
Termi-Luc: a versatile assay to monitor full-protein release from ribosomes
- RNA Therapeutics Institute, University of Massachusetts Medical School, Worcester, Massachusetts 01605, USA
- Corresponding author: andrei.korostelev{at}umassmed.edu
Abstract
Termination of protein biosynthesis is an essential step of gene expression, during which a complete functional protein is released from the ribosome. Premature or inefficient termination results in truncated, nonfunctional, or toxic proteins that may cause disease. Indeed, more than 10% of human genetic diseases are caused by nonsense mutations leading to premature termination. Efficient and sensitive approaches are required to study eukaryotic termination mechanisms and to identify potential therapeutics that modulate termination. Canonical radioactivity-based termination assays are complex, report on a short peptide release, and are incompatible with high-throughput screening. Here we describe a robust and simple in vitro assay to study the kinetics of full-protein release. The assay monitors luminescence upon release of nanoluciferase from a mammalian pretermination complex. The assay can be used to record time-progress curves of protein release in a high-throughput format, making it optimal for studying release kinetics and for high-throughput screening for small molecules that modulate the efficiency of termination.
Keywords
INTRODUCTION
Translation termination occurs when the ribosome recognizes a stop codon in the A site. In eukaryotes, stop codons UAA, UGA, and UAG are decoded by release factor eRF1 aided by GTPase eRF3 (Hellen 2018). Termination defines the lengths of all cellular proteins and is therefore a critical step of gene expression. Indeed, many genetic diseases are caused by nonsense mutations, which result in premature release of truncated proteins that can be nonfunctional or toxic (Mort et al. 2008). Termination is regulated by sequence and structure of the mRNA downstream from the stop codon (Brar 2016; Cridge et al. 2018) and by interactions between release factors and other proteins (Kashima et al. 2006; Ivanov et al. 2016; Mikhailova et al. 2017). For example, poly(A)-binding protein (PABP) and the position of a stop codon relative to the poly(A) tail affect the efficiency of termination, and are associated with the severity of nonsense-mutation diseases (Mort et al. 2008; Wu et al. 2019). Mechanistic understanding of termination, including accuracy, efficiency, and dependence on mRNA sequence and protein factors requires a robust quantitative termination assay. Furthermore, assays amenable to high-throughput drug screening are needed to identify drugs that modulate or prevent termination at premature nonsense codons.
In vitro biochemical assays have extensively informed our understanding of bacterial translation termination. For example, kinetic assays have been used to characterize the efficiency of release factors on different codons (Freistroffer et al. 2000; Hoernes et al. 2018), how release factor mutations affect termination (Nakamura and Ito 1998), and how antibiotics inhibit bacterial termination (Youngman et al. 2007; Svidritskiy et al. 2013). They have also revealed that the efficiency of termination depends on the length of the peptidyl moiety attached to tRNA, suggesting that longer peptides are more efficient than single aminoacyl-tRNA (Zavialov et al. 2001; Alkalaeva et al. 2006). Furthermore, full-protein folding at the end of termination might affect the efficiency of release (Goldman et al. 2015). Nevertheless, release of full-length protein has not been extensively studied using canonical in vitro assays in bacterial systems. Eukaryotic in vitro termination assays are even less accessible. A tour de force mammalian in vitro termination assay requires more than a dozen translation factors, which must be purified and assembled into a pretermination complex (Alkalaeva et al. 2006; Eyler and Green 2011; Ng et al. 2018). These assays usually use an mRNA coding for a short peptide (4–6 amino acids) to monitor release of a radioactively labeled peptide (Alkalaeva et al. 2006). Thus, the kinetics of full-length protein release from eukaryotic ribosomes remains unaddressed. Radioactive labeling is a major limitation of termination assays, as it makes kinetic measurements cumbersome and renders the assays unsuitable for high-throughput, small-molecule screening.
We have developed a robust assay to measure the release of full-length protein from mammalian ribosomes. The assay measures release of a 19-kDa nanoluciferase, whose carboxy-terminal tail must fold into the rest of the protein to form an active catalytic domain (Fig. 1A; Dixon et al. 2016). Nanoluciferase has several advantages over the larger firefly luciferase, which is commonly used as a reporter. Compared to firefly luciferase, nanoluciferase luminescence is at least an order of magnitude greater, more stable over time, and less dependent on temperature and pH (England et al. 2016). Finally, nanoluciferase requires a luciferin-like substrate (furimazine) but does not require ATP, which can chelate Mg2+ and may interfere with translation assays and high-throughput screens for small-molecule modulators of translation (Auld and Inglese 2004).
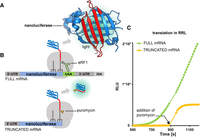
The schematic of the nanoluciferase pretermination complex. (A) Crystal structure of nanoluciferase. Carboxy-terminal amino acids (129–169; red) likely occupy the polypeptide tunnel of the ribosome in the pretermination complex. (B) mRNA constructs used to monitor nanoluciferase release. (C) Progress of nanoluciferase mRNA translation in rabbit reticulocyte lysate: stop codon containing mRNA is shown in green and nonstop mRNA translation followed by addition of puromycin is shown in yellow (relative luminescence units [RLUs] are on the y-axis).
Our assay can be used to record time-progress curves of protein release by release factors and puromycin, an aminoacyl-tRNA mimic that stimulates protein release by covalently attaching to its carboxyl terminus. Release factor eRF1 induces efficient release, which can be inhibited by mutant release factor or small-molecule inhibitor G418. Finally, we demonstrate that the sensitive readout in a microplate luminescence reader can be used to efficiently monitor the inhibition of release, making the assay amenable to high-throughput screens to identify or optimize therapeutics.
RESULTS AND DISCUSSION
Our assay is based on the premise that the nascent tRNA-bound nanoluciferase is inactive because its carboxy-terminal region—essential for luminescence (Dixon et al. 2016)—is occluded by the ribosomal polypeptide tunnel (Fig. 1A,B; Jha and Komar 2011). We hypothesized that the release of nanoluciferase results in the appearance of luminescence upon fast (millisecond range) folding of nanoluciferase into an active state (Dixon et al. 2016). Indeed, in rabbit reticulocyte lysates, nanoluciferase mRNA with a stop codon produced luminescence, but a nanoluciferase mRNA without a stop codon (to prevent protein release) produced no luminescence (Fig. 1C). This result echoes a study of firefly luciferase, showing that the corresponding mRNA without a stop codon produces no luminescence (Kolb et al. 1994). Adding puromycin to the stalled nonstop translation complexes stimulates luminescence, indicating the release of nanoluciferase (Fig. 1C).
We next optimized the purification of pretermination complexes on an in vitro transcribed full-length nanoluciferase mRNA with a UAA stop codon, β-globin 5′- and 3′-UTRs, and a 30-nt poly(A) tail (Fig. 1B). Translation of mRNA was initiated in the commercial nuclease-treated rabbit reticulocyte lysates using a standard set of reagents (Fig. 2A; see Materials and Methods). To stall translation on the stop codon without releasing the protein, we added catalytically inactive eRF1AGQ (catalytic GGQ motif mutated to AGQ) (Frolova et al. 1999; Brown et al. 2015). The stalled 80S was purified by sucrose gradient fractionation (Fig. 2A; see Materials and Methods), which removes eRF1AGQ (Shao et al. 2016) and results in a pretermination 80S complex with nanoluciferase-tRNA in the P site and a vacant A site containing a stop codon (Fig. 1A).
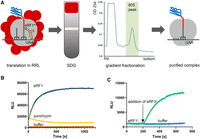
Preparation of the 80S pretermination complex and nanoluciferase release kinetics. (A) Purification of the 80S pretermination nanoluciferase complex stalled on the stop codon in rabbit reticulocyte lysate, using sucrose gradient fractionation. (B) Time-progress curves showing luminescence (in relative luminescence units [RLUs]) of the 80S pretermination nanoluciferase complex upon treatment with recombinant human eRF1 (blue), puromycin (yellow), or buffer (negative control, brown). (C) Time-progress curves showing luminescence (in relative luminescence units [RLUs]) of the 80S pretermination nanoluciferase complex incubated with recombinant yeast eRF1y and treated by yeast eRF3y (green) or buffer (negative control, blue). Exponential fits are shown by black lines. An amount of 20 µL reactions were performed in a microplate reader.
We tested whether adding purified recombinant eRF1 or puromycin to purified pretermination complexes would stimulate release of nanoluciferase and luminescence. The reactions were carried out in 20 µL volumes using a microplate luminescence reader. Indeed, both eRF1 and puromycin stimulated release of nanoluciferase. 0.6 µM human eRF1 released nanoluciferase with an apparent rate constant of ∼0.4 min−1 (obtained by single-exponential fitting; Fig. 2B), similar to rates observed for human eRF1 on pretermination complexes assembled in vitro (Alkalaeva et al. 2006). Puromycin released nanoluciferase faster than eRF1, but the luminescence signal over time remained low (Fig. 2B), suggesting the puromycin adduct may interfere with nanoluciferase activity. Importantly, the pretermination complexes do not spontaneously release nanoluciferase (buffer control; Fig. 2B) in contrast to the complexes with short peptides showing substantial background release that is only a few-fold slower than eRF1-induced release (Alkalaeva et al. 2006). High stability of the nanoluciferase pretermination complex is an excellent property to derive accurate kinetic parameters.
Translation termination in eukaryotes is accelerated by the GTPase protein eRF3 (Zhouravleva et al. 1995; Alkalaeva et al. 2006; Eyler et al. 2013). We therefore asked whether eRF3 synergizes with eRF1 in our nanoluciferase release assay. We used yeast eRF1y and eRF3y, which were shown to function in heterologous eukaryotic systems (Ng et al. 2018). We adjusted eRF1y concentration to 0.01 µM so that nanoluciferase release was negligible (Fig. 2C). Addition of 0.05 µM eRF3y•GTP resulted in fast increase in luminescence, in keeping with efficient protein release by eRF1•eRF3•GTP termination complex (Fig. 2C).
We next tested whether our in vitro release assay is suitable for characterizing termination inhibitors. As an example of a competitive inhibitor, we used the catalytically inactive eRF1AGQ. As expected, release was substantially slower in the presence of equimolar amounts of human eRF1 and eRF1AGQ than eRF1 alone (0.6 µM; Fig. 3A).
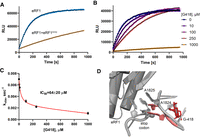
Competitive inhibition of eRF1-induced release of nanoluciferase from purified 80S pretermination complexes. (A) Luminescence in the presence of wild-type eRF1 or equimolar amounts of wild-type eRF1 and catalytically inactive eRF1AGQ (0.6 µM). (B) Luminescence in the presence of wild-type eRF1 and different concentrations of G418. (C) Dependence of catalytic rates (kobs) for eRF1-mediated release of nanoluciferase on G418 concentration. IC50 was determined by hyperbola fitting (red) of kobs values obtained at different concentrations of G418 and 0.6 µM human eRF1. Error bars represent standard deviation of the mean (n = 3). (D) Alignment of G418-bound 80S decoding center (red; Prokhorova et al. 2017) with that of eRF1-bound 80S termination complex carrying a UAA stop codon (gray; Brown et al. 2015) shows steric clash of G418 with A1824.
We then asked whether G418, an aminoglycoside that induces stop codon readthrough (Dabrowski et al. 2018; Wangen and Green 2020), inhibits nanoluciferase release. Stop codon readthrough by G418 and other aminoglycosides could be primarily due to (i) misreading of the stop codon by a near-cognate tRNA (Prokhorova et al. 2017); (ii) inhibition of eRF1; or (iii) a combination of (i) and (ii). On bacterial ribosomes, aminoglycosides both induce miscoding (Davies et al. 1965) and inhibit translation termination (Youngman et al. 2007). But bacterial release factors RF1 and RF2 and their interactions with the stop codons (Korostelev et al. 2008, 2010; Laurberg et al. 2008; Weixlbaumer et al. 2008) substantially differ from those in eukaryotic eRF1-bound termination complexes (Brown et al. 2015; Matheisl et al. 2015). It therefore remains unclear whether G418 efficiently inhibits eukaryotic termination.
We found that G418 inhibited eukaryotic translation termination with an IC50 of 64 ± 20 µM (Fig. 3B,C), similar to that for bacterial termination inhibition by paromomycin (∼35 µM; Youngman et al. 2007). Inhibition of termination is consistent with the structural mechanism of G418 binding to the decoding center of the eukaryotic ribosome (Prokhorova et al. 2017) and inducing the bulged conformations of nucleotides A1824–A1825 (18S rRNA nucleotides of H. sapiens ribosome; corresponding to A1492 and A1493 of E. coli ribosome; Fig. 3D). In this conformation, the adenosines would clash with the U-turn conformation of the stop codon. Furthermore, G418 would clash with A1824 docking within helix 44 upon eRF1 binding (Fig. 3D). These data suggest that modest inhibition of eRF1 may synergize with the ability of G418 to induce decoding of the stop codon by a near-stop tRNA, explaining the readthrough.
Conclusions
We have developed a robust and versatile translation termination assay. The luminescence-based assay is straightforward, more efficient than assays that measure release of radioactive peptides, and suitable for studies of full-protein release on ribosomes from almost any organism. Our assay can be used to study the regulation of translation termination, including how the context of a stop codon—that is, sequences or structures surrounding the stop codon—affects termination. Indeed, nanoluciferase can report on release from mRNAs with and without a 3′-UTR (Figs. 1 and 2), indicating that varying stop codon identities and downstream sequences can be tested. While our manuscript was under review, firefly luciferase was reported to capture the effects of different 3′-UTR sequences and other factors on termination in cell-free translation (Lashkevich et al. 2020), highlighting that distinct luciferase reporters can be harnessed for quantitative termination analyses. Furthermore, sequences upstream of the stop codon can be introduced in our method to study the proposed effects of the ORF on termination (Pierson et al. 2016). The last ∼13 amino acid residues of nanoluciferase are essential for activity, and nanoluciferase retains luminescence upon carboxy-terminal extension (Dixon et al. 2016). Because the polypeptide tunnel can occlude ∼40 amino acids (Jha and Komar 2011), the nanoluciferase carboxyl terminus can be extended by up to 25 varying amino acid sequences to study the effects of the mRNA preceding the stop codon. Lastly, the ability to measure termination in a microplate reader makes the assay suitable for high-throughput screens to identify termination modulators and potential therapeutics for genetic diseases caused by nonsense mutations.
MATERIALS AND METHODS
Plasmids and mRNA constructs
DNA plasmids encoding nanoluciferase constructs were designed as follows. Nanoluciferase sequence from pNL1.1[Nluc] Vector (Promega) was inserted into pUC57(Kan) plasmid (GenScript), flanked by β-globin 5′- and 3′-UTRs. Templates for in vitro transcription were generated using PCR. For FULL mRNA, a pair of primers annealing to the globin UTRs was used: the forward primer (tttttTAATACGACTCACTATAGacacttgcttttgacacaactgtg) containing a T7-promoter and the reverse primer with a 30-nt poly-T-stretch (ttttttttttttttttttttttttttttttGCAATGAAAATAAATTTCCTTTATTAGCC). For the NO-STOP mRNA, a reverse primer annealing to the 3′ end of the nanoluciferase coding sequence was used (CGCCAGAATGCGTTCGCAC). In vitro transcription with recombinant T7 RNA polymerase was done as described (Alkalaeva et al. 2006); transcription products were precipitated with 2.5 M LiCl, washed with 80% ethanol, and dissolved in deionized water. Concentrations of mRNAs were determined using Nanodrop 2000 (Thermo Fisher).
Purification of human eRF1, eRF1AGQ, and yeast eRF1 and eRF3
His-tagged recombinant proteins were expressed in E. coli and purified as described (Frolova et al. 1998, 2000) for human eRF1, eRF1AGQ, and in Eyler et al. (2013) for yeast eRF1 and eRF3.
In vitro translation reaction in RRL
Reaction mixture containing 50% of nuclease-treated rabbit reticulocyte lysate (RRL) (Promega) was supplemented with 30 mM Hepes-KOH (pH = 7.5), 50 mM KOAc, 1.0 mM Mg(OAc)2, 0.2 mM ATP and GTP, 0.04 mM of 20 amino acids (Promega), and 2 mM DTT. Nanoluciferase substrate furimazine (Promega) was added to the mixture at 1%. An amount of 10 µL aliquots of the mixture were placed in a 384-well plate (Corning Low Volume White Round Bottom) and incubated at 30°C for 5 min in a microplate reader (Tecan INFINITE M1000 PRO). Translation reactions were started by addition of mRNA (final concentration 10 ng/µL) and the luminescence signal was recorded. At 900 sec, 1 µL of water (control) or puromycin solution in water were added (100 µM final concentration in the reaction). The resulting data were processed in Microsoft Excel.
Preparation of the pretermination complex and rabbit reticulocyte lysate
Purification of pretermination complexes was done from 1 mL translation mixtures described above. To stall translation progress at the stop codon, reaction mixture was preincubated with 1 µM eRF1AGQ at 30°C for 10 min followed by the addition of FULL mRNA to the final concentration of 8 µg/mL and incubation at 30°C for 25 min. Using this mRNA concentration resulted in predominant formation of monosomic 80S complexes (see representative sucrose gradient profile in Fig. 2A). Then, KoAC concentration was adjusted to 300 mM and the mixture was layered on a 10%–35% linear gradient of sucrose in buffer A (50 mM Hepes-KOH, pH = 7.5, 7.5 mM Mg(OAc)2, 300 mM KOAc, 2 mM DTT), prepared using the Gradient Profiler system (Biocomp). The gradients were centrifuged in SW-41 Ti (Beckman Coulter) rotor at 40,000g for 2.5 h (Beckman Coulter Optima TLX). Gradient Profiler was used to collect fractions corresponding to the monosomic 80S-mRNA ribosomes. The sample was concentrated using Amicon Ultra-15 Centrifugal Filter Units (MilliporeSigma), yielding ∼50 nM ribosome (one A260 unit = 20 nM 80S ribosomes) using Nanodrop 2000 (Thermo Fisher). Ribosome solution was aliquoted, flash-frozen in liquid nitrogen, and stored at −80°C. After 5 mo of storage, the complexes retained ∼70% of the initial signal level (compare Fig. 3A,B).
Kinetic reactions with purified nanoluciferase–ribosome complexes
80S solution was diluted to ∼15 nM with buffer B (50 mM Hepes-KOH, pH = 7.5, 0.25 mM spermidine, 2 mM DTT), supplemented with 1% nanoluciferase substrate (Promega), aliquoted (20 µL), and placed in a 384-well plate. The samples were incubated at 30°C for 5 min in the absence/presence of 0.6 µM eRF1AGQ or 10–1000 µM G418 (Gold Biotechnology) to test inhibition by the eRF1 mutant or G418. Release reaction was started by the addition of 0.6 µM human eRF1, 100 µM puromycin, or 0.01 µM yeast eRF1y followed by the addition of 0.05 µM yeast eRF3y with 0.2 mM GTP, and luminescence was recorded. Buffer C (20 mM Tris-HCI, pH = 7.5, 100 mM KCI, 6 mM 2-Mercaptoethanol, 10% glycerol) or water were used as a negative control. The data were plotted and fitted in GraphPad Prism 8 using single-exponential fits to obtain kobs values, and in Gnuplot using hyperbola fitting to obtain IC50.
Structural analysis and visualization
Structural analysis and visualization were done in PyMol (Schrödinger) using PDB 5IBO for nanoluciferase (S Lovell, in prep.), PDB 5NDG for G418-bound yeast ribosome (Prokhorova et al. 2017), and PDB 3JAG for eRF1-bound mammalian ribosome (Brown et al. 2015). Yeast and mammalian ribosomes were aligned by 18S rRNA. All figures were assembled in Adobe Photoshop.
ACKNOWLEDGMENTS
We thank Darryl Conte Jr., Dmitri N. Ermolenko, and members of the Korostelev laboratory for discussions and comments on the manuscript. This study was supported by National Institutes of Health (NIH) grant R35 GM127094 (to A.A.K.).
Author contributions: D.S. and A.A.K. conceptualized the idea. D.S. and S.E. carried out the methodology and investigation. D.S., S.E., and A.A.K. analyzed the data. A.A.K. provided resources. D.S. and A.A.K. wrote the original draft. All authors contributed to reviewing and editing the manuscript. D.S. provided visualization, A.A.K. the supervision, and A.A.K. the funding acquisition.
Footnotes
-
Article is online at http://www.rnajournal.org/cgi/doi/10.1261/rna.076588.120.
-
Freely available online through the RNA Open Access option.
- Received May 28, 2020.
- Accepted August 11, 2020.
This article, published in RNA, is available under a Creative Commons License (Attribution-NonCommercial 4.0 International), as described at http://creativecommons.org/licenses/by-nc/4.0/.








