
Identification of RNA-binding proteins’ direct effects on gene expression via the degradation tag system
- Corresponding author: hu.wenqian{at}mayo.edu
Abstract
RNA-binding proteins (RBPs) are critical regulators of gene expression. An RBP typically binds to multiple mRNAs and modulates their expression. Although loss-of-function experiments on an RBP can infer how it regulates a specific target mRNA, the results are confounded by potential secondary effects due to the attenuation of all other interactions of the target RBP. For example, regarding the interaction between Trim71, an evolutionarily conserved RBP, and Ago2 mRNA, although Trim71 binds to Ago2 mRNA and overexpression of Trim71 represses Ago2 mRNA translation, it is puzzling that AGO2 protein levels are not altered in the Trim71 knockdown/knockout cells. To address this, we adapted the dTAG (degradation tag) system for determining the direct effects of the endogenous Trim71. Specifically, we knocked in the dTAG to the Trim71 locus, enabling inducible rapid Trim71 protein degradation. We observed that following the induction of Trim71 degradation, Ago2 protein levels first increased, confirming the Trim71-mediated repression, and then returned to the original levels after 24 h post-induction, revealing that the secondary effects from the Trim71 knockdown/knockout counteracted its direct effects on Ago2 mRNA. These results highlight a caveat in interpreting the results from loss-of-function studies on RBPs and provide a method to determine the primary effect(s) of RBPs on their target mRNAs.
Keywords
INTRODUCTION
RNA-binding proteins (RBPs) interact with target mRNAs and control their fate (Moore 2005; Glisovic et al. 2008). Over the past decade, transcriptome-wide targets of an increasing number of RBPs have been identified by high throughput biochemical methods, such as CLIP-seq and RIP-seq (Moore 2005; Hafner et al. 2021). These results reveal that many RBPs interact with numerous mRNAs involved in diverse cellular pathways, strongly arguing that RBPs are prevalent regulators of gene expression. Loss-of-function (e.g., knockdown/knockout the target RBP) and gain-of-function (e.g., overexpression of the target RBP) approaches are commonly used to infer how RBPs modulate their target mRNAs. Although these two types of methods provide consistent results for many RBPs, some RBPs behave incongruously in these two types of experiments, resulting in ambiguity in determining how those RBPs regulate their target mRNA expression. One such protein is Trim71.
Trim71 (Lin41 in Caenorhabditis elegans) is an evolutionarily conserved RBP. It is specifically and abundantly expressed in stem cells. During stem cell differentiation, Trim71 is targeted and down-regulated by the conserved prodifferentiation let-7 miRNAs (Grishok et al. 2001; Roush and Slack 2008). Moreover, genetic studies in C. elegans indicated that developmental defects from mutations in the core components of the miRNA pathway could be suppressed by inhibiting Trim71 (Grishok et al. 2001; Bussing et al. 2010). Although it was unknown how Trim71 regulates miRNAs, such as let-7, these observations led to the hypothesis that Trim71 and let-7 miRNAs reciprocally repress each other, forming a double-negative feedback loop that controls stem cell differentiation (Ecsedi and Grosshans 2013). The recent identification of Ago2 mRNA as a functional target of Trim71 supports this model. Specifically, transcriptome-wide identification of Trim71 targets indicated that Trim71 specifically binds to the 3′UTR of Ago2 mRNA in mouse embryonic stem cells (mESCs) (Welte et al. 2019; Liu et al. 2021a). Moreover, while overexpression of either wild-type (WT) Trim71 or a mutant Trim71 with a catalytic defective E3 ligase domain led to decreased Ago2 protein levels, expressing an RNA-binding mutant Trim71 failed to do so. These indicated that Trim71-mediated repression of Ago2 is dependent on RNA-binding but not the potential E3 ligase activity (Liu et al. 2021a). Furthermore, specific disruption of the interaction between Trim71 and Ago2 mRNA in mESCs via removing Trim71's binding site in the 3′UTR of Ago2 mRNA resulted in increased Ago2 protein levels and let-7-dependent stem cell differentiation defects (Liu et al. 2021a). Altogether, these results strongly argue that Ago2 mRNA is repressed by Trim71 in mESCs. However, inconsistent with these findings, it was puzzling to observe that the Ago2 protein levels were not altered in either Trim71 knockdown or Trim71 knockout mESCs (Chang et al. 2012; Liu et al. 2021a).
To reconcile these different results from gain-of-function and loss-of-function studies on Trim71, here, using the dTAG (degradation tag) system, we generate mESCs in which the endogenous Trim71 can be chemically induced to rapid degradation. We observed that upon inducing Trim71 degradation, Ago2 protein levels first increased, confirming the Trim71-mediated repression, and then returned to the original level after 24 h post-induction, revealing that the secondary effects from the Trim71 knockdown/knockout counteracted its direct effects on Ago2 mRNA. These results not only show that Trim71 indeed represses Ago2 mRNA in mESCs, but also highlight a caveat in interpreting the results from loss-of-function studies on RBPs: direct effects versus secondary effects. Moreover, the method we applied on Trim71 can be used to determine the primary effect(s) of other RBPs on their target mRNAs.
RESULTS AND DISCUSSION
Loss-of-function approaches (e.g., knockout/knockdown of the target RBP) are widely used to infer how an RBP regulates its target mRNAs. However, interpreting the results from these experiments is confounded by two caveats. First, an RBP usually binds to hundreds of target mRNAs. Thus, knocking down/knockout of an RBP will disrupt numerous RBP:mRNA interactions, making it challenging to determine whether the impact on a specific mRNA is due to the direct effect of the RBP or indirect effects from other altered RBP:mRNA interactions. Second, there is a time lag between the knockdown/knockout of the RBP and the measurement of target gene expression. For example, in the siRNA/shRNA-mediated knockdown experiments, the expression of the target gene is typically assayed at 24–48 h post-transfection/transduction. Secondary effects could occur during this lag to alter the expression of the mRNA under examination. Similarly, in the knockout approach, the knockout cells may establish compensatory mechanisms to counteract the potential harmful effects from the RBP knockout, confounding the direct effects of the RBP on its target mRNAs.
To address these issues and to determine the immediate effects of an RBP on its target mRNAs, we adapted the dTAG system for the loss-of-function studies of RBPs. In this method, a small contrived protein dTAG (FKBP12-F36V-2HA, which is ∼14 kd in size) is fused in-frame with the target RBP. Thereby, the target RBP can be induced to rapid and specific degradation by a molecular “glue” (dTAG-13), a small chemical molecule that specifically binds to both the dTAG and a ubiquitous endogenous E3 ligase (Fig. 1A; Nabet et al. 2018). We applied this system to Trim71, a highly conserved stem cell-specific RBP. Independent transcriptome-wide binding studies revealed that Trim71 binds to the 3′UTR of Ago2 mRNA in mESCs (Welte et al. 2019; Liu et al. 2021a). Although overexpression of Trim71 leads to translational repression of Ago2 mRNA and decreased Ago2 protein levels, knockdown or knockout of Trim71 does not alter Ago2 protein levels in mESCs (Chang et al. 2012; Liu et al. 2021a). These inconsistent results make it puzzling to determine how the endogenous Trim71 regulates Ago2 mRNA in mESCs.
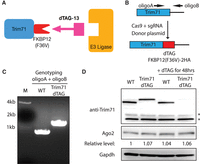
An inducible protein degradation system for Trim71. (A) Schematic depiction of the dTAG system for degrading Trim71. (B) Workflow for knock-in the dTAG degrader to the Trim71 locus. (C) Genotyping of the Trim71-dTAG mESCs using the two primers in (B). (D) Western blotting in the WT and Trim71-dTAG mESCs. Gapdh protein levels were used for normalization in protein quantification. (*) Nonspecific bands.
To clarify Trim71-mediated regulation of Ago2 mRNA, we knocked in the protein dTAG to the carboxyl terminus of Trim71 using genome editing (Fig. 1B) and identified biallelic dTAG knock-in mESCs (Fig. 1C). The knock-in dTAG does not alter the 3′UTR of Trim71 mRNA or Trim71's promoter, and the Trim71-dTAG is expressed at the endogenous level in mESCs (Fig. 1D). Different from the Trim71 knockout mESCs, which have defects in proliferation (Chang et al. 2012), the Trim71-dTAG mESCs proliferated at the same rate as the WT mESCs (Supplemental Fig. S1A). Moreover, the Trim71-dTAG mESCs behaved similarly to the WT mESC in self-renewal and differentiation, as assayed by the colony formation assay and the exit pluripotency assay, respectively (Supplemental Fig. S1B,C). These observations indicated that the Trim71-dTAG mESCs are phenotypically identical to the WT mESCs, and the dTAG does not abolish Trim71's function.
To determine how Trim71 protein degradation influences Ago2 mRNA expression, we examined mESCs cultured with dTAG-13, the molecular “glue” triggering Trim71-dTAG degradation. dTAG-13 induced efficient Trim71 degradation (Fig. 1D). At steady-state, consistent with previous studies of Trim71 knockdown/knockout (Chang et al. 2012; Liu et al. 2021a), the Ago2 protein levels were not altered in the Trim71-dTAG mESCs compared to those in the WT mESCs (Fig. 1D). Next, we performed the kinetic analysis by monitoring protein changes after the dTAG-13 treatment. dTAG-13 induced rapid degradation of Trim71, as at 4 h post-treatment, Trim71 protein levels already reduced substantially (Fig. 2A). Concomitantly, the Ago2 protein levels increased at 4 h and peaked at 8 h post-treatment (Fig. 2A,B). Interestingly, however, the Ago2 protein levels then gradually returned to the original levels despite the dramatically decreased Trim71 levels at later time points (e.g., 16, 24, and 48 h) (Fig. 2A,B). In the WT mESCs, where dTAG-13 did not cause Trim71 degradation, the Ago2 protein levels were not altered by the dTAG-13 treatment over the time course (Fig. 2A,B), indicating that the changes of Ago2 protein levels in the Trim71-dTAG mESCs are due to Trim71 degradation. The increased Ago2 at the early time points (e.g., 4 and 8 h) was not due to the potential inhibition of the global protein degradation system by the dTAG system, because when we applied the same dTAG approach to two other genes that are expressed at higher levels than Trim71, the dTAG-mediated rapid target protein degradation did not lead to increased Ago2 (Supplemental Fig. S2). Consistent with the previous finding that Trim71 represses Ago2 mRNA translation in mESCs (Liu et al. 2021a), Ago2 mRNA levels were not altered during the time course of dTAG-13 triggered Trim71 degradation (Fig. 2C), arguing that the increased Ago2 protein levels at the early time points (e.g., 4 and 8 h) are due to mRNA translational de-repression. Moreover, we performed the polysome profiling on the Trim71-dTAG mESCs harvested at 0, 8, and 24 h post dTAG treatment (Fig. 2D). We observed that the ribosome association of Ago2 mRNA first increased at the 8 h time point and then decreased at the 24 h time point. These changes of ribosome association correlate with the changes in Ago2 protein levels (Fig. 2A,B,D). Altogether, these results confirm that the endogenous Trim71 indeed represses Ago2 expression in mESCs.
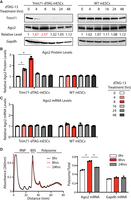
Trim71-mediated repression of Ago2. (A) Western blotting in the WT and Trim71-dTAG mESCs at the indicated time points after dTAG-13 (500 nM) treatment. Gapdh protein levels were used for normalization in protein quantification. (B) Quantification of Ago2 protein levels in the WT and Trim71-dTAG mESCs at the indicated time points after dTAG-13 treatment. (C) Quantification of Ago2 mRNA levels in the WT and Trim71-dTAG mESCs at the indicated time points after dTAG-13 treatment. Gapdh mRNA was used for normalization in the quantification. The results represent the means (±SD) of three independent experiments. (D) Polysome analysis of the Trim71-dTAG mESCs with the dTAG-13 (500 nM) treatment at 0, 8, and 24 h. The target mRNA distribution in the polysome fractions was quantified by qRT-PCR. The quantifications from B to D represent the means (±SD) of three independent experiments. (*) P < 0.05 by one-way ANOVA.
In sum, we show that under a loss-of-function setting, Ago2 protein levels increase upon Trim71 degradation in mESCs. Our kinetic analysis upon rapid Trim71 degradation provides insights into the previous puzzling observations that knockdown/knockout of Trim71 under steady-state does not alter Ago2 levels, even though Trim71 binds to Ago2 mRNA and overexpressed Trim71 reduced Ago2 protein levels in mESCs. The different results from the kinetic analysis upon Trim71 degradation and the steady-state analysis in the Trim71 knockdown/knockout highlight a caveat in interpreting results from the traditional loss-of-function studies on RBPs: discriminating the primary effect versus the secondary effects. The dynamic changes of Ago2 levels (first increase and then go back to normal levels) upon Trim71 degradation (Fig. 2A) strongly argue that in this case (Trim71-mediated regulation of Ago2 mRNA in mESCs), the secondary effects mask the direct effects at the steady-state. Characterizing the specific secondary effects that counteract the direct effects is difficult because numerous RBP:mRNA interactions are disrupted in the loss-of-function approach. However, the time course analysis can discriminate the direct effects from the secondary effects, as the direct effects occur first and then come the secondary effects. Thus, the dTAG approach we applied on Trim71 can be used to determine the direct effects of other RBPs on their target mRNAs.
MATERIALS AND METHODS
Cell culture
E14 mESCs (ES-E14TG2a, ATCC CRL-1821) were used in this study. The cells were cultured in the 2i + Lif medium (DMEM/F-12 supplemented with 2% FBS, 1× nonessential amino acids, 1% penicillin-streptomycin, 2 mM glutamine, 1000 U/mL mLif, 1× N2N27, 0.1 mM β-mercaptoethanol, 3 µM GSK3 inhibitor CHIR99021, and 1 µM MEK inhibitor PD0325901) at 37°C with 5% CO2. The dTAG-13 (Cat. #: 6605, Toris) was used at 500 nmol/mL to induce the target protein degradation.
CRISPR/Cas9-mediated genome editing in mESCs
The sgRNA (GAAGACACAATTAGAAGATG) and the donor plasmid (pT-oWH5986) were cotransfected into mESCs using Fugene6 for dTAG knock-in to the carboxyl terminus of Trim71. At 48 h post-transfection, mESCs were single-cell sorted into each well on 96-well plates, and the resulting clones were then genotyped by PCR and verified by Sanger sequencing.
Western blot analysis
RIPA buffer was used to lysis mESCs, and the protein in the resulting lysate was quantified by using a Pierce 660 nm Protein Assay Kit. Total proteins were resolved by a 10% SDS–PAGE gel and transferred to a PVDF membrane, followed by western blot analysis using anti-Trim71 antibody (Cat. # AF5104, R&D Systems), anti-Ago2 antibody (Cat. # 2896S, Cell Signaling Technology), and anti-Gapdh antibody (Cat. # sc-47724, Santa Cruz Biotechnology).
RT-qPCR
Total RNA was isolated using the TRIzol reagent, followed by DNase1 treatment, phenol extraction and isopropanol precipitation. The cDNA was generated by Superscript3 (Invitrogen) using random primers, and the SsoAdvanced Universal SYBR Green Supermix (Bio-Rad) was used for qPCR on a CFX96 Real-Time-PCR-System (Bio-Rad).
Colony formation assay, exit pluripotency assay, and cell proliferation
The functional assays for mESCs were performed using the methods described previously (Liu et al. 2021a,b).
Polysome analysis
Eight OD260 cell lysates from each sample were resolved on a 5%–50% (w/v) linear sucrose-density gradient by centrifugation at 39,000 rpm in a Beckman SW-41Ti rotor for 2 h at 4°C. The gradient was then unloaded and fractionated using a Gradient Station (BioComp) coupled with an ultraviolet 254 nm detector (Bio-Rad EM-1).
Statistical analysis
Statistical analysis was performed using the Prism software (version 9.5.1).
SUPPLEMENTAL MATERIAL
Supplemental material is available for this article.
ACKNOWLEDGMENTS
This work is supported by the National Institutes of Health (NIH) grant R01GM136869 and the Mayo Foundation for Medical Education and Research.
Author contributions: W.H. conceived the project and supervised the study. K.W., Q.L., K.R.M., and W.H. performed experiments and interpreted the data. W.H. wrote the manuscript with input from all the authors.
Footnotes
-
Article is online at http://www.rnajournal.org/cgi/doi/10.1261/rna.079669.123.
- Received March 24, 2023.
- Accepted June 22, 2023.
This article is distributed exclusively by the RNA Society for the first 12 months after the full-issue publication date (see http://rnajournal.cshlp.org/site/misc/terms.xhtml). After 12 months, it is available under a Creative Commons License (Attribution-NonCommercial 4.0 International), as described at http://creativecommons.org/licenses/by-nc/4.0/.








