al. 2005).
Second, while C(C)UGexp RNA foci are generated as a by-product of MBNL proteins binding to massive reiterations of their preferred RNA motif, are these foci innocent bystanders, pathogenic or protective? In other words, do they add functionality outside of the corresponding soluble RNP pool? The generation of C(C)UGexp RNA foci and MBNL sequestration is clearly a pathogenic event since Mbnl KO mice recapitulate many of the characteristic features of DM1 and DM2 disease, including skeletal muscle myotonia, cardiac conduction defects, insulin insensitivity, subcapsular eye lens cataracts and CNS abnormal sleep cycles and learning/memory deficits (Sznajder and Swanson 2019). Nevertheless, RNP foci formation may also block the export of C(C)UGexp RNAs into the cytoplasm and the production of toxic RAN peptides (Zu et al. 2017). Cumulatively, these observations present a rather interesting mechanistic conundrum with RNA foci serving both pathogenic and protective roles.
Third, do DM1 and DM2 RNA foci escape the nuclear surveillance pathway, or do they simply delay RNA decay and what role do MBNL proteins play in this process? Rapid formation and the dynamics of nascent RNA structures during transcription have been shown to regulate multiple gene expression steps, including transcription rate/pausing, trans-acting factor (e.g., snRNPs, RBPs) accessibility, RNA modifications, and RNA processing designed to generate RNP complexes that are resistant to nuclease degradation and amenable to nuclear trafficking and export into the cytoplasm (Shine et al. 2024). Addition of the 5′ m7G cap and 3′ poly(A) tail inhibits nuclear decay mediated by the XRN2 5′-3′ and nuclear exosome 3′-5′ exoribonucleases while entry into the export pathway requires recruitment of the 5′ cap-binding complex (CBC, heterodimer of NCBP1 and NCBP2), the nuclear poly(A)-binding protein PABPN1 and export factors (ALYREF, ARS2, NXF1) together with a correctly processed RNP architecture that facilitates nuclear export (Garland and Jensen 2020). Early studies of mutant DMPK RNAs in DM1 differentiated myoblasts, or postmitotic myotubes, indicated these foci contained spliced and polyadenylated DMPK mRNA suggesting that the export block occurs following pre-mRNA processing and chromatin release (Davis et al. 1997). Furthermore, nuclear CUGexp RNA turnover is also inhibited by RNP foci formation since MBNL dissociation from these RNAs, induced by either a small molecule, CUG repeat targeted PMO or hU7-(CAG)15 snRNP, results in mutant DMPK RNA turnover (Francois et al. 2011; Rzuczek et al. 2017; Klein et al. 2019). These observations indicate that future studies should revisit the structures of mutant DMPK and CNBP RNAs and RNPs in their respective foci using both cell and animal disease models to clarify the relationships between C(C)UGexp foci composition and structure to disease onset and progression.
RELEVANCE OF RNA GAIN OF FUNCTION TO OTHER STR DISEASES?
RNA gain of function in DM1 and DM2 resulting in RBP sequestration and protein loss of function has served as one of the pathomechanistic models for other STR expansion disorders. As noted previously, the discovery of RAN translation in SCA8 and DM1 (Fig. 2) introduced another RNA gain-of-function mechanism—repeat expansion RNAs can also template protein gain-of-function events upon export into the cytoplasm. This mechanistic duality has played out in other STR expansion diseases, including the most frequent genetic cause of ALS and frontotemporal dementia, C9-ALS/FTD (Fig. 2). C9-ALS/FTD is associated with GGGGCC•CCCGG expansions in intron 1 of the C9orf72 gene, and as described for DM1, RNA gain of function could result in both protein loss of function and gain of function (DeJesus-Hernandez et al. 2011; Renton et al. 2011; Swinnen et al. 2020). For example, C9-ALS/FTD repeat expansions accumulate in nuclear RNA foci although in this case, mutant RNAs have been proposed to trigger a disruption in nucleocytoplasmic transport (NCT) (Freibaum et al. 2015; Jovicic et al. 2015; Zhang et al. 2015). NCT inhibition is linked to the loss of nucleoporins, initially POM121, although mechanistic connections between GGGGCCexp and GGCCCCexp RNA expression and NPC disassembly have not been documented (Coyne et al. 2020; Zaepfel and Rothstein 2021). Moreover, sequestration of NCT factors in stress granules also occurs in this disease and a recent study noted that loss of C9orf72 expression leads to alterations in the Ran-GTPase gradient (Zhang et al. 2018; McGoldrick et al. 2023). Finally, current evidence argues both for, and against, a direct role for RAN protein gain of function in defective C9-ALS/FTD NCT (Zaepfel and Rothstein 2021; Jafarinia et al. 2024). Thus, studies focused on C9-ALS/FTD pathomechanisms have provided, and will likely continue to promote, novel insights into RNA pathways affected by STR expansions.
ONGOING STUDIES AND FUTURE POSSIBILITIES
Another potentially high-impact development in the tandem repeat expansion field are studies showing that mobile genomic elements constitute another type of disease-associated repetitive element polymorphism. For example, the progressive neurodegenerative disease X-linked dystonia-parkinsonism (XDP) has been linked to 5′ hexamer (CCCTCT•GGGAGA)n repeats in a SINE-VNTR-Alu (SVA) retrotransposon that is inserted in an antisense orientation into intron 32 of the TAF1 gene (Fig. 2; Bragg et al. 2017). Several features of these SVA repeats are reminiscent of other STR expansion diseases including: (1) STR repeat length inversely correlates with disease age of onset; (2) (GGGAGA)n repeats form G-quadruplexes; (3) RAN translation has been documented using reporter constructs in HEK293 cells; (4) SVA insertion alters splicing resulting in intron 32 retention and decreased TAF1 full-length expression (Aneichyk et al. 2018; Reyes et al. 2022). In contrast, RNA foci and RBPs that preferentially bind (GGGAGA)n repeats have not been reported. Another intriguing question is why repeat expansions in the SVA 5′, and not the VNTR, region is associated with this disease. Nevertheless, the abundance and genomic mobility of SVA elements suggest that additional hereditary and de novo mutations will be linked to other diseases in the future that may reveal unanticipated RNA connections.
Finally, most of the diseases discussed in this article are located in protein-coding genes, but the STR and disease landscapes may be considerably more extensive for noncoding genes (Johnson and Cooper 2021). Future investigations designed to reveal possible links between STR mutations, both sequence and length variants, in lncRNA and other noncoding genes is warranted and may be even more informative for the RNA research community.
ACKNOWLEDGMENTS
We thank Andy Berglund, James Burke, Tom Cooper, Christopher Pearson, and Phil Wong for informative discussions, Jodi Bubenik for manuscript feedback, and Marina Scotti for assistance with figures. Work in our laboratory is funded by grants from the National Institutes of Health (R01 NS103172, P50 NS132955), the Department of Defense (PR221190), the Marigold Foundation, and a Myotonic Dystrophy Foundation postdoctoral fellowship award to M.L.D.
Footnotes
-
Article is online at http://www.rnajournal.org/cgi/doi/10.1261/rna.080277.124.
-
Freely available online through the RNA Open Access option.
This article, published in RNA, is available under a Creative Commons License (Attribution-NonCommercial 4.0 International), as described at http://creativecommons.org/licenses/by-nc/4.0/.








