
Affinity resins containing enzymatically resistant mRNA cap analogs—a new tool for the analysis of cap-binding proteins
- Sylwia Anna Szczepaniak1,2,3,
- Joanna Zuberek1,
- Edward Darzynkiewicz1,
- Joanna Kufel2,4 and
- Jacek Jemielity1,4
- 1Division of Biophysics, Institute of Experimental Physics, Faculty of Physics, University of Warsaw, 02-089 Warsaw, Poland
- 2Institute of Genetics and Biotechnology, Faculty of Biology, University of Warsaw, 02-106 Warsaw, Poland
- 3College of Inter-Faculty Individual Studies in Mathematics and Natural Sciences, University of Warsaw, 02-089 Warsaw, Poland
Abstract
Cap-binding proteins have been routinely isolated using m7GTP-Sepharose; however, this resin is inefficient for proteins such as DcpS (scavenger decapping enzyme), which interacts not only with the 7-methylguanosine, but also with the second cap base. In addition, DcpS purification may be hindered by the reduced resin capacity due to the ability of DcpS to hydrolyze m7GTP. Here, we report the synthesis of new affinity resins, m7GpCH2pp- and m7GpCH2ppA-Sepharoses, with attached cap analogs resistant to hydrolysis by DcpS. Biochemical tests showed that these matrices, as well as a hydrolyzable m7GpppA-Sepharose, bind recombinant mouse eIF4E(28-217) specifically and at high capacity. In addition, purification of cap-binding proteins from yeast extracts confirmed the presence of all expected cap-binding proteins, including DcpS in the case of m7GpCH2pp- and m7GpCH2ppA-Sepharoses. In contrast, binding studies in vitro demonstrated that recombinant human DcpS efficiently bound only m7GpCH2ppA-Sepharose. Our data prove the applicability of these novel resins, especially m7GpCH2ppA-Sepharose, in biochemical studies such as the isolation and identification of cap-binding proteins from different organisms.
Keywords
INTRODUCTION
Precursors of all RNA classes synthesized by eukaryotic RNA polymerase II (Pol II) are capped at their 5′ ends. The basic cap structure consists of the 7-methylguanosine moiety (m7G) connected to the first nucleoside of the nascent transcript by a 5′–5′ triphosphate bridge, which is atypical for nucleic acids. The role of this structure is to protect RNA from 5′→3′ exonucleases, which are not capable of hydrolyzing pyrophosphate bonds within the 5′–5′ triphosphate bridge. Moreover, cap recognition by specific proteins is essential for several cellular processes, including pre-mRNA splicing, nucleocytoplasmic mRNA export and localization and, finally, translation initiation (Topisirovic et al. 2011). Similarly, the cap structure of snRNA in metazoa plays a role as its localization signal. The m7G cap of an snRNA precursor drives its export to the cytoplasm, whereas a hypermethylated TMG (trimethylguanosine) cap directs snRNP import to the nucleus (Matera et al. 2007).
There are two major cellular cap-binding complexes: the predominantly nuclear cap-binding complex (CBC) and mainly cytoplasmic eukaryotic initiation factor 4F (eIF4F). CBC is a heterodimer composed of Cbp20, which directly binds the cap structure, and Cbp80, which supports this interaction (Izaurralde et al. 1994; Calero et al. 2002; Mazza et al. 2002a). CBC binds the m7G cap immediately following its synthesis and mediates its participation in pre-mRNA processing and nucleocytoplasmic export (Flaherty et al. 1997; Lewis and Izaurralde 1997; Shen et al. 2000). In the cytoplasm, CBC is replaced by eIF4F during the initial round of translation due to the interaction of Cbp80 and the eIF4F subunit, eIF4G (McKendrick et al. 2001; Baron-Benhamou et al. 2003). The cytoplasmic cap-binding protein, eIF4E, plays an essential role in cap-dependent translation initiation and is a crucial target of its regulation (for review, see von der Haar et al. 2004; Richter and Sonenberg 2005).
Cap hydrolysis by specific pyrophosphatases (decapping enzymes) plays a crucial role in mRNA decay, as the 5′→3′ degradation pathway requires the hydrolysis of the pyrophosphate bond. On the other hand, the resulting free cap structure can affect the function of cap-binding complexes by sequestering them from their RNA substrates (Bail and Kiledjian 2008). To circumvent this, two types of decapping enzymes with different substrate specificity hydrolyze the cap structure (Liu and Kiledjian 2006). The Dcp1/Dcp2 complex degrades caps of long deadenylated mRNAs, making them accessible to 5′→3′ exonucleases, while the scavenger decapping pyrophosphatase DcpS acts on the free cap and short-capped RNA molecules resulting from 3′→5′ mRNA degradation by the exosome (Liu et al. 2002). The specificity of the two decapping enzymes toward the cleavage site differs: Dcp1/Dcp2 cleaves between the α and β phosphate groups, resulting in m7GDP and pN-RNA, whereas DcpS hydrolyzes the β–γ pyrophosphate bond to release m7GMP and pp(Np)1–10.
As a key regulator of the free cap structure, and consequently of the pool of available cap-binding proteins, DcpS is considered an important general modulator of cap-dependent processes (Bail and Kiledjian 2008). For example, mammalian DcpS is important for efficient removal of the cap-proximal intron, a process depending on CBC-mediated association of U1 snRNP to the 5′ splice site (Shen et al. 2008). DcpS depletion entails reduced first intron removal by sequestering CBC from the cap structure as a result of an imbalanced cap level. In turn, in Saccharomyces cerevisiae, the lack of DcpS, or its catalytic activity, decreases 5′→3′ mRNA decay at the level of exonucleolytic degradation (Liu and Kiledjian 2005). It has been proposed that either the cap structure or the decapping product serve as a signaling molecule for downstream functions. Recently, hDcpS has been identified as a molecular target of C5-quinazolines, which are potential therapeutics for spinal muscular atrophy (SMA) (Singh et al. 2008). Inhibition of DcpS by these drugs increases the expression of the SMN2 gene, which can complement the defective levels of the survival motor neuron (SMN) protein causing SMA. It has been proposed that the role of DcpS in buffering the active pool of cap-binding proteins is more general and serves as a regulatory feedback mechanism that links the final steps of mRNA decay to earlier events in mRNA biogenesis, namely, mRNA processing, export, and translation initiation (Bail and Kiledjian 2008).
Considering the high specificity of cap recognition, cap-binding proteins can be purified by affinity chromatography using cap-modified resins. The first affinity matrices of this type that used the analogs of m7GDP immobilized by a modified ribose moiety have been used for isolation of eIF4E and associated translation factors, usually from partially fractionated cell lysates (Sonenberg et al. 1979; Altmann et al. 1985; Edery et al. 1988; Ptushkina et al. 1996). The most successful, and up to now the only widely applied approach, utilizes m7GTP-Sepharose, which has been shown to specifically bind both cap-binding complexes (Webb et al. 1984). It has been used for the purification and identification of eIF4E isoforms from different organisms using either endogenous protein extracts or recombinant proteins expressed in bacteria (Browning et al. 1987; Zapata et al. 1994; Jankowska-Anyszka et al. 1998; Ruud et al. 1998; Ramirez et al. 2002). Although eIF4E has a much higher affinity for m7GTP-Sepharose than other cap-binding proteins, this resin also permitted the identification of a cap-specific Nhm1 pyrophosphatase from Schizosaccharomyces pombe, which was found as a contaminant in the eIF4F preparation (Ptushkina et al. 1996; Salehi et al. 2002). Cap-analog affinity resins are also utilized for pull down assays and in vitro binding studies of cap-binding proteins (Mazza et al. 2002b; Kiriakidou et al. 2007; Nojima et al. 2007; Pabis et al. 2010). For example, some new dinucleotide cap analog-modified resins synthesized recently were shown to bind the Caenorhabditis elegans IFE-5 isoform with the capacity comparable to their mononucleotide counterparts (Jankowska-Anyszka and Piecyk 2011).
However, the applications described above are practical mainly for eIF4F or CBC, as the binding of specific pyrophosphatases, such as DcpS, to m7GTP-Sepharose is less efficient, particularly in the presence of the whole-cell extract, where proteins with higher affinities effectively compete out DcpS, thus obstructing its purification (Salehi et al. 2002). More importantly, DcpS can hydrolyze m7GTP, which may significantly reduce resin capacity.
To synthesize an enzymatically stable affinity resin that can be applied for DcpS purification, we have used cap analogs modified with a methylenebisphosphonate moiety in the β/γ position of the 5′–5′ triphosphate bridge, which constitutes the DcpS cleavage site. Analogs of this type were shown to bind human DcpS specifically and to be resistant to hydrolysis by this protein (Kalek et al. 2006). To this end, two nonhydrolyzable cap analogs, mononucleotide m7GpCH2pp or dinucleotide m7GpCH2ppA, were attached directly or via a 1,6-diaminohexane spacer arm, respectively, to the Sepharose. In addition, a hydrolyzable dinucleotide m7GpppA-Sepharose was also synthesized to enhance the affinity of other cap-binding proteins. Binding studies with recombinant proteins and purification of cap-binding proteins from yeast extract showed that the binding capacity of novel affinity resins for known cap-binding proteins was similar to that of standard m7GTP-Sepharose, but only m7GpCH2ppA-Sepharose efficiently bound recombinant or crude yeast extract-derived DcpS.
RESULTS AND DISCUSSION
Chemical synthesis of new affinity resins
We synthesized three novel affinity resins with attached cap analogs (Table 1): m7GpCH2pp-Sepharose (2), m7GpppA-Sepharose (3), and m7GpCH2ppA-Sepharose (4). Enzymatically stable m7GpCH2pp-Sepharose (2) and m7GpCH2ppA-Sepharose (4) are resistant to hydrolysis by DcpS due to methylene group substitution for pyrophosphate oxygen atom closest to the m7G moiety. These two resins, m7GpppA- and m7GpCH2ppA-Sepharoses, are among the first examples of the affinity medium with dinucleotide cap analogs attached to the matrix.
Structures of m7GTP-Sepharose and affinity resins synthesized in this study
The synthesis of m7GpCH2pp-Sepharose (2) (Fig. 1A) was performed using the strategy described in Knorre et al. (1976) and Webb et al. (1984). m7GpCH2pp (8) was obtained using a methodology previously devised by our group (Kalek et al. 2005a, 2006). The bisphosphonate moiety was introduced into the nucleotide by direct phosphonylation of guanosine with methylenebis(phosphonic dichloride) in trimethyl phosphate as the solvent (Kalek et al. 2005b). The resultant GpCH2p (5) was coupled to another phosphate moiety using a two-step procedure. It involved activation of the nucleoside 5′-bisphosphonate with an imidazole exploiting the 2,2′-dithiodipyridine/triphenylphosphine activation system (Mukaiyama and Hashimoto 1972), followed by actual coupling of the obtained imidazole derivative (6) to bis(triethylammonium) phosphate in the presence of ZnCl2 (Kadokura et al. 1997; Stepinski et al. 2001; Jemielity et al. 2003; Kalek et al. 2006). Subsequent methylation of the intermediate 7 with CH3I produced the final cap analog (8). Prior to coupling to Sepharose, m7GpCH2pp (8) was converted into a p-aminophenyl γ-ester derivative (11), following the three-step procedure described in Knorre et al. (1976) and Webb et al. (1984). The first step, activation of 8 by converting it into its tri-n-octylammonium salt and its subsequent cyclization with DCC proceeded easily using the same conditions, but the third reaction, involving the nucleophilic attack of p-nitrophenol on the phosphate group, took place with much slower kinetics than in the case of m7GTP. m7GTP trimetaphosphate hydrolyzes almost immediately in aqueous solutions, but the presence of a methylene group within the triphosphate chain makes the cyclic trimetaphosphate-like intermediate (9) more stable under standard reaction conditions (Trowbridge et al. 1972), which allowed us to monitor the reaction progress using HPLC. The increased stability of intermediate 9 correlates well with its decreased reactivity; therefore, a prolonged incubation time combined with an increased concentration of TEA in the reaction medium were applied to obtain the desired product in a reasonable yield (66% yield by HPLC). Another phenomenon of this reaction is its regioselectivity. There are two possible sites for the nucleophilic attack of p-nitrophenol on either the P2 or P3 phosphorus atom. In theory, attack on the P2 atom, leading to an undesirable by-product, should be preferred with regard to the adjacent methylene group, which makes the P2 atom more electrophilic (Kalek et al. 2005b). Surprisingly, only the required terminal p-nitrophenyl ester formation was observed, meaning that either the attack on P3 is favorable in this case, or the β-ester is labile and converts into the γ-ester (10). The nitro group was subsequently reduced to produce the m7GpCH2pp p-aminophenyl γ-ester (11), which was then coupled to Sepharose 4B using cyanogen bromide according to the protocol developed in Cuatrecasas et al. (1968). Using a similar procedure, we also synthesized Sepharoses substituted with m7GTP (1) and GTP as controls for the biological experiments.
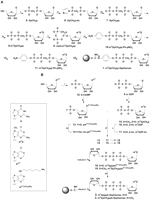
Synthesis of Sepharoses 2–4: (A) Synthesis of m7GpCH2pp-Sepharose: (i) 1. CH2(POCl2)2, (CH3O)3PO, 2. triethylamine bicarbonate (TEAB) aq.; (ii) imidazole, 2,2′-dithiodipiridine, Ph3P, TEA, DMF; (iii) tris(triethylamine) phosphate, ZnCl2, DMF; (iv) CH3I, DMSO; (v) 1. tri-n-octylamine, MeOH, 2. DCC, DMF; (vi) p-nitrophenol, TEA, DMF; (vii) Pd/C, H2, (viii) Sepharose 4B activated witch BrCN, sodium carbonate buffer (pH 9.6). (B) Synthesis of m7GpppA-Sepharose and m7GpCH2ppA-Sepharose: (i) 1. POCl3, (CH3O)3PO, 2. TEA/H2O; (ii) NH2(CH2)6NH2, H2O; (iii, v) imidazole, 2,2′-dithiodipiridine, Ph3P, Et3N, DMF; (iv) CH3I, DMSO; (vi) ZnCl2, DMF; (vii) Sepharose 4B activated witch BrCN, sodium carbonate buffer (pH 9.6).
The attachment of the dinucleotide cap analogs to the resin was performed according to the approach described in Lowe et al. (1972), which was used for the immobilization of nucleotide cofactors for dehydrogenase purification. To our knowledge, this is the first example of applying this synthetic approach to cap analog chemistry. In this case, the dinucleotides 18 and 19 were attached to the Sepharose via a 1,6-diaminohexane linker substituted at position 6 of adenosine (AN6-(CH2)6NH2). The synthetic pathway leading to resins 3 and 4 is depicted in Figure 1B. The first step of the synthesis was the Yoshikawa phosphorylation of 6-chloropurine riboside (Yoshikawa et al. 1967), leading to 6-chloropurinoriboside 5′-monophosphate (6-ClAMP, 12). The introduction of the spacer to the ligand molecule was achieved by the substitution of 12 with 1,6-hexylenediamine as previously described (Huang et al. 2003). The reaction was performed in aqueous solution at room temperature using a 10-fold molar excess of the amine and led to the practically pure desirable product 13 within 2 h (98% yield by HPLC). The introduction of a hydrophobic six-carbon atom chain with primary aliphatic amino group into nucleotide molecule changed its physicochemical properties, but did not significantly affect its reactivity in further synthetic steps, including conversion to the imidazole derivative, coupling to another nucleotide moiety, and immobilization on Sepharose. Dinucleotides 18 and 19 were obtained in a ZnCl2-catalyzed coupling reaction of 2-nt subunits, in which one subunit was activated as P-imidazolide (Mukaiyama and Hashimoto 1972; Kadokura et al. 1997; Stepinski et al. 2001; Jemielity et al. 2003; Kalek et al. 2006). The reaction of 13 with imidazole led to a moderately contaminated imidazolide derivative, which could not be purified due to its low stability in aqueous solution. Therefore, cap analog 18 was synthesized by the coupling of m7GDP-Im (17) to 13. However, m7GpCH2p-Im was even more difficult to obtain (yield below 5% in all tested conditions) and the opposite coupling strategy was used (Fig. 1B, step vi). Although the coupling reaction occurred with a significant accumulation of by-products, we were able to obtain pure cap analog 19 with a good preparative yield (42% total yield). Both nucleotides were then immobilized on cyanogen bromide-activated Sepharose 4B (Cuatrecasas et al. 1968).
The degrees of substitution (DS) to the resin were determined as described previously (Webb et al. 1984) by digestion of the immobilized ligands with two enzymes: alkaline phosphatase and phosphodiesterase I. The concentration of released nucleotides was measured spectrophotometrically at 260 nm. The degree of substitution for Sepharose 2 could not be calculated, as this analog was not cleaved by either of the enzymes. The DS values for all tested resins were within the same range but lower than those described in Webb et al. (1984) (Table 1).
Binding affinities to eIF4E
The binding affinities of synthesized dinucleotide cap analogs for mouse eIF4E(28–217) were determined by fluorescence quenching (Niedzwiecka et al. 2002). Comparison of the equilibrium association constant (KAS) values of different cap analogs with their unmodified counterparts (Table 2) shows that the affinity of the mononucleotide m7GTP was at least 10-fold higher than that of the dinucleotide analogs (18–23). This probably resulted from a combination of increased electrostatic interactions between the pyrophosphate chain and the positively charged residues of the cap-binding pocket due to the additional negative charge on the terminal phosphate group (Zuberek et al. 2004) and the lack of steric hindrance. The presence of the second nucleoside in dinucleotides destabilizes the eIF4E–cap complex (Niedzwiecka et al. 2002). Introduction of the methylene group into the 5′–5′-triphosphate bridge also decreases the affinity between cap analogs and eIF4E (Kalek et al. 2005a), this could be related to a change in the polyphosphate chain conformation and the net charge distribution which may disturb electrostatic interactions and hydrogen bond formation. Lack of a free electron pair is known to prevent methylene groups from forming hydrogen bonds, as observed in the crystal structure of the eIF4E–m7GTP complex (Niedzwiecka et al. 2002). The association constants of novel cap analogs (compounds 18 and 19) were much lower than those of their unmodified equivalents, but comparable to that of the model A6 substituted analog m7Gpppm6A (Zuberek et al. 2003).
Comparison of equilibrium association constants (KAS) for the binding of mouse eIF4E(28–217) to cap analogs as determined by fluorescence quenching
It is noteworthy that the affinity of the immobilized cap analog may differ significantly from that of a free analog. For example, coupling to Sepharose through mononucleotide p-aminophenyl γ-ester eliminates the favorable effect of an additional negative charge on the terminal phosphate and generates steric hindrance by close proximity to the relatively bulky Sepharose bead. Conversely, immobilization of the dinucleotide cap analog via the 1,6-diaminohexane spacer arm may stabilize the eIF4E–cap complex by converting the spacer protonated terminal amino group into an isourea linkage, thus precluding a potentially destabilizing interaction of the linker with one of the cap's negatively charged phosphate groups. Moreover, the introduced linker separates the ligand from the Sepharose bead, which makes it more accessible for interactions with proteins.
m7GpCH2ppA-Sepharose binds DcpS specifically and with high affinity
To determine the properties of the new resins, binding of recombinant mouse eIF4E(28–217) and human DcpS was evaluated. Proteins were incubated with equal volumes of m7GpCH2pp-Sepharose (4), m7GpppA-Sepharose (3), m7GpCH2ppA-Sepharose (2), and control m7GTP-Sepharose (1). Bound fractions were eluted with the free m7GTP cap analog (1 mM) and precipitated from pooled eluates using pyrogallol red (Aguilar et al. 1999).
The efficiency of mouse eIF4E(28–217) binding was comparable for m7GpCH2ppA-Sepharose (4) and standard m7GTP-Sepharose (1) and significantly lower for the two other resins (2, 3) (Fig. 2). This result was not consistent with the association values for different cap analogs (see Table 2); however, as discussed above, immobilized nucleotides do not necessarily have the same properties as their free counterparts. Remarkably, the only resin that bound human DcpS with high efficiency was the m7GpCH2ppA-Sepharose (4) with a hexylene spacer linking the cap to the resin (Fig. 2). The other nonhydrolyzable, but mononucleotide, m7GpCH2pp-Sepharose (2) had a much lower affinity for hDcpS, probably due to the restricted access to the enzyme binding pocket, which is located within a deep cleft (Gu et al. 2004). The binding capacity of the m7GpCH2ppA-Sepharose (4) was probably further increased by the incorporation of a dinucleotide analog, based on the structural data showing that hDcpS interacts not only with the 7-methylguanosine moiety, but also with the second nucleobase of the cap structure (Gu et al. 2004).
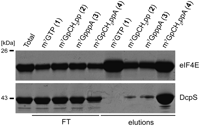
Binding of recombinant mouse eIF4E(28–217) and human DcpS to m7GTP-Sepharose (1), m7GpCH2pp-Sepharose (2), m7GpppA-Sepharose (3), and m7GpCH2ppA-Sepharose (4). Coomasie blue-stained SDS-PAGE of total, flow-through (FT), and bound fractions eluted with 1 mM m7GTP.
Purification of cap-binding proteins from yeast extract
To further test the new resins, their applicability for the purification of cap-binding proteins from cellular extracts was assessed. The m7GTP-Sepharose (1), GTP-Sepharose, and unmodified CNBr-activated Sepharose 4B were used as positive and negative controls.
Resins were incubated with crude yeast extract under saturating conditions in the presence of 0.1% Triton X100 and 100 μM of GTP to minimize unspecific interactions. Bound proteins were eluted with the free m7GTP cap analog (100 μM), precipitated with pyrogallol red, and analyzed by tandem mass spectrometry (Brooks et al. 2010).
Approximately 200 different proteins were identified in each eluate (Supplemental Table S1) by tandem mass spectrometry. The highest scored hits specific for m7GTP-Sepharose (1), m7GpppA-Sepharose (3), and m7GpCH2ppA-Sepharose (4) that were not represented in both negative control eluates are listed in Table 3.
Purification of cap-binding proteins from yeast extract
All eluates from cap analog modified resins contained both major cap-binding complexes, eIF4F consisting of the cap-binding protein eIF4E, one of the two eIF4G isoforms and one of the two eIF4A isoforms, as well as CBC composed of cap-binding proteins Cbp20 and Cbp80, with either Cbp80 or eIF4G1 as the highest scored hits. Only m7GpCH2ppA-Sepharose (4) was able to bind the yeast DcpS homolog, Dcs1 (Table 3), which is consistent with binding studies for recombinant DcpS (see Fig. 2).
Although several nucleotide-binding proteins and some housekeeping proteins were also retained on cap analog resins, this probably resulted from unspecific binding, as they were also present in eluates from GTP-Sepharose and unmodified Sepharose 4B controls (Supplemental Table S1).
The remaining proteins that were specifically recovered from cap analog-modified Sepharoses were either well-known or possible binding partners of cap-binding complexes. Most of them are involved in RNA metabolism, including regulation of translation (eIF4E associated protein Eap1) (Cosentino et al. 2000), mRNA nuclear export (Npl3, reported to interact with CBC) (Shen et al. 2000), or transcription termination (Nrd1/Nab3 complex, potentially interacting with CBC) (Vasiljeva and Buratowski 2006). Unexpectedly, the m7GpCH2pp-Sepharose had the lowest specificity for yeast cap-binding complexes (Supplemental Table S1) but efficiently bound the L-A yeast virus capsid protein, which was recovered using all resins (Szczepaniak et al. 2008) (Supplemental Table S1), most likely due to its decapping activity (Blanc et al. 1992).
To obtain more quantitative information concerning the relative efficiency of binding to different cap analogs we performed similar experiments using yeast strains expressing tagged yeast cap-binding proteins, eIF4E and Cbp20. eIF4E was expressed from a plasmid under the control of the PGAL promoter as a C-terminal fusion with a triple affinity tag consisting of a His6-HA epitope, a protease 3C cleavage site, and the IgG domain of protein A (Gelperin et al. 2005). Cbp20 was expressed under its endogenous promoter in a fusion with the C-terminal Myc epitope (Wong et al. 2007). Eluted proteins were resolved by SDS-PAGE and subjected to Western blot analysis (Fig. 3).

A Western blot of eIF4E-His6/HA/ProtA and Cbp20-Myc purified from yeast extract using m7GTP-Sepharose, m7GpCH2pp-Sepharose, m7GpppA-Sepharose, eluted with 1 mM m7GTP and detected with PAP and anti-Myc antibodies, respectively. Western blot was performed on total lysate (Total), flow trough (FT), and elutions from each resin.
The level of eIF4E was similar in all four eluates (Fig. 3), which is consistent with the MS-MS data, assuming that the number of identified peptides corresponds to the amount of protein (see Table 3; Supplemental Table S1). These results differ significantly from binding studies with mouse eIF4E(28–217) that show a comparably high capacity of resins 1 and 4 and comparable, but lower, capacity of resins 2 and 3. This discrepancy may result from structural differences between yeast and mammalian eIF4E, which in turn affects protein-cap complex stability (Niedzwiecka et al. 2002; Kiraga-Motoszko et al. 2003).
In turn, Cbp20 bound with a comparable high affinity to the control m7GTP-Sepharose (1) and the newly synthesized m7GpppA-Sepharose (3). The eluates from m7GpCH2pp-Sepharose (2) and m7GpCH2ppA-Sepharose (4) showed a much lower level of Cbp20, which is in agreement with the MS-MS data (maximum only one peptide identified for Cbp20) (see Table 3; Supplemental Table S1). This feature of resins 2 and 4 may be regarded as an advantage, as these resins were designed especially for purification of DcpS.
General conclusions
Efficient syntheses of three novel affinity resins with attached cap analogs are described and as the developed methodology is general, it can be applied to the synthesis of other resins carrying different cap analogs, which may specifically and differentially bind various cap-binding proteins. One such example is a group of dinucleotide analogs with increased length of the 5′–5′ triphosphate bridge, which has been shown to bind eIF4E with higher affinity (Niedzwiecka et al. 2002). All synthesized resins specifically bind cap-binding proteins, either recombinant or from complex biological samples such as crude yeast extract. m7GpCH2pp-Sepharose (2) and m7GpCH2ppA-Sepharose (4) are both resistant to hydrolysis by DcpS due to the methylene modification. However, only m7GpCH2ppA-Sepharose (4) efficiently binds this protein due to the introduction of a hexylene spacer arm that separates the cap analog from the bulky Sepharose bead. On the other hand, resins 2 and 4 seem to be relatively less specific toward other cap-binding proteins, especially CBC, which in turn makes them good tools for the isolation or identification of cap-specific pyrophosphatases from different organisms.
MATERIALS AND METHODS
Chemical synthesis
General procedures
Solvents and other reagents (excluding guanosine and 6-chloropurine riboside) were purchased from Sigma-Aldrich and used without further treatment unless otherwise stated. Sodium salts of commercially available nucleotides were converted into triethylammonium salts by passing through Dowex 50 WX8/TEA form, evaporating collected fractions to dryness, and drying in a vacuum over P2O5. Tris(triethylamine) phosphate was prepared in the same way from sodium phosphate. Guanosine 5′-diphosphate (TEA salt), GTP-Sepharose, and m7GTP-Sepharose (1) were synthesized as described previously (Webb et al. 1984; Darzynkiewicz et al. 1985; Jemielity et al. 2003).
The intermediate nucleotides were purified by ion-exchange chromatography on a DEAE-Sephadex A-25 (HCO3− form) column using a linear gradient of triethylammonium bicarbonate (TEAB) in deionized water. The eluates containing the product, as followed by UV adsorption measurement at 260 nm, were pooled and after evaporation under reduced pressure with repeated addition of ethanol and drying in a vacuum dessicator over P2O5, were isolated as triethylammonium salts.
Monitoring of the reaction progress as well as the purity of intermediates was performed by analytical HPLC on the Agilent Technologies Series 1200 apparatus using Supelcosil LC-18-T RP column (4.6 × 250 mm, flow rate 1.3 mL/min) developed with a linear gradient 0%–100% of methanol in 0.05 M ammonium acetate buffer (pH 5.9) within 30 min (gradient 1) or 15 min (gradient 2), UV-detection at 260 nm, and fluorescence detection (excitation at 280 nm and detection at 337 nm).
The final nucleotide products (18, 19) were purified by the Semi-preparative HPLC Waters 600E Multisolvent Delivery System using Waters HR-C-18 HPLC column (300 × 19 mm, 6 μm, flow rate 5.0 mL/min) with linear gradient of methanol in 0.05 M ammonium acetate buffer (pH 5.9) and UV-detection at 260 nm. After repeated freeze-drying, the products were isolated as ammonium salts.
The yields were calculated based on the optical density miliunits at 260 nm (mODU260) of substrates and isolated products, measured in 0.1 M phosphate buffer (pH 6) for m7Guo mononucleotides, or pH 7 for cap dinucleotides and Guo nucleotides. The optical density miliunits mODU260 are defined as absorption of compound solution in 0.1 M phosphate buffer of appropriate pH at 260 nm, multiplied by the volume of the solution (milliliters). The extinction coefficients taken for calculations were ɛ260(pH = 7) = 10,400 M−1 cm−1 or ɛ260(pH = 6) = 11,400 M−1 cm−1 for m7Guo mononucleotides, ɛ260(pH = 7) = 12,000 M−1 cm−1 for Guo mononucleotides, ɛ260(pH = 7) = 9750 M−1 cm−1 for 6-ClAMP (12), and ɛ260(pH = 7) = 15,000 M−1 cm−1 for N6 substituted adenosine derivatives. For cap dinucleotides the extinction coefficient was calculated from the equation: ɛ = 0.9 × (ɛ1 + ɛ2), where ɛ1 and ɛ2 represent extinction coefficients of both nucleotides subunits.
The structure and homogeneity of each final product was confirmed by rechromatography on RP HPLC, mass spectrometry using negative electrospray ionization (MS ESI-), and 1H NMR and 31P NMR spectroscopy. 1H NMR and 31P NMR spectra were recorded in D2O at 25°C on a Varian UNITY-plus spectrometer at 399.94 MHz and 161.90 MHz, respectively. 1H NMR chemical shifts in ppm were reported to sodium 3-trimethylsilyl-[2,2,3,3-D4]-propionate (TSP) in D2O as an internal standard. 31P NMR chemical shifts in ppm were reported to 20% phosphorus acid in D2O as an external standard. Coupling constants (J) are given in hertz. Mass spectra were recorded on a Micromass QToF 1 MS spectrometer.
P1-(guanosin-5′-yl) 1,2-methylenediphosphate, GpCH2p (5).
Phosphonylation of guanosine with methylenebis-(phosphonic dichloride) was achieved by the modified Yoshikawa procedure as described in the literature (Kalek et al. 2005b). Guanosine (1.27 g, 4.5 mmol) was suspended in 10 mL of trimethyl phosphate and cooled to 0°C. Pre-cooled solution of methylenebis(phosphonic dichloride) (2.26 g, 9 mmol) in trimethyl phosphate (2 mL) was added to the stirring mixture. Reaction was performed at 0°C, and after 1.5 h, when the reaction was completed (assigned by analytical HPLC), it was stopped by the addition of 0.7 M TEAB to pH 7. Product purification was carried out by ion-exchange chromatography on DEAE-Sephadex with 0–1.05 M linear gradient of TEAB to yield 21,400 mODU260 (1.78 mmol, 39%) of 5 as a TEA salt. HPLC (gradient 1) Rt = 3.7 min, ESI MS (-) m/z 440.0 (calc. for C11H16N5O10P2: 440.0).
P1-guanosin-5′-yl 1,2-methylenediphosphate P2-imidazolide, GpCH2p-Im (6).
Imidazolides were prepared according to the previously described procedure (Jemielity et al. 2003) with several modifications. GpCH2p (5) (225 mg; 0.51 mmol, TEA salt), imidazole (699 mg, 10.3 mmol), 2,2′-dithiodipyridine (678 mg, 4.1 mmol) were mixed in anhydrous DMF (6 mL). Triethylamine (569 μL, 3.1 mmol) and triphenylphosphine (415 mg, 4.1 mmol) were added and the mixture was stirred for 24 h. The product was precipitated from a reaction mixture with a solution of anhydrous NaClO4 (3 eq. per one negative charge) in dry acetone (∼8 mL/1 mL of DMF). After cooling at 4°C, the precipitate was settled by centrifugation (4000g at 4°C), washed repeatedly with cold, dry acetone and dried in a vacuum over P4O10. A total of 210 mg of GpCH2p-Im (6) (0.39 mmol, 77% yield) was obtained. Analytical RP HPLC (gradient 1) Rt = 4.8 min.
P1-guanosin-5′-yl 1,2-methylenetriphosphate, GpCH2pp (7).
GpCH2pp (7) was prepared as previously described (Rydzik et al. 2009). GpCH2p-Im (6) (250 mg, 0.47 mmol), tris(triethylamine) phosphate (575 mg, 1.86 mmol), and ZnCl2 (1.1 g, 7.4 mmol) were mixed in anhydrous DMF (4 mL) and stirred at room temperature for 3 h until the HPLC analysis showed the reaction was completed. To stop the reaction, a solution of EDTA (1 eq./1 eq. of ZnCl2) in water was added and pH was brought to 7 by addition of solid NaHCO3. The product (7) was purified as TEA salt using DEAE-Sephadex in a linear 0–1.2 M gradient of TEAB to yield 3070 mODU260 (0.26 mmol, 54%). Analytical HPLC (gradient 1) Rt = 2.4 min.
P1-(7-methylguanosin-5′-yl) 1,2-methylenetriphosphate, m7GpCH2pp (8).
Methylation of guanosine moiety leading to m7G derivatives was performed as previously described (Jemielity et al. 2003). A total of 3070 mODU260 GpCH2pp (7) (0.26 mmol) and 303 μL (682 mg, 4.8 mmol) of CH3I was dissolved in DMSO (8 mL) and stirred for 5 h; when the reaction was completed (assigned by analytical HPLC), the mixture was diluted with water (200 mL) and extracted with diethyl ether (4 × 150 mL). Solid Na2S2O5 was added until the solution turned colorless and the remaining ether was evaporated under reduced pressure. The product (8) (886 mODU260, 0.09 mmol, 35% yield) was isolated as TEA salt on DEAE Sephadex using a linear 0–1.1 M gradient of TEAB. HPLC (gradient 1) Rt = 2.9 min.
P1-(7-methylguanosin-5′-yl) 1,2-methylenetrimetaphosphate, meta-m7GpCH2pp (9).
Introduction of reactive nitro group into the nucleotide was obtained following procedure of Webb et al. (1984). A solution of m7GpCH2pp (8) (1310 mODU260, 0.126 mmol) in methanol (3 mL) was treated with tri-n-octylamine (0.17 mL, 0.38 mmol) and stirred at room temperature until a clear solution was obtained (∼1 h). The solvent was evaporated under reduced pressure and the tri-n-octylammonium salt of m7GpCH2pp dried under vacuum with dry DMF (3 × 1 mL). The dried product was dissolved in anhydrous DMF/methanol (9:1 v/v, 4.4 mL), treated with dicyclohexylcarbodiimide (189 mg, 0.9 mmol), and stirred overnight at room temperature in a desiccator. The solvent was evaporated under reduced pressure at the end of the reaction; 9 was obtained as a dense, oily liquid that was directly used in the next step of the synthesis. HPLC (gradient 1) Rt = 5.4 min and 5.8 min.
P1-(7-methylguanosin-5′-yl) 4-nitrophenyl-(1,2-methylenetriphosphate), p-NO2-Ph-m7GpCH2pp (10).
Synthesis of 10 was performed as previously described (Webb et al. 1984) with some major modifications. meta-m7GpCH2pp (9) (∼0.126 mmol), p-nitrophenol (425 mg, 0.3 mmol), and triethylamine (406 μL, 0.3 mmol) were dissolved in dry DMF (4.4 mL). The reaction mixture was tightly stoppered and stirred for 6 d at 50°C (the reaction progress was monitored by HPLC). The reaction mixture was then diluted with water (15 mL), cooled, and the pH was adjusted to 3.5 with HCl. The mixture was extracted with ether (2 × 25 mL) and the aqueous layer was neutralized with NaHCO3. The product was purified on DEAE-Sephadex in a linear 0–0.7 M gradient of TEAB; 10 was obtained as TEA salt to yield 603 mODU260 (0.06 mmol, 46%). HPLC (gradient 1) Rt = 8.15 min. ESI MS (-) m/z 655.0 (calc. for C18H22N6O15P3: 655.0). δH 8.20 (2H, d, J2Ph,3Ph = J5Ph,6Ph 8.6, H3Ph, H5Ph); 7.35 (2H, d, J2Ph,3Ph = J5Ph,6Ph 8.6, H2Ph, H6Ph); 5.99 (1H, d, J1′,2′ 4.0, H1′); 4.63 (1H, dd, J1′,2′ 4.0, J2′,3′ 4.7, H2′); 4.52 (1H, dd, J2′,3′ 4.7, J3′,4′ 5.3, H3′); 4.36 (1H, m, H4′); 4.32 (1H, m, H5′); 4.20 (1H, m, H5′′); 4.08 (3H, s, -CH3); 2.42 (2H, dt, JPβ,-CH2- = JPγ,-CH2- 20.6, -CH2-). δP 17.38 (1P, m, Pγ); 8.01 (1P, m, Pβ); -16.55 (1P, d, Jα,β 25.6, Pα).
P1-(7-methylguanosin-5′-yl) 4-aminophenyl-(1,2-methylenetriphosphate), p-NH2-Ph-m7GpCH2pp (11).
Synthesis of 11 was performed as previously described (Webb et al. 1984) with few modifications. p-NO2-Ph-m7GpCH2pp (10) (600 mODU260, 0.06 mmol) was dissolved in H2O (8.4 mL) and reduced by the method described previously (Berglund and Eckstein 1972). Argon was bubbled through the solution, after which palladium on charcoal (150 mg) was added. Hydrogen gas was bubbled through the suspension for 3 h. The reaction mixture was then filtered and the catalyst washed repeatedly with water. The filtrate and combined washings were concentrated by partial evaporation of water under reduced pressure; 11 was obtained to yield 355 mODU260 (0.034 mmol, 59%), and without further purification was coupled to Sepharose 4B. HPLC (gradient 1) Rt = 4.4 min. ESI MS (-) m/z 625.1 (calc. for C18H24N6O13P3: 625.1). δH 9.37 (s, H8m7G); 7.02 (2H, d, J2Ph,3Ph = J5Ph,6Ph 8.2, H3Ph, H5Ph); 6.88 (2H, d, J2Ph,3Ph = J5Ph,6Ph 8.2, H2Ph, H6Ph); 6.02 (1H, d, J1′,2′ 3.5, H1′); 4.71 (1H, m, H2′); 4.55 (1H, m, H3′); 4.39 (1H, m, H4′); 4.27 (1H, m, H5′); 4.17 (1H, m, H5′′); 4.09 (3H, s, -CH3); 2.39 (2H, t, JPß,-CH2- = JPγ,-CH2- 20.3, -CH2-). δP 17.61 (1P, m, Pγ); 7.89 (1P, m, Pβ); -14.81 (1P, d, Jα,β 24.6 Pα).
Activation of Sepharose 4B with CNBr.
Activation reaction was performed by the procedure described before (Cuatrecasas et al. 1968; Webb et al. 1984). Washed and settled Sepharose 4B (GE Healthcare) was suspended in an equal volume of water. The suspension was stirred at 4°C. A solution of cyanogen bromide (1.8 mL of a 1 g/mL solution for 15 mL of settled Sepharose) in acetonitrile was added, and the pH was maintained between 11 and 11.5 by addition of 1 M NaOH for ∼30 min. The resin was then filtered and washed with H20 (1 L for 15 mL of Sepharose) and cold 0.1 M NaHCO3-Na2CO3 buffer (pH 9.0, 1 L). The washed resin was suspended in an equal volume of buffer and treated with the appropriate cap analog.
P1-(7-methylguanosin-5′-yl) 1,2-methylenetriphosphate Sepharose, m7GpCH2pp-Sepharose (2).
Coupling reaction was performed as described before (Cuatrecasas et al. 1968, Webb et al. 1984). A total of 6 mL (settled volume) of CNBr activated Sepharose 4B resuspended in cold 0.1 M NaHCO3–Na2CO3 buffer (pH 9.0, 8 mL) was treated with 100 mODU260 (9.6 μmol) of p-NH2-Ph-m7GpCH2pp (11). The slurry was gently stirred overnight at 4°C, after which the resin was filtered, washed with 50 mL of water, and suspended in 6 mL of 20% ethanol.
6-chloropurineriboside 5′-monophosphate, 6-ClAMP (12).
Phosphorylation of 6-chloropurineriboside was performed as described previously (Jemielity et al. 2003) using Yoshikawa reaction (Yoshikawa et al. 1967). 6-chloropurineriboside (515 mg, 1.8 mmol) was suspended in 12 mL of trimethyl phosphate and cooled to 0°C. Freshly distilled POCl3 (670 μL, 1.12 g, 7.3 mmol) was added to the stirred mixture and stirring was continued for ∼2 h, at which time the HPLC analysis showed the reaction was completed. Reaction was stopped by addition of 0.7 M TEAB to pH 7. The product was purified by ion–exchange chromatography on DEAE-Sephadex in linear 0–0.7 M gradient of TEAB; 12 was obtained as TEA salt to yield 12,760 mODU260 (1.31 mmol, 72%). Analytical RP HPLC (gradient 1) Rt = 9.5 min.
N6-(6-aminohexyl)adenosine 5′-monophosphate, pAN6H(CH2)6NH2 (13).
Synthesis of 13 was performed as previously described (Huang et al. 2003). 6-ClAMP (12760 mODU260 (1.3 mmol) was suspended in 18 mL of water and treated with 1,6-diaminohexane (2.5 g, 19.3 mmol). Reaction was stirred for 1.5 h at room temperature until it was completed (assigned by analytical HPLC), after which it was stopped by neutralization with 10% HCl. The product was purified by ion-exchange chromatography on DEAE-Sephadex in a linear 0–0.7 M gradient of TEAB to yield 18,500 mODU260 (1.23 mmol, 95%). HPLC (gradient 1) Rt = 14.2 min. ESI MS (-) m/z 445.2 (calc. for C16H26N6O7P: 445.2).
N6-(6-aminohexyl)adenosine 5′-monophosphate P-imidazolide, Im-pAN6H(CH2)6NH2 (14).
Imidazole derivative of 13 was synthesized following the procedure used for compound 6 with several modifications. A total of 35 mg of 14 (0.068 mmol, yield 68%) was obtained from pAN6H(CH2)6NH2 (55 mg, 0.1 mmol), imidazole (68 mg, 1 mmol), 2,2′-dithiodipyridine (66 mg, 0.3 mmol), triethylamine (28 μL, 0.2 mmol), and triphenylphosphine (78 mg, 0.3 mmol) mixed in anhydrous DMF (2 mL) and stirred for 24 h. Analytical RP HPLC (gradient 1) Rt = 18.7 min. ESI MS (-) m/z 495.2 (calc. for C19H28N8O6P: 495.2).
P1-(7-methylguanosin-5′-yl) 1,2-methylenediphosphate, m7GpCH2p (15).
Methylation of GpCH2p (5) was performed similarly as for GpCH2pp (7). GpCH2p (5) (12,650 mODU260, 1.05 mmol) was dissolved in DMSO (27 mL) and treated with CH3I (591 μL, 1.35 g, 9.6 mmol). The mixture was stirred for 8 h. 15 was obtained as triethylammonium salt after purification on DEAE-Sephadex in a linear 0–1.05 M TAEB gradient to yield 6460 mODU260 (0.57 mmol, 54%). HPLC (gradient 1) Rt = 4.6 min.
P1-(7-methylguanosin-5′-yl) diphosphate, m7GDP (16).
m7GDP (16) was obtained using the same procedure. GDP (12,700 mODU260, 1.06 mmol) was dissolved in DMSO (27 mL) and treated with CH3I (527 μL, 1.2 g, 8.5 mmol). The mixture was stirred for 3 h. 16 was purified on DEAE-Sephadex in a linear 0–1 M TAEB gradient to yield 5450 mODU260 (0.48 mmol, 45%). HPLC (gradient 1) Rt = 4.8 min.
m7-guanosine 5′-diophosphate P2-imidazolide, Im-m7GDP (17).
A total of 120 mg of 17 was obtained from m7GDP (3000 mODU260, 0.26 mmol), imidazole (270 mg, 3.98 mmol), 2,2′-dithiodipyridine (263 mg, 1.19 mmol), triethylamine (181 μL, 1.05 mmol), triphenylphosphine (208 mg, 0.79 mmol) mixed in 4 mL of DMF, and stirred for 3 d following the procedure described for compound 6. Analytical RP HPLC (gradient 1) Rt = 7.2 min.
P1-(7-methylguanosin-5′-yl) P3-(N6-(6-aminohexyl)adenosin-5′-yl) triphosphate, m7GpppAN6H(CH2)6NH2 (18).
Coupling of m7GDP-Im (17) (55.1 mg, 0.1 mmol) and pAN6H(CH2)6NH2 (13) (1500 mODU260, 0.1 mmol) was performed following the procedure described for compound 7. Both nucleotides and ZnCl2 (109 mg, 0.8 mmol) were mixed in anhydrous DMF (2 mL) and stirred for 24 h (the reaction progress was monitored by HPLC). The product was isolated on DEAE-Sephadex using 0–1.1 M gradient of TEAB, and further purified using preparative HPLC. 18 was obtained as NH4+ salt with a yield of 849 mODU260 (0.036 mmol, 36%). HPLC (gradient 2) Rt = 9.2 min. ESI MS (-) m/z 884.2 (calc. for C27H41N11O17P3: 884.2). δH 8.92 (<1H, H8m7G), 8.38 (1H, s, H2A); 8.13 (1H, s, H8A); 6.01 (1H, d, J1′,2′ 6.2, H1′A); 5.86 (1H, d, J1′,2′ 3.5, H1′m7G); 4.69 (1H, dd, J1′,2′ 6.2, J2′,3′ 5.0, H2′A); 4.50 (2H, m, H2′m7G, H3′A), 4.40 (2H, m, H3′ m7G, H4′A); 4.38 (1H, m, H5′) 4.34 (1H, m, H4′m7G);4.26 (3H, m, H5′′m7G, H5′A, H5′′A); 3.98 (3H, s, -CH3); 3.49 (2H, bs, -NHCH2); 2.96 (2H, t, J 7.6, CH2NH2,); 1.65 (4H, m, NHCH2CH2(CH2)2CH2CH2NH2); 1.41 (4H, m, NHCH2CH2(CH2)2CH2CH2NH2). δP -11.25 (2P, m, Pα, Pγ), -22.74 (1P, t, Jα,β = Jβ,γ 18.5, Pβ).
P1-(7-methylguanosin-5′-yl) P3-(N6-(6-aminohexyl)adenosin-5′-yl) 1,2-methylenetriphosphate, m7GpCH2ppAN6H(CH2)6NH2 (19).
Synthesis of 19 was obtained in similar way. Im-pAN6H(CH2)6NH2 (14) (35 mg, ∼0.04 mmol regarding contamination), m7GpCH2p (15) (500 mODU260, 0.045 mmol) and ZnCl2 (95 mg, 0.7 mmol) were mixed in anhydrous DMF (2 mL) and stirred for 24 h. Obtained product 19 was purified on DEAE-Sephadex in a linear 0–1 M TAEB gradient and further purified using preparative HPLC to yield 896 mODU260 (0.017 mmol, 42%). HPLC (gradient 1) Rt = 14.4 min. ESI MS (-) m/z 882.2 (calc. for C28H43N11O16P3: 882.2). δH 8.41 (1H, s, H2A); 8.17 (1H, s, H8A); 6.04 (1H, m, J1′,2′ 5.5, H1′A); 5.91 (1H, m, J1′,2′ 3.7, H1′m7G); 4.67 (1H, dd, J1′,2′ 5.5, J2′,3′ 4.5, H2′A); 4.58 (1H, m, H2′m7G); 4.48 (2H, m, H3′m7G, H3′A); 4.38–4.21 (6H, m, H4′AH4 ′m7G, H5′m7GH5′ A, H5′′A, H5′′m7G); 4.02 (3H, s, CH3); 3.53 (2H, bs, -NHCH2); 2.96 (2H, m, CH2NH2); 2.39 (2H, m, JPβ,-CH2- = JPγ,-CH2- 20.2, p-CH2-p); 1.66 (4H, m, NHCH2CH2(CH2)2CH2CH2NH2); 1.42 (4H, m, NHCH2CH2(CH2)2CH2CH2NH2). δP 17.60 (1P, m, Pγ); 7.46 (1P, m, Pβ); -10.74 (1P, d, Jα,β 23.3, Pα).
P1-(7-methylguanosin-5′-yl) P3-adenosine-5′-yl triphosphate, m7GpppA-Sepharose (3).
A total of 6 mL (settled volume) of resin 3 was obtained from 155 mODU260 (6.27 μmol) of m7GpppAN6H(CH2)6NH2 (18), following the procedure described for compound 2.
P1-(7-methylguanosin-5′-yl) P3-adenosine-5′-yl 1,2-methylenetriphosphate, m7GpCH2ppA-Sepharose (4).
Similarly, 6.5 mL (settled volume) of resin 4 was obtained from 164 mODU260 (6.9 μmol) of m7GpCH2ppAN6H(CH2)6NH2 (19).
Biological assays
Used reagents were purchased from Sigma-Aldrich unless otherwise stated.
Determination of degrees of substitution (DS)
A total of 250 μL of resin (settled volume) was suspended in 1 mL of buffer P (75 mM Tris-HCl at pH 8.8, 5 mM MgCl2) and treated with Alkaline phosphatase (4 U, TaKaRa) and phosphodiesterase I (5 U, Pharmacia Biotech) (Webb et al. 1984). The change in absorbance of the supernatant fluid was followed at 260 nm. The absorbance value obtained after 4 h of incubation was used for calculation of DS after subtraction of value measured for a negative control (unmodified Sepharose 4B, GE Healthcare). Each reaction was performed in three replicas.
Expression and purification of recombinant proteins
Mouse eukaryotic initiation factor eIF4E (residues 28–217) was expressed in E. coli BL21(DE3) as inclusion bodies. The guanidinium-solubilized protein was renatured by one-step dialysis and purified by ion-exchange chromatography on a HiTrapSP column without contact with cap analogs (Marcotrigiano et al. 1997). The concentration of eIF4E was determined spectrophotometrically assuming ɛ280 = 53,400 M−1 cm−1. Human DcpS was expressed in E. coli Rosetta DE3 according to the procedures described previously (Cohen et al. 2004) with several modifications. His-tagged DcpS was purified by a two-step procedure on Ni-NTA Syperflow Cartridge (QIAGEN) followed by gel filtration on Superdex 200 column (GE Healthcare) using the AKTA Purifier system (GE Healthcare). The concentration of DcpS was determined spectrophotometrically assuming ɛ280 = 30,495 M−1 cm−1.
Binding affinities for eIF4E
Fluorescence titration measurements were carried using a LS-55 spectrofluorometer (Perkin Elmer Co.) in 50 mM HEPES/KOH (pH 7.2), 100 mM KCl, 0.5 mM EDTA, 1 mM DTT at 20.0 ± 0.2°C. Aliquots of 1 μL of solutions with increasing concentration of cap analog were added to 1.4 mL of 0.1-μM protein solutions. Fluorescence intensities (excitation at 280 nm with 2.5 nm bandwidth and detection at 337 nm with 4 nm bandwidth and 290 nm cut-off filter) were corrected for sample dilution and the inner filter effect. Equilibrium association constants (KAS) were determined by fitting the theoretical dependence of the fluorescence intensity on the total concentration of cap analog to the experimental data points according to the equation described previously (Niedzwiecka et al. 2002). The concentration of protein was fitted as a free parameter of equilibrium equation showing the amount of “active” protein. The final KAS was calculated as a weighted average of three to five independent titrations, with the weights taken as the reciprocals of the numerical standard deviations squared. Numerical nonlinear least-squares regression analysis was performed using ORIGIN 6.0 (Microcal Software, Inc.).
Yeast extract preparation
Wild-type yeast strains BY4741 and CBP20-Myc (Wong et al. 2007) were grown at 30°C in 2 L of YPD medium (20 g/L Bacto Tryptone, Difco; 10 g/L Bacto Yeast Extract, Difco; 2% D-glucose, 50 μg/mL Ampicilin) to 1–2 OD600. Strain Y258 expressing eIF4E-His6/HA/ZZ under the control of the PGAL promoter from a plasmid BG1805 (Open Biosystems) (Gelperin et al. 2005) was grown in l.3 L of complete synthetic medium without uracil (0.77 g/L CSM–URA, MP; 6.7 g/L YNB without amino acids, Difco, 2% sodium-L- lactate, 50 μg/mL Ampicilin) at 30°C to 0.5–1 OD600. Induction with 650 mL of 3× induction medium (60 g/L Bacto Tryptone, Difco; 30 g/L Bacto Yeast Extract, Difco; 6% D-galactose, 50 μg/mL Ampicilin) was carried out for 6 h at 30°C. Cells were harvested, resuspended in an equal volume of lysis buffer (40 mM Hepes at pH 8.0, 250 mM NaCl, 1 mM DTT), frozen in liquid nitrogen, and homogenized in a laboratory blender (Waring) with dry ice (4 × 2 min). The lysate was then centrifuged for 20 min at 50,000g at 4°C. The extract was cleared by ultracentrifugation at 120,000g for 1 h, 30 min at 4°C, and dialyzed for 4 h at 4°C in buffer D (40 mM Hepes at pH 8.0, 150 mM NaCl, 1 mM DTT, 1 mM PMSF, 20% glycerol).
Large-scale purification of yeast cap-binding proteins
Yeast extract prepared from BY4741 and CBP20-Myc cells was supplemented with reduced triton X100 (0.1% final concentration) and GTP (100 μM final concentration). A total of 7–10 mL of extract was incubated for 3 h at 4°C with slow rotation with 200 μL (settled volume) of resins (1–4) or control GTP-Sepharose and unmodified CNBr activated Sepharose 4B (GE Healthcare), prewashed in IPP150 buffer (10 mM Tris-HCl at pH 8.0, 150 mM NaCl, 0.1% rTX100). Flow-through fractions were then separated by centrifugation (5 min at 400g, at 4°C). The resins were resuspended in 10 mL of buffer IPP150 with 100 μM GTP and washed twice with 10 mL of buffer IPP150/100 μM GTP and twice with 10 mL of buffer B (10 mM Tris-HCl at pH 8.0, 150 mM NaCl) in polyprep chromatography columns (10 mL, BioRad). Bound proteins were eluted with buffer E (10 mM Tris-HCl at pH 8.0, 150 mM NaCl, 0.1–1 mM m7GTP; 6 × 200 μL) by incubation with the resin for 10 min at 4°C. The experiments were repeated two to three times.
Protein precipitation and analysis by mass spectrometry
Proteins from combined eluates were precipitated using pyrogallol red (Aguilar et al. 1999) prior to further analysis. For mass spectrometry analysis, proteins were separated by electrophoresis performed on NuPAGE 4%–12% gradient gels using MES buffer gel system (Invitrogen) and stained with SimplyBlue SafeStain (Invitrogen). Mass spectrometry was performed on proteins purified from wild-type strain (BY4741) both with eluates in solution and from bands excised from gels. Samples were processed by standard procedures with trypsin digestion and cysteine alkylation. The obtained peptide mixtures were separated on a nano-HPLC system and the column outlet was coupled to the ion source of an LTQ FTICR spectrometer (Brooks et al. 2010).
Small-scale purification of cap-binding proteins
Yeast extract (containing eIF4E-His6/HA/ZZ) or protein samples (520 μg/mL eIF4E and 200 μg/mL DcpS) were supplemented with reduced triton X100 (0.1% final concentration) and GTP (100 μM final concentration). A total of 300–500 μL of extract or protein sample was incubated for 2 h with 50 μL (settled volume) of resin (1–4) equilibrated with IPP150 buffer in 4°C. Flow-through fractions were separated and the resins were washed twice with 0.5 mL of buffer IPP150/100 μM GTP and three times with 0.5 mL of buffer B. Bound proteins were eluted with buffer E2 (10 mM Tris-HCl at pH 8.0, 150 mM NaCl, 1 mM m7GTP; 4 × 40 μL) by incubation with the resin for 5 min. The purification procedure was carried out in 0.8-mL spin columns (Mo Bi Tec). The experiments were repeated two to three times.
Western blot analysis
Western blot analysis was carried out with horseradish peroxidase-anti-peroxidase (PAP, Sigma) in a 1:2000 dilution, mouse anti-Myc antibody (Santa Cruz Biotechnology) in a 1:5000 dilution, and horseradish peroxidase-conjugated anti-mouse antibody (Calbiochem) in a 1:10000 dilution. Visualization was performed using a CCD-camera (Fluorchem SP, Gel Biosciences).
SUPPLEMENTAL MATERIAL
Supplemental material is available for this article, showing the list of all proteins bound to m7GTP-, m7GpppA-, m7GpCH2ppA-, GTP-, and 4B-Sepharoses identified by tandem mass spectrometry.
ACKNOWLEDGMENTS
We thank Mike Kiledjian (Rutgers University) for providing the hDcpS encoding plasmid; Zbigniew M. Darzynkiewicz (Institute of Experimental Physics, University of Warsaw), and Krystian Stodus (Institute of Biochemistry and Biophysics of the Polish Academy of Sciences, IBB PAS, Warsaw) for expressing and purifying the protein; Alan Hinnebusch (National Institute of Child Health and Human Development) for providing the CBP20-Myc strain; Andrzej Dziembowski (Institute of Biochemistry and Biophysics of the Polish Academy of Sciences, IBB PAS, Warsaw) for discussions; the Laboratory of Biological NMR (IBB PAS, Warsaw) for access to the NMR apparatus, and the Laboratory of Mass Spectrometry (IBB PAS, Warsaw) for recording MS spectra. This work was supported by grants from the Polish Ministry of Science and Higher Education to J.J. and E.D. (N N204 089438 and N N301 096339), the Howard Hughes Medical Institute to E.D. (55005 604), the Wellcome Trust to J.K. (067504/Z/02/Z), the European Social Fund (UDA-POKL.04.01.01-00-072/09-00), by the EU through the European Social Fund (contract number UDA-POKL.04.01.01-00-072/09-00) and the Ministry of Science and Higher Education intramural BW through the Faculty of Biology, University of Warsaw to S.A.S.
Footnotes
-
↵4 Corresponding authors
E-mail jacekj{at}biogeo.uw.edu.pl
E-mail kufel{at}ibb.waw.pl
-
Article published online ahead of print. Article and publication date are at http://www.rnajournal.org/cgi/doi/10.1261/rna.032078.111.
- Received December 22, 2011.
- Accepted April 12, 2012.








