
iSnoDi-LSGT: identifying snoRNA-disease associations based on local similarity constraints and global topological constraints
- Wenxiang Zhang1 and
- Bin Liu1,2
- 1School of Computer Science and Technology, Beijing Institute of Technology, Beijing 100081, China
- 2Advanced Research Institute of Multidisciplinary Science, Beijing Institute of Technology, Beijing 100081, China
- Corresponding author: bliu{at}bliulab.net
Abstract
Growing evidence proves that small nucleolar RNAs (snoRNAs) have important functions in various biological processes, the malfunction of which leads to the emergence and development of complex diseases. However, identifying snoRNA-disease associations is an ongoing challenging task due to the considerable time- and money-consuming biological experiments. Therefore, it is urgent to design efficient and economical methods for the identification of snoRNA-disease associations. In this regard, we propose a computational method named iSnoDi-LSGT, which utilizes snoRNA sequence similarity and disease similarity as local similarity constraints. The iSnoDi-LSGT predictor further employs network embedding technology to extract topological features of snoRNAs and diseases, based on which snoRNA topological similarity and disease topological similarity are calculated as global topological constraints. To the best of our knowledge, the iSnoDi-LSGT is the first computational method for snoRNA-disease association identification. The experimental results indicate that the iSnoDi-LSGT predictor can effectively predict unknown snoRNA-disease associations. The web server of the iSnoDi-LSGT predictor is freely available at http://bliulab.net/iSnoDi-LSGT.
Keywords
- snoRNA-disease association identification
- network embedding technology
- local similarity constraint
- global topological constraint
INTRODUCTION
Small nucleolar RNAs (snoRNAs) are a type of small noncoding RNAs that are 60–300 nt in length (Esteller 2011; Wang et al. 2021b). According to snoRNA structures, snoRNAs are mainly divided into two categories: C/D box snoRNAs and H/ACA box snoRNAs (Maxwell and Fournier 1995; Balakin et al. 1996; Kiss 2002). SnoRNAs are associated with specific proteins to form small nucleolar ribonucleoprotein (snoRNP) complexes, which participate in various biological processes (Watkins et al. 2004; Krastev et al. 2011; Bragantini et al. 2021; Wang et al. 2021b). With the development of computer technology and experimental approaches, more and more snoRNAs are identified and their functions revealed, such as gene expression, RNA synthesis, transport, and nucleotide modifications, etc. (Lowe and Eddy 1999; Huttenhofer et al. 2001; Zhao et al. 2017; Dong et al. 2020; Hasler et al. 2020, 2021; Yang et al. 2020; Deryusheva et al. 2021; Shi et al. 2021; Trucks et al. 2021).
In the process of revealing the functions of snoRNAs, more and more evidence indicated that snoRNAs play crucial roles in the emergence and spread of diseases (Esteller 2011; Williams and Farzaneh 2012; Jiang et al. 2013; Gong et al. 2017). For example, Okugawa et al. systematically analyzed the role of snoRNAs in colorectal cancer and found that SNORA42 can serve as a prognostic biomarker in patients with colorectal cancer (Okugawa et al. 2017; Dong et al. 2020). SNORA23 can promote SYNE2 overexpression to influence pancreatic ductal adenocarcinoma cell survival and invasion (Cui et al. 2017). Cui et al. revealed that specific H/ACA box snoRNAs and the corresponding snoRNPs play an oncogenic role in non-small cell lung cancer (NSCLC), providing a new perspective of the biomarkers and therapeutic targets of NSCLC (Cui et al. 2021).
Various snoRNA databases were constructed to reveal the functions of snoRNAs. For example, the snoRNA-LBME-db database collected numerous C/D box snoRNAs and H/ACA box snoRNAs (Lestrade and Weber 2006). The SnOPY database mainly provided many types of snoRNA gene locus and major targets (Yoshihama et al. 2013). The SNORic database investigated the snoRNA expression profiles in 31 cancers and found that snoRNAs are highly expressed in various cancers (Gong et al. 2017). The snoatlas database listed the genomic loci for snoRNA and snoRNA-like genes (Jorjani et al. 2016). The snoDB database integrated various human snoRNA data from established databases (Bouchard-Bourelle et al. 2020). Although these databases revealed the functions of snoRNAs in detail, the research on snoRNA-disease associations was still in its infancy. The main reason is that current studies of snoRNA-disease associations are mainly based on biological experiments. In recent years, the MNDR (Mammalian ncRNA-Disease Repository) v3.0 database integrated considerable experimentally supported and predicted ncRNA-disease associations (Ning et al. 2021), such as miRNA-, lncRNA-, circRNA-, piRNA-, and snoRNA-disease associations. If the snoRNA was proved to cause the corresponding disease reported in literatures, such as high-expression in cancer tissue (Wu et al. 2018; Tian et al. 2021), down-regulating in cancer tissue (Xia et al. 2020), deteriorating cancer by regulating pathway (Zhu et al. 2019), etc., the snoRNA was considered to be associated with the diseases, which will be collected into the MNDR v3.0 database as a snoRNA-disease association. This definition of RNA-disease association is also adopted for constructing the miRTarBase (Chou et al. 2016) and the RAID v2.0 database (Yi et al. 2017). Therefore, the MNDR v3.0 database provided an opportunity to efficiently identify disease-related snoRNAs by computational methods. In recent years, some computational methods have been proposed to identify unknown RNA and disease associations, such as miRNA-disease associations (Chen et al. 2018; Tang et al. 2018; Huang et al. 2019), lncRNA-disease associations (Lu et al. 2020; Zeng et al. 2020a; Zhang et al. 2021) and circRNA-disease associations (Wang et al. 2020; Zeng et al. 2020b; Lei et al. 2021; Wang et al. 2021a; Yang and Lei 2021). These methods demonstrated that it is efficient for identifying RNA-disease associations by computational methods.
In this study, a computational method named iSnoDi-LSGT is proposed to identify unknown snoRNA-disease associations via local similarity constraints and global topological constraints. The iSnoDi-LSGT predictor has the following advantages: (i) To the best of our knowledge, the iSnoDi-LSGT predictor is the first computational predictor for snoRNA-disease association identification. (ii) The local similarity constraint, global topological constraint, snoRNA and disease global topological features are incorporated into iSnoDi-LSGT predictor to accurately identify the snoRNA-disease associations. (iii) A web server of iSnoDi-LSGT predictor has been constructed at http://bliulab.net/iSnoDi-LSGT.
RESULTS AND DISCUSSION
Evaluation criteria
To comprehensively analyze the performance of iSnoDi-LSGT predictor, 10-fold cross-validation is employed to optimize its parameters on Sbenchmark. Sindependent is employed to evaluate the performance of iSnoDi-LSGT predictor. The area under the receiver operating characteristics curve (AUC) and area under the precision-recall curve (AUPR) are employed to analyze the performance of iSnoDi-LSGT predictor for predicting potential snoRNA-disease associations.
The impact of parameters on the predictive performance of iSnoDi-LSGT predictor
There are four parameters r, α, β, and γ in matrix factorization (see Equation 11). r represents the subspace dimensionality of a latent feature matrix. α, β, and γ are three regularization parameters, where
β and γ adjust local the similarity constraint and global topological constraint, respectively. Therefore, we mainly analyze
the impact of β and γ on the predictive performance, and the values of r and α are assigned as 70 and 2 × 10−3, respectively, following Wei and Liu (2020). The parameters of node2vec are set as default values following Grover and Leskovec (2016). The final parameter combinations of β and γ are optimized in terms of the AUC score via 10-fold cross-validation on the
Sbenchmark data set in the following ranges:
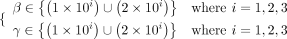
The results are listed in Figure 1, from which the results achieved by different iSnoDi-LSGT predictors based on different parameters are not obviously different, indicating that iSnoDi-LSGT is insensitive with parameters. Finally, iSnoDi-LSGT predictor achieves the best performance when β = 1 × 10−2 and γ = 1 × 10−2.
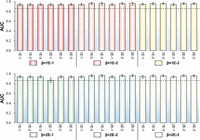
The influence of parameters on the performance of iSnoDi-LSGT predictor via 10-fold cross-validation on Sbenchmark. Different bar charts denote different AUC values based on all predicted results via 10-fold cross-validation on Sbenchmark. X-axis, y-axis, and different color bars represent the values of γ, AUC, and β, respectively. Error bars represent deviation across the different 10-fold cross-validation on Sbenchmark. The black dotted line denotes a horizontal line at the level of the minimum value.
The influence of local similarity constraint and global topological constraint
In this section, we evaluate the influence of local similarity constraint and global topological constraint on the identification of snoRNA-diseases associations. Their influences on the performance of iSnoDi-LSGT for predicting snoRNA-disease associations are listed in Table 1, from which we can see the following: (i) iSnoDi-GT predictor is better than iSnoDi-LS predictor in terms of AUC and AUPR. The reason is that iSnoDi-LS predictor uses snoRNA sequence similarity and disease semantic similarity to constrain matrix factorization, treating snoRNAs and diseases as separate objects. However, snoRNA topological similarity and disease topological similarity used in iSnoDi-GT predictor are calculated by network embedding technology based on heterogeneous network, where snoRNAs and diseases are connected together by snoRNA-disease associations. Therefore, they contain more prior information. (ii) iSnoDi-LSGT predictor achieves the best performance compared with iSnoDi-LS predictor and iSnoDi-GT predictor. The main reason is that iSnoDi-LSGT predictor considers both the local information and the global topological similarity.
The effect of local similarity constraint and global topological constraint for identifying snoRNA-disease associations on Sindependenta
Case study for detecting potential snoRNA-disease associations
To illustrate the capability of iSnoDi-LSGT predictor for identifying potential snoRNA-disease associations, it is further evaluated for identifying potential associated diseases for the query snoRNA SNORD33 (NCBI: 26818) based on Sall. Table 2 lists the top 10 diseases with the highest predicted scores for SNORD33 (NCBI: 26818), from which we can see that the top eight predicted results listed in Table 2 are supported by the literature. The sixth predicted associated disease (breast cancer) is supported by very recent literature (Wang et al. 2022), which is even not recorded in the MNDR v3.0 database. Therefore, the proposed iSnoDi-LSGT predictor is useful for identifying snoRNA-disease associations.
The top 10 diseases associated with SNORD33 (NCBI: 26818) predicted by iSnoDi-LSGT predictor
Conclusion
In this study, we propose the first computational predictor iSnoDi-LSGT for snoRNA-disease association identification with the following advantages: (i) It provides a novel insight into the identification of snoRNA-disease associations by nonnegative matrix factorization. (ii) iSnoDi-LSGT predictor combines local similarity constraint and global topological constraint to effectively identify potential snoRNA-disease associations. (iii) iSnoDi-LSGT predictor can serve as a powerful tool for snoRNAs and disease association prediction. (iv) The corresponding web server of iSnoDi-LSGT predictor can be accessed at http:// bliulab.net/iSnoDi-LSGT/.
MATERIALS AND METHODS
Data sets
A stable and reliable data set is essential for training a model to identify unknown snoRNA-disease associations. Therefore,
a benchmark data set and an independent data set are constructed based on the MNDR v3.0 database (Ning et al. 2021), only containing the experimentally supported snoRNA-disease associations without the predicted snoRNA-disease associations.
The snoRNA sequences are extracted from the snoDB database (Ning et al. 2021). To ensure data standardization, we extract 571 snoRNAs with the term “NCBI id” from the snoDB database (Bouchard-Bourelle et al. 2020), and we extract associations between 571 snoRNAs and diseases from the MNDR v3.0 database (Ning et al. 2021). To facilitate the calculation of disease semantic similarity, associations with diseases without DOID (https://disease-ontology.org/) (Kibbe et al. 2015) or MeSH terms (http://www.nlm.nih.gov/) are removed. Finally, we extract 722 experimentally validated associations between 60 diseases and 302 snoRNAs. The other
269 snoRNAs are not associated with any disease. The data set Sall is represented as:
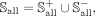 (1)
where
(1)
where 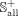 contains 722 experimentally validated positive associations between 302 snoRNAs and 60 diseases,
contains 722 experimentally validated positive associations between 302 snoRNAs and 60 diseases, 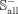 is composed of 16,140 (269 × 60) negative associations between 269 snoRNAs and 60 diseases. To avoid over-fitting, the benchmark
and independent data sets are constructed based on Sall. The benchmark data set is utilized to adjust parameters and train the model. The independent data set is employed to evaluate
the predictive performance of models. Therefore, Sall can be further divided as:
is composed of 16,140 (269 × 60) negative associations between 269 snoRNAs and 60 diseases. To avoid over-fitting, the benchmark
and independent data sets are constructed based on Sall. The benchmark data set is utilized to adjust parameters and train the model. The independent data set is employed to evaluate
the predictive performance of models. Therefore, Sall can be further divided as:
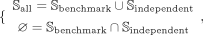 (2)
where Sbenchmark and Sindependent are benchmark data set and independent data set, respectively. They can be represented as:
(2)
where Sbenchmark and Sindependent are benchmark data set and independent data set, respectively. They can be represented as:
 (3)
where
(3)
where 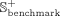 and
and 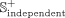 represent positive subsets of the benchmark and independent data sets, respectively. We select 20% of positive snoRNA-disease
associations as the independent positive subset
represent positive subsets of the benchmark and independent data sets, respectively. We select 20% of positive snoRNA-disease
associations as the independent positive subset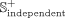 from the
from the 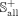 data set, and the remaining known associations are selected as the benchmark positive subset
data set, and the remaining known associations are selected as the benchmark positive subset 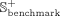 .
. 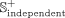 is used to imitate potential unverified snoRNA-disease associations. Similarly,
is used to imitate potential unverified snoRNA-disease associations. Similarly, 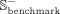 and
and 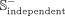 represent negative subsets of the benchmark and independent data sets extracted from
represent negative subsets of the benchmark and independent data sets extracted from 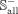 in a ratio of 8:2, respectively. Besides, many snoRNAs have copies resulting from retrotransposition or recombination events,
leading to snoRNAs with similar or identical sequences encoded in the same host gene. To ensure that such redundancy is removed,
no snoRNA in the independent set is encoded in the same host gene as any snoRNA in the benchmark set. All these data sets
can be downloaded from http://bliulab.net/iSnoDi-LSGT/dataset/.
in a ratio of 8:2, respectively. Besides, many snoRNAs have copies resulting from retrotransposition or recombination events,
leading to snoRNAs with similar or identical sequences encoded in the same host gene. To ensure that such redundancy is removed,
no snoRNA in the independent set is encoded in the same host gene as any snoRNA in the benchmark set. All these data sets
can be downloaded from http://bliulab.net/iSnoDi-LSGT/dataset/.
Method overview
The framework of the iSnoDi-LSGT predictor is shown in Figure 2 with four main steps, including local similarity constraint, global topological constraint, nonnegative matrix factorization and candidate disease-associated snoRNA ranking. These steps will be introduced in the following sections.
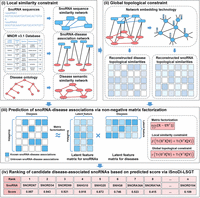
The framework of iSnoDi-LSGT predictor. The four main steps are as follows: (i) Local similarity constraint. The snoRNA sequence similarity network, snoRNA-disease association network and disease semantic similarity network are constructed, in which snoRNA sequence similarity network and disease semantic similarity network are used as local similarity constraint. (ii) Global topological constraint. Disease topological similarity and snoRNA topological similarity are calculated as global topological constraint based on network embedding technology and heterogeneous network constructed by snoRNAs, diseases and their associations. (iii) Nonnegative matrix factorization. Nonnegative matrix factorization with local similarity constraint and global topological constraint is employed to identify potential snoRNA-disease associations. (iv) Candidate disease-associated snoRNA ranking. The candidate disease-associated snoRNAs are ranked according to the scores calculated by nonnegative matrix factorization.
Problem formulation
Let 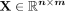 be the snoRNA-disease association matrix, where n and m represent the number of snoRNAs and diseases, respectively. If a snoRNA si is associated with a disease dj recorded in
be the snoRNA-disease association matrix, where n and m represent the number of snoRNAs and diseases, respectively. If a snoRNA si is associated with a disease dj recorded in 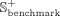 ,
, 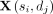 is equal to 1, otherwise 0.
is equal to 1, otherwise 0.
Local similarity constraint
The snoRNA sequence similarity network and the disease semantic similarity network are used as local similarity constraints. The following sections will introduce how to construct the snoRNA sequence similarity network and the disease similarity network.
SnoRNA sequence similarity
In this study, snoRNA sequences are downloaded from the snoDB database (Bouchard-Bourelle et al. 2020). The pseudo K-tuple nucleotide composition (PseKNC) feature (Chen et al. 2015) is extracted by using the Pse-in-One2.0 web server with default values (Liu et al. 2017). SnoRNA sequence similarity is calculated by:
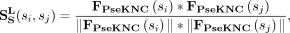 (4)
where
(4)
where 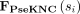 represents the PseKCN feature, and the similarity between snoRNA si and snoRNA sj is represented as
represents the PseKCN feature, and the similarity between snoRNA si and snoRNA sj is represented as 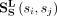 .
.
Disease semantic similarity
Because the snoRNAs have complex biology and genomic contexts (Bouchard-Bourelle et al. 2020), it is insufficient to identify snoRNA-disease associations only based on snoRNA sequence similarities. Therefore, we incorporate the disease semantic similarity into iSnoDi-LSGT for snoRNA-disease association identification. Because the structures of the directed acyclic graph (DAG) can effectively describe the semantic values of diseases (Wang et al. 2007; Li et al. 2019; Li et al. 2021), we employ two types of DAG from the Disease Ontology database (Kibbe et al. 2015) and the MeSH database (http://www.nlm.nih.gov/). In DAG, nodes and edges represent different disease terms and associations between them, respectively, based on which disease semantic similarity can be calculated as (Li et al. 2019):
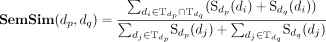 (5)
(5)
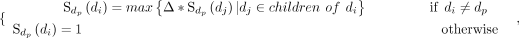 (6)
where the semantic similarity between disease dp and dq is denoted as
(6)
where the semantic similarity between disease dp and dq is denoted as 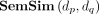 .
. 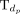 is the set of disease terms including all ancestor nodes of dp and itself.
is the set of disease terms including all ancestor nodes of dp and itself. 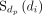 represents the semantic contribution value of disease term di to disease term dp. Δ is the semantic contribution factor. As an effective disease semantic similarity calculation method, it has been successfully
applied to the identification of the other unknown disease-associated RNAs, such as miRNAs (Chen et al. 2018; Yu et al. 2018; Huang et al. 2019), lncRNAs (Lu et al. 2020; Wei et al. 2021) and circRNAs (Wei and Liu 2020; Yang and Lei 2021). Finally, the disease semantic similarity can be calculated as:
represents the semantic contribution value of disease term di to disease term dp. Δ is the semantic contribution factor. As an effective disease semantic similarity calculation method, it has been successfully
applied to the identification of the other unknown disease-associated RNAs, such as miRNAs (Chen et al. 2018; Yu et al. 2018; Huang et al. 2019), lncRNAs (Lu et al. 2020; Wei et al. 2021) and circRNAs (Wei and Liu 2020; Yang and Lei 2021). Finally, the disease semantic similarity can be calculated as:
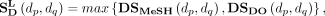 (7)
where
(7)
where 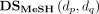 and
and 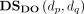 are disease semantic similarities calculated by Equation 5 based on DAG from the MeSH database and the Disease Ontology database, respectively.
are disease semantic similarities calculated by Equation 5 based on DAG from the MeSH database and the Disease Ontology database, respectively.
Global topological constraint
Based on snoRNAs, diseases and their associations, a heterogeneous network is constructed to extract global features by network embedding technology. SnoRNA topological similarity and disease topological similarity are constructed as global topological constraints based on extracted features. The detailed processes are as follows:
Heterogeneous network
Based on the snoRNA sequence similarity network, the disease semantic similarity network and the snoRNA-disease association network, a heterogeneous network can be constructed as:
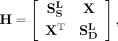 (8)
where
(8)
where 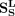 ,
, 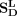 , and X are the adjacency matrix of snoRNA sequence similarity, disease semantic similarity and snoRNA-disease associations,
respectively.
, and X are the adjacency matrix of snoRNA sequence similarity, disease semantic similarity and snoRNA-disease associations,
respectively. 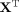 is the transpose of X.
is the transpose of X.
Network embedding technology
Network embedding is an effective feature extraction method for each node of a given network (Goyal and Ferrara 2017; Zhai et al. 2020; Zhang et al. 2020; Li et al. 2021), and it can incorporate the global topological information of a network into a low-dimensional feature space, which has
been successfully applied to various tasks, such as network compression, node classification and link prediction (Goyal and Ferrara 2017). In this study, a network embedding technology called node2vec (Grover and Leskovec 2016) is employed to learn global topological features of snoRNAs and diseases based on constructed heterogeneous network H. Based
on extracted global topological features for snoRNAs and diseases, snoRNA topological similarity and disease topological similarity
can be represented as:
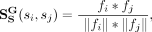 (9)
(9)
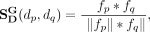 (10)
where
(10)
where 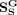 and
and 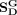 are global topological similarities of snoRNAs and diseases, respectively. fi, fj, fp, and fq are global topological features of snoRNA si, snoRNA sj, disease dp, and disease dq, respectively.
are global topological similarities of snoRNAs and diseases, respectively. fi, fj, fp, and fq are global topological features of snoRNA si, snoRNA sj, disease dp, and disease dq, respectively.
Prediction of snoRNA-disease associations via nonnegative matrix factorization
Nonnegative matrix factorization (Ding et al. 2022) is used to identify potential snoRNA-disease associations, which is efficient for a recommendation task (Hao et al. 2020; Zeng et al. 2020a). Cai et al. proposed a graph regularized nonnegative matrix factorization method, which converts the graph structure into
constraints (Cai et al. 2011). In this study, we incorporate a local similarity constraint and a global topological constraint into matrix factorization,
and the objective function is defined as:
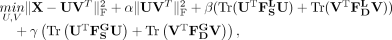 (11)
(11)
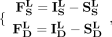 (12)
(12)
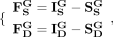 (13)
where
(13)
where 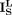 ,
, 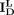 ,
, 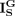 , and
, and 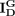 are a diagonal matrix whose corresponding values are the row sum of
are a diagonal matrix whose corresponding values are the row sum of 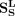 ,
, 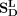 ,
, 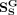 , and
, and 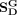 , respectively. α, β, and γ are regularization factors, where the local similarity constraint and the global topological constraint
are adjusted by β and γ, respectively. U and V are the latent feature matrices for snoRNAs and diseases, respectively.
, respectively. α, β, and γ are regularization factors, where the local similarity constraint and the global topological constraint
are adjusted by β and γ, respectively. U and V are the latent feature matrices for snoRNAs and diseases, respectively.
Based on Lagrange multipliers and Karush–Kuhn–Tucker (KKT) conditions (Facchinei et al. 2014), the Lagrange function for Equation 11 can be represented as:
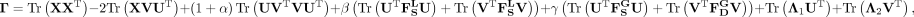 (14)
where
(14)
where 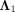 and
and 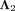 represent the Lagrange multiplier. The partial derivatives of Equation 14 with respect to U and V are:
represent the Lagrange multiplier. The partial derivatives of Equation 14 with respect to U and V are:
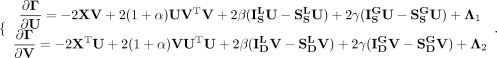 (15)
To confirm
(15)
To confirm 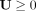 and
and 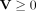 , we make
, we make 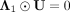 and
and 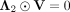 based on KKT conditions (Facchinei et al. 2014). Equation 15 can be represented as:
based on KKT conditions (Facchinei et al. 2014). Equation 15 can be represented as:
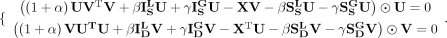 (16)
Therefore, the updating rules for U and V are:
(16)
Therefore, the updating rules for U and V are:
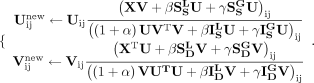 (17)
(17)
Candidate disease-associated snoRNA ranking
Based on Equation 17, the matrix U and V can be updated until convergence. Predicting a score matrix for snoRNA-disease associations can be defined
as 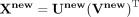 . Candidate disease-associated snoRNAs are ranked based on
. Candidate disease-associated snoRNAs are ranked based on 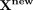 , in which a larger value indicates a higher probability of association between the corresponding snoRNA and disease.
, in which a larger value indicates a higher probability of association between the corresponding snoRNA and disease.
ACKNOWLEDGMENTS
We are very much indebted to the reviewers, whose constructive comments were very helpful for strengthening the presentation of this paper. This work was supported by the National Natural Science Foundation of China (no. 62271049), the National Key R&D Program of China (no. 2018AAA0100100) and the Beijing Natural Science Foundation (no. JQ19019).
Footnotes
-
Article is online at http://www.rnajournal.org/cgi/doi/10.1261/rna.079325.122.
- Received June 22, 2022.
- Accepted September 26, 2022.
This article is distributed exclusively by the RNA Society for the first 12 months after the full-issue publication date (see http://rnajournal.cshlp.org/site/misc/terms.xhtml). After 12 months, it is available under a Creative Commons License (Attribution-NonCommercial 4.0 International), as described at http://creativecommons.org/licenses/by-nc/4.0/.
REFERENCES
MEET THE FIRST AUTHOR

Meet the First Author(s) is a new editorial feature within RNA, in which the first author(s) of research-based papers in each issue have the opportunity to introduce themselves and their work to readers of RNA and the RNA research community. Wenxiang Zhang is the first author of this paper, “iSnoDi-LSGT: identifying snoRNA-disease associations based on local similarity constraints and global topological constraints.” Wenxiang is currently pursuing a PhD degree at the Beijing Institute of Technology, focusing on disease-related noncoding RNA identification.
What are the major results described in your paper and how do they impact this branch of the field?
Three major points are described in the paper. (i) It provides a novel insight into the identification of snoRNA-disease associations by nonnegative matrix factorization. (ii) iSnoDi-LSGT predictor combines the local similarity constraint and global topological constraint to effectively identify potential snoRNA-disease associations. (iii) iSnoDi-LSGT predictor can serve as a powerful tool for snoRNAs and disease association prediction.
What led you to study RNA or this aspect of RNA science?
RNAs have important functions in various biological processes, related to the emergence and development of complex diseases. However, identifying disease-related RNAs is an ongoing challenging task due to the high cost of biological experiments, and as a result, most associations between RNAs and diseases are unknown. Therefore, it is urgent to design efficient computational methods for the identification of snoRNA-disease associations. This led me to study the complex associations between RNAs and diseases so as to improve understanding of the pathogenesis of disease by the use of computational methods.
During the course of these experiments, were there any surprising results or particular difficulties that altered your thinking and subsequent focus?
When this work started, we only considered disease-associated snoRNAs that were contained in the snoRNA-disease association database. However, this was not adequate. Most snoRNAs are not included in snoRNA-disease association database because the effect of these snoRNAs on disease is unknown. Therefore, only focusing on snoRNAs contained in the snoRNA-disease association database is not adequate. In the end, we not only focused on identifying disease-related snoRNAs contained in the snoRNA-disease association database, but also considered the other snoRNAs.
What are some of the landmark moments that provoked your interest in science or your development as a scientist?
When I started studying disease pathogenesis by computational method. I found I could contribute to the research of pathogenic mechanisms.
If you were able to give one piece of advice to your younger self, what would that be?
I would tell my younger self that if you don't give up easily when you want to do something, you will succeed in the end. For example, I encountered many difficulties in the experiments of this paper. I kept on working on it, and I finally succeeded.
Are there specific individuals or groups who have influenced your philosophy or approach to science?
There are many outstanding researchers influencing my philosophy and approach to science, such as my supervisor Professor Dr. Bin Liu.
What are your subsequent near- or long-term career plans?
My near-term career plan is to further study pathogenic mechanisms and obtain my doctoral degree. For my long-term career plan, I want to obtain an academic position as an independent research scientist to explore the challenging tasks in computational biology.








