
Protein binding in an mRNA 5′-UTR sterically hinders translation
- Corresponding author: weeks{at}unc.edu
Abstract
Structures in the 5′ untranslated regions (UTRs) of mRNAs can physically modulate translation efficiency by impeding the scanning ribosome or by sequestering the translational start site. We assessed the impact of stable protein binding in 5′- and 3′-UTRs on translation efficiency by targeting the MS2 coat protein to a reporter RNA via its hairpin recognition site. Translation was assessed from the reporter RNA when coexpressed with MS2 coat proteins of varying affinities for the RNA, and at different expression levels. Binding of high-affinity proteins in the 5′-UTR hindered translation, whereas no effect was observed when the coat protein was targeted to the 3′-UTR. Inhibition of translation increased with coat protein concentration and affinity, reaching a maximum of 50%–70%. MS2 proteins engineered to bind two reporter mRNA sites had a stronger effect than those binding a single site. Our findings demonstrate that protein binding in an mRNA 5′-UTR physically impedes translation, with the effect governed by affinity, concentration, and sterics.
Keywords
INTRODUCTION
Translation is carried out by the ribosomal complex, which catalyzes protein synthesis directed by a messenger RNA (mRNA) template. Translation involves three central steps: initiation, elongation, and termination. The initiation step is rate-limiting, with recognition of the start codon being the most influential event in modulating protein expression levels (Jackson et al. 2010; Merrick and Pavitt 2018). During initiation, the preinitiation complex, comprising initiation factors and the ribosomal small subunit, is recruited to the 5′ end of the mRNA. The resulting ribonucleoprotein complex scans the mRNA from the 5′ end until it encounters an AUG start codon. Upon recognition of a start codon, the ribosomal large subunit is recruited, initiation factors are released, and peptide chain elongation begins (Gebauer and Hentze 2004; Hinnebusch et al. 2016). This model implies that features at the 5′ end of an mRNA that interfere with ribosome movement or with interaction with the AUG region can modulate translation.
Extensive prior work shows that stable RNA structures upstream of and overlapping the translation start site influence translation (Fig. 1A), through two main mechanisms. First, RNA structures that overlap the translation start site and adjacent regulatory elements reduce ribosomal recognition and translation in both prokaryotes and eukaryotes (Kozak 2005; Wachter 2014; Leppek et al. 2018; Mustoe et al. 2018b). In prokaryotes, this feature is exploited in riboswitches where small molecule metabolites bind and alter the stability of RNA structures overlapping the start codon region (Mandal and Breaker 2004). Second, both highly stable engineered (Kozak 2005; Babendure et al. 2006; Beaudoin and Perreault 2010) and natively occurring (Mustoe et al. 2018a) RNA secondary structures can physically impede movement or scanning of the ribosome along the 5′-UTR, thereby reducing translation (Bao et al. 2022; Wang et al. 2022). Stable RNA structures can also upregulate translation by preventing formation of inhibitory RNA folds (Grayeski et al. 2022) or by promoting ribosome pausing at an AUG start site (Hedaya et al. 2023; Xiang et al. 2023).
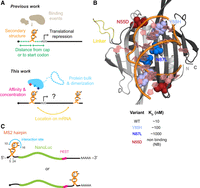
Exploiting MCP–RNA interactions to investigate the influence of mRNA-binding proteins on gene expression. (A, top) Summary of the impact of RNA secondary structure and small molecule or protein binding upstream of the start codon, as established in prior work. (Bottom) Effects of protein binding on translation evaluated here as a function of binding affinity, size, dimerization potential, and concentration. (B) Model of the MCP-M protein–RNA complex. Location of interdomain linker and sites of mutation are emphasized. Affinities are estimated based on prior studies (Peabody 1993). (C) Architecture of reporter mRNAs.
Translation is also extensively modulated by RNA-binding proteins (RBPs) (Fig. 1A), which are numerous, highly expressed, and bind with varying specificity based on RNA sequence and structure (Dominguez et al. 2018; Van Nostrand et al. 2020; Gebauer et al. 2021). RBPs can recruit additional factors to an mRNA, alter the structure and accessibility of specific regions, and influence splicing and localization, thereby affecting translation (Castello et al. 2012; Polymenis 2022). RBPs circularize mRNAs via interactions between the 5′ and 3′ ends resulting in broad effects on translation (Kuersten and Goodwin 2003; Szostak and Gebauer 2013). Engineered protein binding in the 5′-UTR of yeast and human mRNAs hinders translation and ribosome processivity (Stripecke et al. 1994).
In this study, we developed a reporter system to explore the effects of protein binding to mRNA, using the coat protein from bacteriophage MS2 as a model. The native MS2 coat protein (MCP) homodimer binds a hairpin in the MS2 RNA to repress translation and direct genome packaging (Peabody and Ely 1992; Koning et al. 2016). We engineered a monomeric MCP, called MCP-M, which binds a single RNA hairpin, and a set of MCP-M mutants with a range of affinities for RNA (Fig. 1B; Peabody 1993). We targeted these proteins to an mRNA reporter expressing NanoLuc with the target hairpin in the 5′- or 3′-UTR (Fig. 1C). We show that protein binding has large impacts on translation, and the effects are tunable by protein affinity, concentration, size, multisite binding, and target site location (Fig. 1A).
RESULTS
Design of a monomeric coat protein-target hairpin reporter system
The MCP homodimer binds specifically to its cognate RNA hairpin with an affinity of ∼10 nM (Peabody 1993). We created a version of the MCP consisting of two individual MCP domains connected by a (His)6 linker (Fig. 1B). The engineered monomer, which binds a single recognition site, is termed MCP-M. Mutant versions of MCP, Y85H and N87L, with measured affinities of ∼100 and ∼1000 nM, respectively, and a nonbinding (NB) variant, N55D, have been previously described (Peabody 1993). We engineered MCP-M to incorporate these mutations in both domains (Fig. 1B). mRNA reporters encoding NanoLuc (England et al. 2016) were constructed to contain the target hairpin in either the 5′ or 3′-UTR (Fig. 1C). The 5′-UTR hairpin was positioned 5 nt downstream from the transcription start site and 40 nt upstream of the start codon, with the nucleotides forming the core MCP-M interaction site at positions 10–16. In the 3′-UTR reporter, the MS2 hairpin was placed 30 nt upstream of the poly(A) signal region. A control reporter mRNA lacked the MS2 hairpin. All mRNA reporters encode NanoLuc with a C-terminal PEST sequence to promote protein turnover and limit accumulation effects (García-Alai et al. 2006).
Effect of MCP-M affinity on gene expression
Plasmids encoding MCP-M, NanoLuc, and firefly luciferase protein were cotransfected into HEK293T cells. After 48 h, NanoLuc expression was measured and normalized to firefly luciferase expression. Fold-change in translation was calculated by comparing NanoLuc luminescence in the presence of binding MCP-M proteins with the nonbinding (NB) MCP-M mutant. MCP-M expression did not affect the translation levels of the reporter lacking the MS2 hairpin (Fig. 2A; Supplemental Fig. S1). Targeting MCP-M to the 3′-UTR also showed no significant change in NanoLuc expression levels, at any concentration (5 or 15 ng plasmid/well) (Fig. 2A; Supplemental Fig. S1).
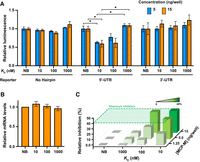
Effect of MCP-M protein binding on reporter translation. (A) Luciferase signal from reporter genes without and with MS2 hairpins, as a function of MCP-M proteins with different RNA-binding affinities. NanoLuc luminescence was normalized to firefly luciferase signal and plotted relative to signal in the presence of the NB mutant (Hatfield et al. 2024). Protein-encoding plasmids were transfected at 5 or 15 ng of plasmid per well. Mean ± SEM of biological triplicates are shown. (*) P-value < 0.05; Student's t-test. (B) Relative reporter mRNA levels in the presence of nonbinding MCP-M (NB) or MCP-M with KD of 10 nM. Protein-encoding plasmids were transfected at 15 ng/well. Mean ± SEM of biological duplicates, two technical measurements each, are shown. (C) Relative inhibition of NanoLuc signal normalized to signal in the presence of the nonbinding MCP-M (NB), as a function of protein amount (based on protein-encoding plasmid transfected) and affinity. Complete data sets are presented in Supplemental Figures S1 and S2.
In strong contrast, targeting the native-like MCP-M to the 5′-UTR decreased NanoLuc expression by up to 40% relative to the NB variant. At 5 ng/well, NanoLuc expression was lower upon coexpression of the MCP-M protein with dissociation constant (KD) of 10 nM (at 60%) compared to the mutant with a KD of 100 nM (80%) (Fig. 2A; Supplemental Fig. S2). The weakest binding MCP-M protein (KD 1000 nM) had no effect. Thus, gene expression decreases when a stable protein–mRNA complex forms in the 5′-UTR, and tighter-binding yields greater suppression.
We used RT-qPCR to measure reporter mRNA levels in cells expressing the 5′-UTR NanoLuc reporter and MCP-M protein variants. Reporter mRNA levels were similar across MCP-M variants (Fig. 2B), indicating that MCP-M binding does not lead to reporter mRNA degradation, and that reduction in NanoLuc expression reflects decreased translation efficiency. Maximal inhibition (40%) was achieved with the 10 nM-binding protein at 5 ng/well or, comparably, with the 100 nM binder at 15 ng/well (Fig. 2C; Supplemental Fig. S2). Inhibition thus follows mass-action behavior such that weaker-binding MCP-M proteins affect translation as effectively as higher-affinity MCP-M proteins when expressed at higher concentrations.
Role of protein steric bulk and dimerization in translation inhibition
To investigate steric effects of protein-mediated occlusion on translational efficiency, for proteins targeted to the 5′-UTR, we constructed dimerized forms of MCP-M, which we term MCP-D. MCP-D consists of two MCP-M units linked by a rigid (EAAAK)3 helical linker (Chen et al. 2013b), forming a four-MS2 domain protein capable of binding two target hairpins. In principle, MCP-D should be able to bind simultaneously two cognate hairpins in two mRNAs, bringing their 5′-UTRs into proximity. We created multiple versions of MCP-D: a nonbinding mutant (NB-NB), a native sequence dimer (10-10), and weaker-binding dimers (100-100 and 1000-1000, respectively). In addition, we made heterodimers tethering one native or binding mutant MCP-M domain to an NB MCP-M domain: 10-NB, 100-NB, and 1000-NB.
Cells expressing the NB-NB homodimer showed luminescence activity at approximately the same level as that of the cells that expressed the MCP-M NB variant (Fig. 3A; Supplemental Figs. S1 and S2). Similarly, heterodimers and homodimers of the lowest affinity variant (1000-NB and 1000-1000) did not affect translation (Fig. 3A; Supplemental Figs. S1 and S2), confirming that weak or NB variants have no impact on reporter translation.
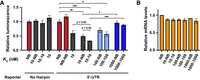
Effect of steric bulk and potential RNA dimerization on translation. (A) Luciferase signal from reporter without and with the target hairpin in the 5′-UTR upon expression of MCP-D variants with similar or different RNA-binding domains (homo- and heterodimers, respectively). NanoLuc luminescence was normalized to firefly luciferase signal and plotted relative to signal in the presence of the NB variant. Protein-encoding plasmids were transfected at 15 ng/well. Means ± SEMs of biological triplicates are shown. (*), (**), and (***) P-values < 0.05, 0.01, and 0.001, respectively; Student's t-test. Complete data set presented in Supplemental Figures S1 and S2. (B) Relative reporter mRNA levels upon expression of MCP-D variants normalized to expression in the presence of NB MCP-M. Protein-encoding plasmids were transfected at 15 ng/well. Means ± SEM of biological duplicates, two technical measurements each, are shown.
In contrast, the expression of binding heterodimers (10-NB, 100-NB) and homodimers (10-10, 100-100) significantly reduced luminescence levels compared to the NB variants. Homodimers consistently showed a larger decrease in luminescence signal than heterodimers. The homodimers showed a 30% greater reduction in luciferase activity compared to an equivalent number of monomeric units, achieved through twofold higher transfection levels of plasmids encoding the monomer (Supplemental Table S1). These results suggest that dimerization, or bringing two 5′-UTRs into proximity, increases translation inhibition beyond that caused by the MCP-M monomer alone. RT-qPCR showed no significant differences in mRNA levels in cells expressing the NB variant or any homo- or heterodimers (Fig. 3B), indicating that the increased translation inhibition reflected direct effects on translation, rather than mRNA degradation. Maximum inhibition reached 50% for 10-NB and 70% for 10-10 expression.
DISCUSSION
Translation initiation involves multiple steps, including an early ribosome scanning phase to identify the start codon (Hinnebusch 2017; Gu et al. 2021) and potential movement from the 3′- to the 5′-end of a pseudo-circularized mRNA (Brito Querido et al. 2024). Here, we characterized the impact of protein binding on translation in human cells. Proteins were targeted either to the 5′-UTR, immediately downstream from the transcription start site, or to the 3′-UTR, upstream of the polyadenylation signal. Our study is consistent with and extends a much earlier study on targeting proteins to 5′-UTRs (Stripecke et al. 1994). We observed significant inhibition of translation upon protein binding in the 5′-UTR of the mRNA, but no effect for protein binding in the 3′-UTR. Protein binding did not affect mRNA levels, consistent with a model in which stable protein binding upstream of the start codon inhibits translational initiation. Translational repression occurred to a similar degree at a low relative concentration of proteins with high-affinity RNA binding (5 ng/µL transfected plasmid; KD 10 nM) and at higher concentration of weaker-binding proteins (15 ng/µL; KD 100 nM). Engagement with the RNA is the most important factor, with repression modestly enhanced by larger proteins.
Both our findings and earlier work (Stripecke et al. 1994), emphasize the significant translational repression caused by protein binding in the 5′-UTR, with effects in our study reaching up to 70%. The mechanism likely involves interference with ribosome scanning. The strong intrinsic helicase activity of the ribosome complex (Takyar et al. 2005), coupled with associated DEAD-box helicases like initiation factor eIF4A, helps overcome intervening RNA structures and RNA–protein complexes (Sweeney et al. 2021; Ryan and Schröder 2022). Stable hairpins can resist the unwinding process by helicases (Bleichert and Baserga 2007; Yang et al. 2007), which correlates with reduced translation efficiency (Chen et al. 2013a; Desai et al. 2019). This study extends the correlation between translation inhibition and stable structural features in an mRNA to include protein binding events. The inhibitory effect of protein binding is tunable, as revealed by the observation of larger reductions in translation by tighter-binding MCP-M proteins. While the impact of MCP-D-mediated dimerization of mRNA on translation efficiency is modest (P = 0.08), the observed effect suggests that macroscopic cellular events, such as mRNA proximity and dimerization, have the potential to influence translation.
Our engineered system for repressing translation highlights opportunities for RNA–protein interactions to control or modulate gene expression and supports further analysis of mRNA–protein interactions that occur upstream of the start codon. Our study specifically demonstrates that translation inhibition effects are pronounced at an affinity of ∼10 nM, consistent with the affinities of a subset of RBPs (Helder et al. 2016; Corley et al. 2020). Translational downregulation is clearly tunable, and we show protein-mediated control that varies by affinity, concentration, and sterics. There are broad opportunities to regulate translation via stable protein binding in mRNA 5′-UTRs, in both endogenous and engineered systems.
MATERIALS AND METHODS
Plasmid construction
Reporter plasmids are based on the backbone of NanoLuc plasmid pNL3.2 (N1411, Promega), which served as the no-hairpin control. Reporters containing 5′- or 3′-UTR hairpins were constructed by introducing the MS2 hairpin sequence into the NanoLuc backbone. MCP-M plasmids contained two copies of an MCP variant separated by a (His)6 linker. MCP-D plasmids were constructed by combining two MCP-M backbones separated by a rigid helical linker (EAAAK)3. All mutants are based on these backbones; plasmid maps and construction fragments are provided in the Supplemental Material. For all dual-luciferase assays, the control plasmid was firefly pGL4.50 vector (E1310, Promega), and contained transfection carrier plasmid DNA (E4881, Promega). All plasmids were transformed in DH5α Escherichia coli cells (C2987, NEB), grown in LB medium supplemented with 100 µg/mL of ampicillin for 24 h, extracted, purified (PureYield Midiprep; A2495, Promega), and stored at −20°C. Plasmid quality was confirmed by linearization and agarose gel electrophoresis and quantified by NanoDrop; full-plasmid sequencing was also performed.
Cell culture and plating
HEK-293T/17 cells were cultured in Dulbecco's Modified Eagle Medium (DMEM) supplemented with 10% fetal bovine serum (FBS) at 37°C in a 5% CO2 atmosphere. Cell stocks (UNC Tissue Culture Facility) were stored at −80°C in a medium containing 10% FBS and 10% DMSO. Cultures were passaged at 80%–90% confluency every 36–60 h and kept below 20 passage cycles. New stocks were passaged at least three times before use. Biological replicates were performed on different days using distinct cell cultures or the same culture separated by at least two generations. For dual-luciferase assays or RT-qPCR experiments, cells at 80% confluency in a 10 cm dish were washed twice with DPBS, trypsinized (TrypLE Express; Gibco), resuspended in DMEM with 10% FBS, and homogenized by gentle pipetting. Cell counts were performed using Trypan Blue on a Countess 3 automated cell counter, to confirm live cell counts above 95%. The suspension was diluted to 100,000 cells per mL and seeded into plates using 100 µL per well for 96-well plates or 1 mL per well for 12-well plates. The resulting plates were incubated for 24 h at 37°C in a 5% CO2 atmosphere before transfection.
Three-plasmid transfection and dual-luciferase assays
The dual-luciferase assay followed a recent protocol established by our team (Hatfield et al. 2024). No-hairpin, 5′-UTR, and 3′-UTR hairpin reporter plasmids were used at 1, 5, and 2 ng/µL, respectively (in their linear ranges). A plasmid mix was prepared, including 5 ng/µL of firefly luciferase plasmid, the desired reporter plasmid concentration, 0–15 ng/µL MCP-M or MCP-D plasmids, and carrier DNA to achieve a total plasmid concentration of 50 ng/µL. Transfection reagent (FuGENE 6; E2692, Promega) was added to FBS-free DMEM and incubated for 5 min (typically, 1.2 µL FuGENE to 14.8 µL medium per sample). Transfection mixes were prepared by addition of the transfection reagent to the DNA plasmid solution in a 6:1 ratio of FuGENE to DNA (typically, 16 µL of FuGENE and medium were added to 4 µL of plasmid mix; 1.2 µL FuGENE for 0.2 µg DNA). The resulting solutions were incubated for 15–20 min. For treatments in 96-well plates, 5 µL of the FuGENE:DNA solution (50 ng DNA; final concentration of ∼0.5 ng/µL) was added per seeded well. For each condition, transfections were performed in technical triplicate, and plates were incubated for 48 h at 37°C in 5% CO2. For the dual-luciferase luminescence readout (Nano-Glo Dual-Luciferase Reporter Assay System N1630; Promega), reagents and buffers were thawed at room temperature for 2 h (except NanoLuc reagent was kept at −20°C until use). Next, 48 h after transfection, plates were equilibrated to room temperature, and 50 µL per well was carefully removed. Firefly luciferase reagent and buffer were mixed, 50 µL was added to each well, and the plate was incubated for 3 min with shaking before measuring luminescence. Then, NanoLuc reagent and buffer were mixed in a 1:100 ratio, and 50 µL was added to each well of the same plate. The plate was incubated for 3 min with shaking and 7 min without before luminescence measurements. Luminescence was measured on a BMG Labtech CLARIOStar Plus plate reader. Results were averaged over three technical replicates, background-subtracted, and then the NanoLuc/firefly luminescence ratio (NL/FF) was calculated for each condition on the plate (Hatfield et al. 2024).
Quantification of mRNA levels
HEK293T cells were seeded in 12-well plates and incubated for 24 h before transfection with 500 ng of DNA per well, as outlined above. RNA was extracted with TRIzol (15596026, Invitrogen), precipitated with isopropanol, washed with 75% ethanol, treated with DNase (Turbo DNase; Thermo Scientific), and purified by spin column (R1013, Zymo). Reverse transcription was carried out using Superscript II (18064071, Invitrogen) and random hexamer primers (N8080127, Invitrogen). The resulting cDNA was used directly, on the same day, without further purification. qPCR was performed using a SYBR Green/ROX Master Mix Kit (K0221, Thermo Scientific; Applied Biosystems QuantStudio 6 Flex Real-Time PCR System), with primers used within their quantitative ranges. Each reverse transcription sample was analyzed in triplicate qPCR reactions; no-RT samples were used once for each primer set. PCR was performed for 40 cycles, and CT values were collected and analyzed. ΔCT values were calculated by subtracting firefly CT from the average NanoLuc CT, and ΔΔCT values were obtained by subtracting the ΔCT of the reference (NB variant) sample for each condition. Finally, relative mRNA levels were quantified as 2−ΔΔCT, normalizing all treatments to firefly and the reference sample.
SUPPLEMENTAL MATERIAL
Supplemental material is available for this article.
COMPETING INTEREST STATEMENT
K.M.W. is a founder of ForagR Medicines, Ribometrix, and A-Form Solutions.
ACKNOWLEDGMENTS
This work was funded, in part, by a sponsored research agreement with Ribometrix. The study was supported by the National Institutes of Health (NIH, R35 GM122532 to K.M.W. and R21 AG084970 to S.F. and K.M.W.). I.M.N. participated in this project as part of the Undergraduate Transcriptome Project (National Science Foundation [NSF] Award MCB-2027701 to K.M.W.) and was supported by an Undergraduate Packard Research Fellowship.
Footnotes
-
Article is online at http://www.rnajournal.org/cgi/doi/10.1261/rna.080136.124.
- Received June 11, 2024.
- Accepted November 21, 2024.
This article is distributed exclusively by the RNA Society for the first 12 months after the full-issue publication date (see http://rnajournal.cshlp.org/site/misc/terms.xhtml). After 12 months, it is available under a Creative Commons License (Attribution-NonCommercial 4.0 International), as described at http://creativecommons.org/licenses/by-nc/4.0/.








