
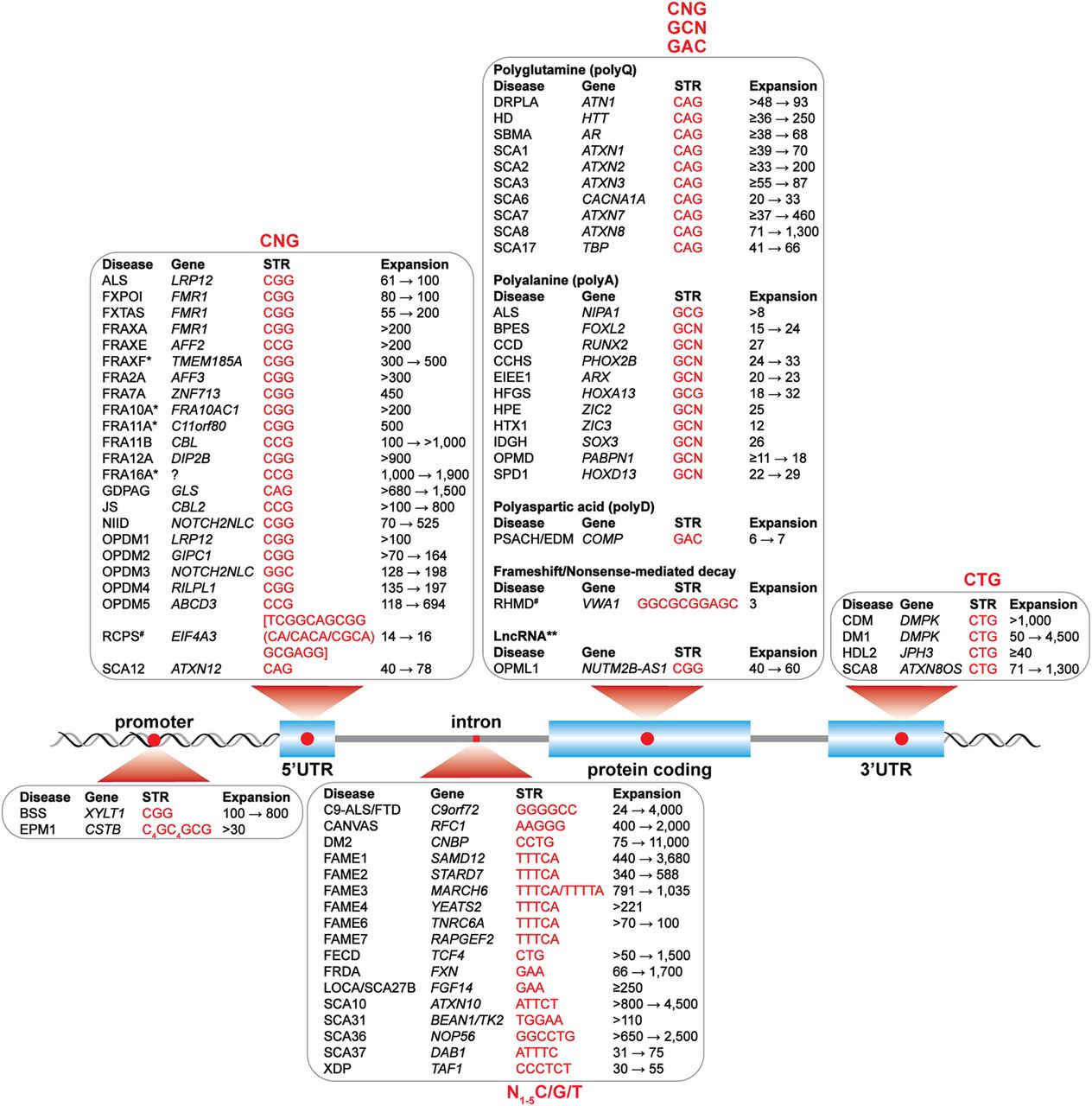
STR-mediated diseases. Shown are STRs (red font) in the promoter, 5′ untranslated (5' UTR), intron, coding, and 3′ untranslated (3' UTR) regions together with the disease acronym, gene name, and pathogenic repeat range (black font). Listed diseases are: amyotrophic lateral sclerosis (ALS); blepharophimosis, ptosis, and epicanthus inversus syndrome (BPES); Baratela–Scott syndrome (BSS); C9orf72-linked ALS and frontotemporal dementia (C9-ALS/FTD); cerebellar ataxia, neuropathy, and vestibular areflexia syndrome (CANVAS); cleidocranial dysplasia (CCD); congenital central hypoventilation syndrome (CCHS); congenital myotonic dystrophy (CDM); myotonic dystrophy type 1 (DM1) and type 2 (DM2); dentatorubral-pallidoluysian atrophy (DRPLA); early infantile epileptic encephalopathy 1 (EIEE1); progressive myoclonus epilepsy (EPM1); familial adult myoclonic epilepsy types 1–4,6,7 (FAME1-4,6,7); Fuchs endothelial corneal dystrophy (FECD); intellectual disability-associated fragile sites 2A, 7A, 10A*, 11A*, 11B, 12A, 16A* (FRA2A, FRA7A, FRA10A*, FRA11A*, FRA11B, FRA12A, FRA16A*) and fragile X syndromes A,E,F (FRAXA, FRAXE, FRAXF*), (*) indicates the disease link is questionable, and for FRA16A* a gene link has not been documented; fragile X-associated primary ovarian insufficiency (FXPOI); fragile X-associated tremor/ataxia syndrome (FXTAS); Friedreich's ataxia (FRDA); global developmental delay, progressive ataxia, elevated glutamine (GDPAG), Huntington's disease (HD); Huntington's disease-like 2 (HDL2); hand-foot genital syndrome (HFGS); holoprosencephaly (HPE); X-linked heterotaxy VACTERLX syndrome (HTX1); intellectual disability with growth hormone deficiency (IDGH); Jacobsen syndrome (JS); late-onset cerebellar ataxia/spinocerebellar ataxia 27B (LOCA/SCA27B); neuronal intranuclear inclusion disease (NIID); oculopharyngodistal myopathy types 1–5 (OPDM); oculopharyngeal muscular dystrophy (OPMD); oculopharyngeal myopathy with leukoencephalopathy (OPML1); pseudoachondroplasia/multiple epiphyseal dysplasia-1 (PSACH/EDM); Richieri-Costa-Pereira syndrome (not the common 5′-UTR pattern so noted as RCPS#); recessive hereditary motor neuropathy (not the common CDS pattern so noted as RHMD#); spinal-bulbar muscular atrophy (SBMA); spinocerebellar ataxia (SCA) types 1–3, 6–8, 10, 12, 17, 31, 36, 37 (SCA1–3, 6–8, 10, 12, 17, 31, 36, 37); synpolydactyly (SPD1); X-linked dystonia-parkinsonism (XDP). See Depienne and Mandel (2021), Malik et al. (2021), Mirceta et al. (2022) for references except ALS LRP12 (Kume et al. 2023), ALS NIPA1 (Tazelaar et al. 2019), FRA10A (Sarafidou et al. 2004), FRA11A (Debacker et al. 2007), FRA11B (Jones et al. 1995), FRAXF (Shaw et al. 2002), HTX1 (Wessels et al. 2010), LOCA/SCA27B (Pellerin et al. 2023; Rafehi et al. 2023), OPDM3 (Yu et al. 2021), OPDM4 (Yu et al. 2022; Zeng et al. 2022), OPDM5 (Cortese et al. 2024), RCPS (Favaro et al. 2014), and RHMD (Pagnamenta et al. 2021).








