
A temporal difference in the stabilization of two mRNAs with a 3′ iron-responsive element during iron deficiency
- Department of Pharmacology, Medical School, University of Minnesota, Minneapolis, Minnesota 55455, USA
- Corresponding author: conne018{at}umn.edu
-
↵1 These authors contributed equally to this work.
Abstract
The interactions of iron regulatory proteins (IRPs) with mRNAs containing an iron-responsive element (IRE) maintain cellular iron homeostasis and coordinate it with metabolism and possibly cellular behavior. The mRNA encoding transferrin receptor-1 (TFRC, TfR1), which is a major means of iron importation, has five IREs within its 3′ UTR, and IRP interactions help maintain cytosolic iron through the protection of the TfR1 mRNA from degradation. An IRE within the 3′ UTR of an mRNA splice variant encoding human cell division cycle 14A (CDC14A) has the potential to coordinate the cellular iron status with cellular behavior through a similar IRP-mediated mechanism. However, the stability of the CDC14A splice variant was reported earlier to be unaffected by the cellular iron status, which suggested that the IRE is not functional. We labeled newly synthesized mRNA in HEK293 cells with 5-ethynyl uridine and found that the stability of the CDC14A variant is responsive to iron deprivation, but there are two major differences from the regulation of TfR1 mRNA stability. First, the decay of the CDC14A mRNA does not utilize the Roquin-mediated reaction that acts on the TfR1 mRNA, indicating that there is flexibility in the degradative machinery antagonized by the IRE–IRP interactions. Second, the stabilization of the CDC14A mRNA is delayed relative to the TfR1 mRNA and does not occur until IRP binding activity has been induced. The result is consistent with a hierarchy of IRP interactions in which the maintenance of cellular iron through the stabilization of the TfR1 mRNA is initially prioritized.
Keywords
INTRODUCTION
Iron regulatory proteins (IRPs) are an essential means of maintaining cellular iron homeostasis and mediating some of the adaptive changes required for survival during the stress resulting from either iron depletion or overload (Anderson et al. 2013; Ghosh et al. 2013; Wilkinson and Pantopoulos 2013; Santos et al. 2020; Bonadonna et al. 2022; Maio et al. 2022). The major homeostatic effects of the two IRPs, IRP-1 (ACO1) and IRP-2 (IREB2), result from their interactions with a subset of mRNAs containing iron-responsive elements (IREs), which are highly conserved bulged hairpin loops. An IRE–IRP interaction near the 5′ end of the mRNA inhibits ribosome assembly and translation. This interaction was initially characterized within the mRNAs encoding both the light chain (FTL) and heavy chain (FTH1) subunits of the iron storage protein ferritin (Hentze et al. 1987). Under iron-rich conditions, the RNA binding of the IRPs is attenuated through either modification or degradation, which relieves the blockage to ferritin translation and ultimately protects the cell from oxidative damage through increased iron storage. Other mammalian mRNAs with a functional canonical 5′ IRE include erythroid aminolevulinate synthase (ALAS2), ferroportin (SLC40A1), hypoxia-inducible factor 2 alpha (EPAS1, HIF2α), and mitochondrial aconitase (ACO2).
An alternative IRP-mediated mechanism was identified for the mRNA encoding the transferrin receptor (TFRC, TfR1), which is a major mechanism for iron importation in proliferating mammalian cells. The TfR1 mRNA contains five IREs within its 3′ UTR, and the IRE–IRP interactions protect the TfR1 mRNA from degradation under iron-poor conditions, facilitating homeostasis through increased iron importation (Casey et al. 1988, 1989). When IRP protection is lost under iron-rich conditions, degradation is mediated by the two paralogs of Roquin (RC3H1 and RC3H2) in several human and mouse cell types (Corral et al. 2021). The Roquins were initially identified as being required for the decay of mRNAs encoding components of the adaptive immune system through their recruitment of deadenylation and decapping complexes (Vinuesa et al. 2005; Glasmacher et al. 2010; Mino et al. 2015), and they have subsequently been implicated with the instability of several other mRNAs (Braun et al. 2018). Although additional iron-responsive degradative activities are likely operating in other cell types or conditions (Bayeva et al. 2012; Yoshinaga et al. 2017; Babu and Muckenthaler 2019), two independent studies do not support a role for two microRNAs previously proposed to mediate the TfR1 mRNA instability (Corral et al. 2019; Shi et al. 2021). The divalent metal transporter 1 (SLC11A2, DMT1) mRNA also has a 3′ IRE that modulates mRNA stability (Qatato et al. 2022), and several additional IREs, including noncanonical structures, have been proposed to further increase the number of mRNAs and processes regulated by the IRPs (Sanchez et al. 2011; Corley et al. 2020).
There are cellular conditions that require IRP interactions to occur preferentially with only a subset of the IRE-containing mRNAs. This is exemplified during terminal erythropoiesis when the demand for high iron availability requires IRP interactions with the 5′ IRE on both ferritin mRNAs to limit storage but not with the 5′ IRE of ALAS2, which encodes the enzyme catalyzing the initial step of heme synthesis (Schranzhofer et al. 2006). The affinity of the IRPs for an IRE is impacted by both the stability of the lower stem and by variations within internal bulge/loops (Henderson et al. 1994; Meehan and Connell 2001; Ke and Theil 2002; Walden et al. 2006; Goforth et al. 2010; Garza et al. 2020). A hierarchy of interactions was proposed in which the 5′ IREs making the highest affinity interactions with the IRPs in vitro would be preferentially bound within the cell, resulting in preferential translational inhibition (Theil and Eisenstein 2000; Goforth et al. 2010). This hierarchy is supported by the translation of the mRNAs with a relatively low-affinity IRE (Aco2, Alas2, and Hif2α) being repressed in vivo to a lesser extent during iron-poor conditions than mRNAs containing a higher affinity IRE (Schranzhofer et al. 2006; Anderson et al. 2013; Garza et al. 2020; Shen et al. 2023).
An IRE within the 3′ UTR of a human cell division cycle 14A (CDC14A) mRNA splice variant (plus-IRE variant) is intriguing as it provides a potential adaptive response through which cellular iron status could be coordinated with cellular behavior (Sanchez et al. 2006). Although the precise function of CDC14A is still unclear, it is a tyrosine phosphatase that controls the exit from stemness in pluripotent cells, modulates actin dynamics, and is essential for both hearing and male fertility (Imtiaz et al. 2018; Partscht et al. 2021; Villarroya-Beltri et al. 2023). The steady-state abundance of the CDC14A plus-IRE variant increases in response to iron depletion, which is consistent with IRP-mediated stabilization analogous to the regulation of the TfR1 mRNA. However, the stability of the plus-IRE variant was reported to be unaffected by the iron status of the growth media under conditions in which the stability of the TfR1 mRNA was responsive (Fig. S3 of Sanchez et al. 2006), which put into question whether the CDC14A IRE was truly functional. We show here that the half-life of the CDC14A plus-IRE variant does in fact increase in response to iron-poor growth conditions, but its iron-responsive degradation differs from the Roquin-mediated decay of the TfR1 mRNA. The stabilization of the CDC14A plus-IRE variant is also delayed relative to the TfR1 mRNA and only occurs after the induction of IRP binding activity in response to prolonged iron depletion. The study extends the proposed hierarchy of IRP interactions to include mRNAs with a 3′ IRE in which the stabilization of the TfR1 mRNA that works to maintain cytosolic iron is initially prioritized.
RESULTS AND DISCUSSION
Steady-state changes to the abundance of the TfR1 and CDC14A mRNAs
The effect of iron availability was initially tested on the regulation of the TfR1 mRNA in HEK293 cells, which is an immortalized human embryonic kidney cell type that was used for the initial characterization of the CDC14A IRE (Sanchez et al. 2006). The steady-state abundance of the TfR1 mRNA, which is the net difference between production and degradation, is ∼10-fold greater with cells grown in the presence of deferoxamine (DFO), an iron chelator, than in the presence of ferric ammonium citrate (FAC), an iron source (Fig. 1A). This ratio of DFO/FAC abundance reflects the iron-responsive range of the TfR1 mRNA within the HEK293 cells and is reached after 6 h of treatment and maintained at 12 h. Although the response to DFO treatment is greater at 12 h than at 6 h, the impact of the FAC treatment is the predominant effect at both time points.
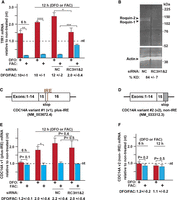
Steady-state changes to TfR1 and the CDC14A mRNAs in response to the iron status of the growth media and a Roquin KD. (A) Changes to the TfR1 mRNA abundance in response to iron-poor (red) or iron-rich (blue) treatments for 6 and 12 h relative to normal growth media (nt). The iron regulation of TfR1 mRNA abundance is attenuated by the Roquin KD in the cells compared to the negative control (NC) siRNA treatment. This is indicated by the ratio of the TfR1 mRNA abundance under DFO and FAC conditions being reduced to two. (B) Western analysis of the Roquin KD efficiency in the HEK293 cells. (C) Schematic of the CDC14A variant #1 exons indicating the location of the IRE within its 3′ UTR. (D) Schematic of the exons of CDC14A variant #2, which does not contain an IRE. (E) Changes to the CDC14A plus-IRE variant in response to the iron status of the growth media and the Roquin KD. (F) Changes to the CDC14A non-IRE variant in response to the iron status of the growth media. The ΔΔCq method was used to quantify changes in the mRNA amplicons relative to an RPL4 reference. Statistical significance was analyzed by two-tailed Student's t-test. (*) P < 0.01; (**) P < 0.001; (***) P < 0.0001; (****) P < 0.00001. All error bars represent ±SEM of three biological replicates.
The siRNA knockdown (KD) of both Roquin isoforms (RC3H1 and RC3H2) decreased the iron-responsiveness of the TfR1 mRNA by ∼80%, as reflected by the DFO/FAC ratio being decreased from 10 to 2 (Fig. 1A). The decreased DFO response by the KD is consistent with IRP protection not being as relevant when the degradation activity is eliminated, and the effect on the FAC response is consistent with less degradation activity even while IRP protection is minimized. The result is consistent with a Roquin KD also decreasing most of the iron-responsive changes to TfR1 mRNA in all other cell types that have been tested (Corral et al. 2021), which includes human HAP1, human umbilical vein endothelial (HUVEC), mouse embryonic fibroblast (MEF), and mouse L–M cells. The extent of the iron-responsive attenuation among the earlier tested cell types correlates well with the extent of the KD at the protein level. The KD in the HEK293 cells reduced the Roquin protein abundance by 84% (Fig. 1B), which is also consistent with the similar loss of iron-responsiveness (Fig. 1A).
The effect of iron availability on the changes to the steady-state level of the CDC14A splice variants with and without an IRE was also investigated (Fig. 1C–F). There are three major differences between the iron-responsiveness of the CDC14A plus-IRE and TfR1 mRNAs (contrast Fig. 1A,E). First, an increased steady-state abundance of the CDC14A plus-IRE variant is not evident after 6 h of growth under iron-poor conditions, but it is after 12 h (Fig. 1E). Second, the steady-state abundance of CDC14A plus-IRE mRNA is not impacted by growth under iron-rich conditions. Third, whereas the majority (80%) of the TfR1-mRNA iron-responsiveness is blunted after KD of Roquin (Fig. 1A), the KD has no impact on the iron-responsiveness of the CDC14A plus-IRE mRNA (Fig. 1E). This is also consistent with the three RNA loops required for Roquin-mediated degradation of the TfR1 mRNA not being present within the CDC14A mRNA (Rupani and Connell 2016; Corral et al. 2021). In contrast to the CDCD14A plus-IRE and TfR1 mRNAs, the steady-state abundance of the CDC14A non-IRE variant does not change in response to the iron status of the cellular growth conditions at either time point (Fig. 1F), which is consistent with the earlier study (Sanchez et al. 2006).
Iron-responsiveness of TfR1 and CDC14A mRNA stability
The half-life of the TfR1 mRNA was measured to test whether the steady-state changes observed in response to iron availability could be attributed to changes in mRNA stability. Newly synthesized mRNA was labeled with 5-ethynyl uridine (EU) in the HEK293 cells, which enabled degradation to be monitored with minimal impact on the transcriptome (Bao et al. 2018). The TfR1 mRNA is stable under growth in normal media and does not significantly differ from the DFO-treated cells until 7.5 h of treatment (Table 1; Fig. 2A). In contrast, transfer of cells to iron-rich conditions resulted in a rapid decrease in mRNA stability with the half-life decreasing to 1.6 h, which could account for the observed steady-state changes (Fig. 1A). Both results are consistent with the TfR1 mRNA being mostly protected from degradation through IRE–IRP interactions under the standard growth conditions.
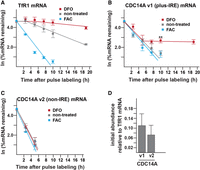
The impact of iron availability on the TfR1 and CDC14A mRNA stability. Cells were treated with either 100 µg/mL FAC or 100 µM DFO following the pulse labeling with the EU as described in Materials and Methods. (A) Impact of the DFO and FAC treatments on the TfR1 mRNA stability. (B) Impact of the DFO and FAC treatments on the stability of the CDC14A plus-IRE variant. The difference between the stability of the variant under iron-poor and iron-rich conditions reaches statistical significance by 10 h of treatment. (C) Impact of the DFO and FAC treatments on the stability of the CDC14A non-IRE variant. (D) Relative abundance of the CDC14A and TfR1 mRNAs immediately following the EU pulse labeling. Statistical significance was analyzed by two-tailed Student's t-test. (**) P = 0.003. All error bars represent ±SEM of three biological replicates except the 5 h time point for the CDC14A non-IRE variant which could only be detected within two sets of the biological replicates. The t1/2 values calculated from the first-order rate constants obtained from the slopes are summarized in Table 1.
mRNA half-lives calculated for the conditions indicated in Figure 2
The stabilities of the CDC14A mRNA splice variants were also measured under the different conditions of iron availability (Fig. 2B,C). In contrast to the TfR1 mRNA, the half-life of the CDC14A plus-IRE mRNA is not altered by the abundance of iron in the HEK293 growth media during the first 5 h of treatment, with the half-life under iron-poor, iron-rich, and normal conditions being ∼2 h (Table 1; Fig. 2B). The failure of the FAC treatment to impact stability is suggestive that there is no significant IRP-mediated protection under normal growth conditions. However, the CDC14A plus-IRE mRNA becomes stable after a 5 h exposure of the cells to DFO, with a significant difference (P = 0.003) from the nontreated cells obtained at 10 h and no further decrease in mRNA abundance detected up to 19 h, the last time point of the assay. The results are consistent with the steady-state measurements in which an increase in the CDC14A plus-IRE RNA in response to DFO is delayed relative to that observed with the TfR1 mRNA (Fig. 1A,E). In contrast, the CDC14A non-IRE variant has a half-life of 1 h independent of the iron status of the growth media which is also consistent with the steady-state measurements (Figs. 1F, 2C); the short half-life in combination with the relatively low starting abundance made it challenging to detect this variant even at the 5 h timepoint (Fig. 2C,D).
The increased stabilization of the CDC14A plus-IRE variant that occurs after prolonged DFO exposure was not detected by an earlier study (Sanchez et al. 2006). The earlier measurement of the mRNA half-life relied on the treatment of the cells with the transcription inhibitor 5,6-dichloro-1-β-d-ribofuranosylbenzimidazole (DRB) to enable stability changes to be distinguished from transcriptional changes. This could have complicated the analysis as the inhibition of Pol II-mediated transcription can have both direct and indirect effects on cellular metabolism and mRNA processing, including some decay processes (Harrold et al. 1991; Sun et al. 2013). In contrast to the CDC14A mRNA, the earlier DRB treatment did not negate the iron-responsive instability of the TfR1 mRNA (Sanchez et al. 2006), which is consistent with the two decay processes being different (Fig. 1A,E).
The time course for increased IRP binding activity in response to iron deprivation
An electromobility shift assay (EMSA) was performed to test whether the increased stability of the CDC14A plus-IRE mRNA during prolonged DFO treatment is consistent with the time course for the induction of IRP binding activity within the HEK293 cells (Fig. 3). A radiolabeled ferritin IRE was used in the EMSA because it is a well-established probe for both cytosolic iron and IRP binding activity (Li et al. 2004; Henderson et al. 2005). A shifted band that formed with the extract prepared from cells grown under iron-poor conditions but not under iron-rich conditions is consistent with an IRE–IRP complex (Leibold and Munro 1988; Rouault et al. 1988). This complex with the radioactive probe is effectively competed by a 20-fold molar excess of unlabeled wild-type IRE (Fig. 3A,B). In contrast, it is not competed by a 20-fold molar excess of the unlabeled mutated IRE (ΔC) that is missing two nucleotides that are critical for IRP binding, which further supports the specificity and identity of the complex. There is only one shifted band consistent with an IRP complex rather than two because the complexes formed with the human IRP-1 and IRP-2 comigrate in the EMSA (Henderson et al. 1993; Guo et al. 1995). The IRP binding activity of the HEK293 cells increases with prolonged DFO treatment up to ∼7.5 h (Fig. 3B,C). This time course for the increased IRP activity is similar to what had initially been described in mouse L–M fibroblast-like cells (Mullner et al. 1989), and it is consistent with the increased stabilization of the CDC14A plus-IRE mRNA that occurs within the same time interval (Fig. 2B).
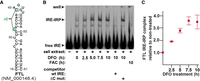
The time course for increased IRP binding activity in response to iron deprivation. (A) Secondary structure of the FTL IRE used for the EMSA and the location of two deletions (ΔC) that inhibit IRP interactions. (B) Representative EMSA indicating the complexes formed between the radiolabeled FTL IRE and the extracts prepared from cells grown under the indicated iron-poor or iron-rich conditions. A 20-fold molar excess of unlabeled wild-type (wt) IRE but not the IRE with the ΔC mutation effectively competes out the interaction. (C) Quantitation of the IRE–IRP complex formed during iron deprivation relative to the nontreated cells. All error bars represent ±SEM of three biological replicates.
A hierarchy of 3′ IRE interactions
The delayed stabilization of CDC14A plus-IRE mRNA relative to the TfR1 mRNA is analogous to an earlier study describing the differential translational regulation of the ferritin H chain mRNA and succinate dehydrogenase subunit b mRNAs from Drosophila melanogaster, both of which have a 5′ IRE (Kohler et al. 1995). Whereas wt IRP-1 only mediated translational repression of the Drosophila ferritin mRNA, an IRP that was genetically altered to be constitutively active also repressed translation of the succinate dehydrogenase subunit b mRNA. Both the Drosophila and HEK293 studies are consistent with IRP binding activity being limiting under normal growth conditions resulting in a hierarchy of IRE interactions.
Although the CDC14A IRE has a high affinity for both IRPs (Sanchez et al. 2006), there are several additional factors that could contribute to preferential interactions with the TfR1 mRNA. The relative concentration of the two mRNAs is likely a significant factor shifting the chemical equilibria toward the formation of specific IRE–IRP complexes, as the TfR1 mRNA is 10-fold more abundant (Fig. 2D), and it has five IREs in contrast to the single CDC14A IRE. The hierarchy of IRE interactions could also be influenced in vivo by differences in the folding of the IREs in the context of the full-length mRNAs, competing protein interactions or different cellular localizations having different access to the IRPs. An RNA-CLIP strategy that was earlier developed to include these possibilities failed to identify the CDC14A IRE, but it may not have had the sensitivity to detect the lower abundant interactions (Connell et al. 2018). It is also possible that direct inhibition of the CDC14A mRNA degradation activity during prolonged iron depletion contributes to the increased mRNA stability, but this cannot be tested until the activity is identified. In the case of the TfR1 mRNA regulation, Roquin protein abundance does not change in response to iron availability (Corral et al. 2021), indicating that the IRPs and Roquins are required as iron-sensing and TfR1 mRNA destabilizing trans-acting factors, respectively.
Conclusion
This study demonstrates that the stability of the CDC14A plus-IRE mRNA is iron-responsive, and it is consistent with the presence of a hierarchy of 3′ IRE interactions in which the stabilization of the TfR1 mRNA is initially prioritized. The CDC14A iron-responsive degradation activity does not appear to involve Roquin, which indicates that there is flexibility in the identity of the degradation activity that can be antagonized by an IRE–IRP interaction on different mRNAs. It is possible that the increased stabilization of the CDC14A plus-IRE mRNA is a means to modulate cellular behavior in response to its iron status, potentially facilitating cellular survival during prolonged exposure to iron-poor conditions. However, determining the function of the stabilization is complicated by the precise role of CDC14A within vertebrates still being unclear (Imtiaz et al. 2018; Partscht et al. 2021; Villarroya-Beltri et al. 2023).
MATERIALS AND METHODS
Cell treatment
HEK293 cells were obtained directly from ATCC (CRL-1573) and maintained in DMEM (Thermo Scientific, 11995-065) plus 10% FBS (Millipore-Sigma, F0926). Cells were plated at a density of 120,000 per 3.5 cm diameter plate. After 24 h, the media was replaced, and cells were either transfected with the appropriate siRNAs or left for an additional 24 h prior to treatment with either 100 µM DFO or 100 µg/mL FAC in fresh media. Transfections were with 20 nM of the appropriate siRNAs using Lipofectamine RNAiMAX (Thermo Scientific) in a final volume of 1.2 mL (Corral et al. 2021). The transfections were repeated 24 h later and left for an additional 30 h prior to treatment with either the 100 µM DFO or 100 µg/mL FAC in fresh media. The siRNAs targeting RC3H1 mRNA (GAUCGAGAGUUACUAUCCA; cat # 4427037, s45244), RC3H2 (CAAGGACUCAGAUACCCUU; cat # 4427037, s29171), and two NCs (cat # 4390843 and 4390846) were obtained from Thermo Scientific. Both Roquin siRNAs had earlier been validated using a CRISPR KO of each target in HAP1 cells (Corral et al. 2021).
RNA purification and RT-qPCR
Total RNA for the RT-PCR was purified from cells using TRI-reagent (Chomczynski and Sacchi 2006), as previously described (Corral et al. 2021). Annealing reactions for the cDNA consisted of 150–200 ng of the total RNA and 90 ng of a random decamer. After denaturation and buffer addition, the samples were equilibrated at 25°C, and 150 units of Superscript III were added to initiate the reverse transcriptions in a final volume of 10 µL. The reverse transcriptions were initiated at 25°C for 10 min prior to transferring to 42°C for an additional 50 min (Corral et al. 2021). The qPCR of the cDNA was performed in a 10 µL volume within 96 well plates, and each reaction contained 200 nM of the appropriate primers (Table 2), and 5 µL SYBR Green Mix (Thermo Scientific, 4309155). Amplification was with a QuantStudio 3 thermal cycler (Thermo Scientific), and the thermal profile consisted of an initial 10 min 95°C denaturation followed by 40 cycles of a 15 sec 95°C denaturation and a 1 min 65°C extension. Minus reverse transcriptase controls consistently indicated <1% genomic DNA contamination. The ΔΔCq method was used to quantify changes in the mRNA amplicons relative to an RPL4 reference.
Oligodeoxynucleotides used for the RT-qPCR
Western analysis
HEK293 cells were plated on 3.5 cm diameter plates and transfected as already described. Cells were briefly rinsed with PBS (4°C) prior to directly scraping into 100 µL lysis buffer (100 mM Hepes pH 7.5, 2 mM MgCl2, 100 mM NaCl, 0.4% deoxycholate, 0.8% NP40, and 0.08% sodium dodecyl sulfate) containing 8 mg/mL protease inhibitors (Thermo Scientific, A32955). The lysate was left on ice for 10 min and then centrifuged at 16,000g for 5 min. The clarified lysate, containing ∼30 µg protein, was mixed with an equal volume of 2× Tris-glycine SDS loading buffer and resolved on a 10% Tris-glycine gel (Corral et al. 2021). Electroblotting onto Immobilon transfer membrane (Millipore-Sigma, IPFL07810) was for 2 h at 30 V in Tris-glycine transfer buffer. Membranes were treated for 1 h with TBS blocking buffer (Li-Cor, 927-50000) prior to incubation with either anti-Roquin-1/2 (3F12) rat monoclonal antibody (Millipore-Sigma, MABF288) or with anti-β-Actin mouse monoclonal antibody (Santa Cruz, sc-47778); both primary antibodies were diluted 1000-fold. Goat anti-rat or anti-mouse secondary antibodies containing an infrared-linked dye were diluted 10,000-fold (Li-Cor, 925-32219, 925-32210). Blots were scanned with an Odyssey (Li-Cor) infrared imager at several intensities to avoid saturation of the actin normalization signal. Quantification was with Image Studio Lite software (Li-Cor).
mRNA half-life measurements
HEK293 cells were plated at 120,000 cells per 3.5 cm diameter plates. Media was replaced after 24 h and incubated for a further 20 h prior to replacement with 1 mL fresh media containing 200 µM EU (Bao et al. 2018). After a 4 h treatment, the media was removed, and the plates were rinsed with fresh media containing 2 mM uridine prior to replacing with this same media containing 100 µg/mL FAC or 100 µM DFO where specified. Total RNA was isolated at the indicated intervals over the following 19 h (Fig. 2), and a biotin group added through click chemistry (Thermo Scientific, C10365). Biotin-labeled RNA was enriched on Streptavidin conjugated magnetic beads, and the beads were resuspended in 14 µL dH2O containing 180 ng of a random decamer and 10 nmol of each dNTP (Corral et al. 2021). The resuspended beads were heated at 65°C for 5 min, briefly cooled on ice and then a mixture containing 4 µL of the Superscript IV 5× buffer, 2 µL of 0.1 M dithiothreitol and 200 units of Superscript IV (Thermo Scientific) was added. The reverse transcription reactions were incubated for 10 min at 25°C and then transferred to a rotation wheel within a 55°C hybridization oven for 1 h. The reactions were terminated, and the cDNA eluted from the magnetic beads through heating at 85°C for 5 min. The qPCR was performed on the released cDNA as described above. Control reactions using RNA isolated from cultures not treated with the EU indicated that there was <1% contamination arising from nonspecific binding of the RNA to the Streptavidin beads.
EMSA
The wt and mutated IREs were transcribed from synthetic oligodeoxynucleotide templates encoding the RNAs described in Figure 3 and purified on a denaturing 8% polyacrylamide gel (Milligan and Uhlenbeck 1989). The specific activity of the radiolabeled RNAs was ∼2 × 104 dpm/pmol. HEK293 cells were plated at 120,000 cells per 3.5 cm diameter plates and treated as indicated. Cells were rinsed briefly with PBS (4°C) and directly scraped into 100 µL EMSA buffer (1 mM DTT, 5% glycerol, 10 mM Hepes pH 7.5, 40 mM KCl, 3 mM MgCl2, 0.2% NP40, and 8 mg/mL protease inhibitors). The lysate was left on ice for 10 min prior to clarification at 15,000g for 5 min. RNA solutions, containing 0.5 µM radiolabeled RNA and 10 µM unlabeled competitor where indicated, were heated at 65°C for 3 min. The binding reactions consisted of 1 µL of the RNA solution and 9 µL of the appropriate cellular extract containing ∼15 µg protein. The RNA binding reactions were incubated for 25 min at room temperature prior to the addition of 1 µL 500 µg/mL heparin and 1 µL 95% glycerol (Corral et al. 2021). Samples were loaded onto a 6% acrylamide/0.074% bis-acrylamide gel (0.75 mm thick), and electrophoresis was in 0.5× TBE (0.045 M Tris-borate, 0.5 mM EDTA) at 10 V/cm for 1 h. Dried gels were scanned using a Typhoon FLA 9500 (GE Healthcare). The density corresponding to the IRE–IRP complexes measured under each condition was normalized to that formed with the extract prepared from the nontreated cells. Quantification was with Image Studio Lite software (Li-Cor).
Statistical analysis
The t1/2 values were calculated from the slope of each line in Figure 2, which corresponds to the negative value of the first-order rate constant, and under this condition t1/2 = ln(2)/k. The R2 for the linear regression was reported as an indicator of the goodness of the fit, and the 95% confidence interval of the slope was used to derive a confidence interval for the t1/2 (Table 1). Statistical significance for all other assays was analyzed by a two-tailed Student's t-test. All assays were performed with three biological replicates unless explicitly indicated otherwise.
ACKNOWLEDGMENTS
This work was supported in part by the University of Minnesota Grant-in-Aid from the Office of the Vice President for Research to G.J.C. We also acknowledge the University of Minnesota Imaging Centers for use of the phosphor scanner.
Footnotes
-
Article is online at http://www.rnajournal.org/cgi/doi/10.1261/rna.079665.123.
- Received March 19, 2023.
- Accepted April 21, 2023.
This article is distributed exclusively by the RNA Society for the first 12 months after the full-issue publication date (see http://rnajournal.cshlp.org/site/misc/terms.xhtml). After 12 months, it is available under a Creative Commons License (Attribution-NonCommercial 4.0 International), as described at http://creativecommons.org/licenses/by-nc/4.0/.








