
Comprehensive evaluation of canonical versus Dicer-substrate siRNA in vitro and in vivo
- Donald J. Foster1,4,
- Scott Barros1,
- Rick Duncan1,
- Sarfraz Shaikh1,
- William Cantley1,
- Amy Dell1,
- Elena Bulgakova1,
- Jonathan O'Shea1,
- Nate Taneja1,
- Satya Kuchimanchi1,
- Christopher B. Sherrill2,
- Akin Akinc1,
- Gregory Hinkle1,
- Amy C. Seila White1,
- Bo Pang1,
- Klaus Charisse1,
- Rachel Meyers1,
- Muthiah Manoharan1 and
- Sayda M. Elbashir3
- 1Alnylam Pharmaceuticals, Cambridge, Massachusetts 02142, USA
- 2EraGen Biosciences, Madison, Wisconsin 53717, USA
- 3ModeRNA Therapeutics, Cambridge, Massachusetts 02142, USA
Abstract
Since the discovery of RNA interference (RNAi), researchers have identified a variety of small interfering RNA (siRNA) structures that demonstrate the ability to silence gene expression through the classical RISC-mediated mechanism. One such structure, termed “Dicer-substrate siRNA” (dsiRNA), was proposed to have enhanced potency via RISC-mediated gene silencing, although a comprehensive comparison of canonical siRNAs and dsiRNAs remains to be described. The present study evaluates the in vitro and in vivo activities of siRNAs and dsiRNAs targeting Phosphatase and Tensin Homolog (PTEN) and Factor VII (FVII). More than 250 compounds representing both siRNA and dsiRNA structures were evaluated for silencing efficacy. Lead compounds were assessed for duration of silencing and other key parameters such as cytokine induction. We identified highly active compounds from both canonical siRNAs and 25/27 dsiRNAs. Lead compounds were comparable in potency both in vitro and in vivo as well as duration of silencing in vivo. Duplexes from both structural classes tolerated 2′-OMe chemical modifications well with respect to target silencing, although some modified dsiRNAs demonstrated reduced activity. On the other hand, dsiRNAs were more immunostimulatory as compared with the shorter siRNAs, both in vitro and in vivo. Because the dsiRNA structure does not confer any appreciable benefits in vitro or in vivo while demonstrating specific liabilities, further studies are required to support their applications in RNAi therapeutics.
Keywords
INTRODUCTION
The potential for short duplex oligonucleotides to mediate RNAi was first demonstrated in 2001 (Elbashir et al. 2001a,b,c). Dicer initiates endogenous RNAi by cleaving long double-stranded RNA substrates into smaller fragments of 21–25 nt (Bernstein et al. 2001b; Elbashir et al. 2001a). These fragments are incorporated into the RNA Induced Silencing Complex (RISC) with Argonaute 2 mediating sequence-specific mRNA cleavage (Rand et al. 2004; Matranga et al. 2005). A 21-nt duplex with 2-bp overhangs at the 3′ end, termed “small interfering RNA” (siRNA), is the natural structure for RNAi triggers (Elbashir et al. 2001a,b). Because exogenously derived siRNAs are effective in gene silencing, siRNAs are used for functional genomics studies and are being developed for clinical applications (Vaishnaw et al. 2010).
Subsequent to the characterization of siRNA, a variety of other short duplex oligonucleotide structures were described in the literature. One such design is the 25/27-mer Dicer-substrate siRNA (dsiRNA), which is cleaved by Dicer to yield fragments of a similar size and structure as canonical siRNA (Kim et al. 2005). While it has been postulated that engaging Dicer may confer certain advantages in potency for dsiRNAs, this assertion has not been thoroughly investigated (Kim et al. 2005). Here we evaluate a panel of siRNAs and dsiRNAs against two genes, Factor VII and Phosphatase and Tensin Homolog (PTEN). We find highly active compounds of both siRNA and dsiRNA designs, with similar activity in vitro and in vivo, as well as similar duration of effect in vivo. Introduction of 2′-OMe into select duplexes reduces activity with dsiRNAs as compared with siRNAs. Unmodified dsiRNAs were found to be more immunostimulatory than siRNAs in vitro and in vivo, with some sequences continuing to show immunostimulation in vivo after chemical modification. The results of our comprehensive evaluation did not reveal any substantive benefits to the dsiRNA structure; therefore, the simpler canonical siRNA is preferred.
RESULTS
Evaluation of in vitro efficacy
To determine performance characteristics of canonical siRNAs and dsiRNAs, we tested a series of 63- and 67-sequence-matched duplex pairs targeting FVII and PTEN, respectively. A comparison of FVII suppression by 0.1 nM canonical and Dicer-substrate FVII-targeted duplexes revealed no statistically significant difference for either mRNA (p = 0.1482) or protein (p = 0.1164) (Fig. 1A) silencing. The same comparison for the PTEN-targeting duplex set indicated a small but statistically significant (p < 0.0001) improvement in mRNA silencing by the Dicer-substrate duplexes (Fig. 1B). The 10 top-performing duplexes from each structure type were selected for dose-response evaluation and IC50 determination (Supplemental Table S3). The top canonical siRNAs were not substantially different from the top dsiRNAs for either FVII- or PTEN-targeting sequences. For FVII-targeting sequences, canonical siRNAs yielded an average IC50 of 7.4 ± 3.1 pM, while dsiRNAs produced an average IC50 of 11.0 ± 3.2 pM (average and standard deviation, respectively). For PTEN-targeting sequences, canonical siRNAs yielded an average IC50 of 13.6 ± 6.5 pM compared with 7.1 ± 1.8 pM for dsiRNAs (Fig. 1C). It became apparent while conducting these studies that dsiRNA-treated cells appeared less healthy. We selected 12 pairs of sequence-matched compounds of equal efficacy targeting either FVII or PTEN (six each). Between Days 2 and 4 post-transfection with 5 nM duplex, dsiRNAs significantly reduced viability compared with their canonical counterparts as determined by a comparison of the sum of viability values over 3 d (Fig. 1D).
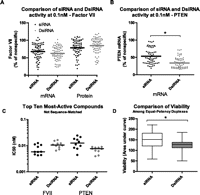
In vitro activity and viability. (A,B) HeLa cells stably transfected with FVII (A) or parental HeLa cells (B) were reverse-transfected using Lipofectamine RNAiMax with 0.1 nM duplex. FVII protein (A) was quantified by chromogenic assay, while mRNA (A–C) was quantified by branched DNA assay. Values were normalized to percent of nonspecific 21-mer control. (C) Transfection and assay as in A and B, but a sixfold dilution series containing eight points was prepared, ranging from 20 nM to 75 fM. After normalization to nonspecific 21-mer, IC50 values were determined. Log-transformed IC50 values were compared by a two-tailed Student's t-test. (D) Viability assay in HeLa using sequence-matched compounds of equal efficacy targeting FVII (six pairs) or PTEN (six pairs). Reverse transfection was performed with Lipofectamine RNAiMax and 5 nM duplex. Samples were evaluated by CellTiter Blue on Days 2, 3, and 4 after transfection. Data were normalized to mock-transfected, and area under the curve (AUC) values were calculated as a measure of cumulative viability. Data were compared by two-way ANOVA (effects of structure and sequence). Data (A–D) are averages of at least two independent experiments, each with a minimum of two biological replicates. (*) p < 0.05.
Chemical modification of siRNA can impart many beneficial properties, including attenuated immunostimulation and increased serum stability (Bumcrot et al. 2006). To test the impact of chemical modifications on immunostimulation, lead siRNAs and dsiRNAs targeting FVII or PTEN, together with sequence-matched structural counterparts, were modified as described in Materials and Methods. In addition to the unmodified forms denoted as UU, chemical variants containing 2′-OMe, phosphorothioate (PS), and deoxy thymidine (dT) residues were prepared (Table 1). Modification followed one of three paradigms, termed heavy/light (HL), unmodified/alternating (UA), or heavy/alternating (HA) (Fig. 2A). The components of the HL modification paradigm are effective in reducing cytokine stimulation by siRNAs while retaining silencing activity (Soutschek et al. 2004; Nguyen et al. 2010). Similarly, the UA modification motif was selected based on data indicating this 2′-OMe pattern generally preserves dsiRNA activity and mitigates immunostimulation (Collingwood et al. 2008). These motifs allowed us to evaluate each structure with a chemical modification pattern that is well tolerated with respect to silencing activity and reduces immunostimulation. At the same time, the compatibility of the HL modification motif with dsiRNA could be assessed. Consistent with previous reports, the unmodified siRNAs and dsiRNAs stimulated TNFα and IFNα production in vitro, as evaluated in human PBMC (Fig. 2B,C). A two-way ANOVA (structure × sequence) indicated an effect of structure for unmodified compounds (99.5 ± 72.4 vs. 187.8 ± 129.3 pg/mL IFNα for siRNA and dsiRNA, respectively). The introduction of systematic chemical modification patterns eliminated cytokine induction in vitro for both siRNA and dsiRNA structures (Fig. 2B,C).
Sequences of compounds tested in vivo
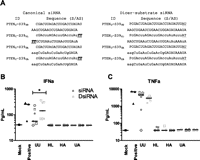
Chemical modifications and human PBMC assay. (A) Example of chemical modification motifs. Lowercase letters represent 2′-OMe-modified nucleotides. Underline denotes DNA residues, underline with italics indicates DNA residues with phosphorothioate linkage, and “P” designates a 5′ phosphate. (B,C) 133 nM canonical or Dicer-substrate siRNA listed in Table 1 was transfected into human PBMC using 8 μg/mL DOTAP. Supernatant was collected 24 h after transfection and analyzed for IFN-α (B) and TNF-α (C) by ELISA. Canonical siRNAs are depicted in dark gray and dsiRNAs in light gray, with chemical variants of each sequence sharing the same symbol. (*) Effect of structure as determined by ANOVA, p < 0.05. Modifications are (UU) unmodified/unmodified, (HL) heavy/light, (HA) heavy/alternating, and (UA) unmodified/alternating.
As the therapeutic potential of compounds is best evaluated in vivo, the efficacy of select siRNA and dsiRNA pairs was tested in mouse. Lead unmodified compounds with sequence-matched structural counterparts, their chemically modified variants, and two additional sequence-matched pairs of unmodified (UU) compounds were formulated in lipid nanoparticles (LNPs) for hepatic delivery (Table 1). For assessment of potency, equal molar amounts of each duplex were administered i.v. at one of three doses. FVII-targeting siRNAs were tested at 0.6, 0.1, and 0.02 mg/kg, while PTEN-targeting siRNAs were assessed at 0.4, 0.1, and 0.02 mg/kg. The dosage of dsiRNA was adjusted (increased) to account for the larger size of the molecule, such that dosing was equimolar across structures. Blood was collected from animals dosed with FVII-targeted compounds 2, 9, and 21 d after injection to evaluate the extent and duration of protein silencing. Animals dosed with PTEN-targeted compounds were sacrificed at the same time points, and livers were obtained for evaluation of the extent and duration of mRNA knockdown.
Unmodified siRNA and dsiRNA pairs showed comparable knockdown activity for five of six sequences on Day 2 (Figs. 3, 5A below). For Factor VII protein, the average ED50 values for active compounds were 0.061 ± 0.011 mg/kg for canonical siRNAs and 0.087 ± 0.045 mg/kg for dsiRNAs. The unmodified canonical siRNA match to the lead FVII-targeted dsiRNA (FVIIC-S52UU) was inactive. For PTEN, the average ED50 values were 0.037 ± 0.006 mg/kg for canonical siRNAs and 0.021 ± 0.013 mg/kg for dsiRNAs. Examining for more subtle differences, two-way ANOVA analyses indicated a significant effect of structure for compounds targeting either FVII or PTEN. Bonferroni post tests reveal two sequence pairs performed equally (FVIIc-S41UU, PTENC-S49UU, and respective dsiRNA matches), one siRNA (FVIIC-S14UU) performed better than the corresponding dsiRNA, and three dsiRNAs performed better than the corresponding siRNA (FVIID-S52UU, PTEND-S39UU, PTEND-S15UU). However, the differences were generally quite small: The median difference in ED50 for these six pairs was 0.013 mg/kg (canonical–Dicer-substrate).
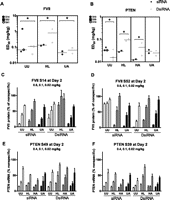
Female C57/BL6 mice were injected with LNPs containing siRNA or dsiRNA. Mice were dosed with 0.6, 0.1, or 0.02 mg/kg for FVII-targeting compounds and controls. The dose of dsiRNA was adjusted such that the amount of 21-mer produced by Dicer cleavage in vivo (assuming 100% accurate and complete cleavage) was equal to the amount of canonical siRNA administered. For PTEN-targeting compounds and controls, mice received 0.4, 0.1, or 0.02 mg/kg with dosing adjusted for dsiRNA as described. Blood (FVII) or liver (PTEN) was collected 2 d after injection. Factor VII protein was quantified by chromogenic assay, while PTEN mRNA was quantified by branched DNA. Data were normalized to Luciferase-targeted siRNA control. (A,B) ED50 values were determined by nonlinear regression of Day 2 activity data. Log-transformed ED50 data were compared by two-way ANOVA (structure × dose). (*) p ≤ 0.05. (C–F) Dose-response data obtained 2 d after injection. Canonical leads with dsiRNA matches (C,E), as well as dsiRNA leads with canonical siRNA matches (D,F), are shown. Modifications are (UU) unmodified/unmodified, (HL) heavy/light, (HA) heavy/alternating, and (UA) unmodified/alternating.
Chemically modified siRNAs and dsiRNAs produced similar ED50 values in vivo. In the chemically modified Factor VII-targeting set, the average ED50 across all active variants was 0.090 ± 0.053 mg/kg for canonical siRNAs and 0.109 ± 0.080 mg/kg for dsiRNAs (Figs. 3, 5A below). One dsiRNA was rendered inactive by chemical modification (FVIID-S14HL). Notably, the canonical siRNA sequence that was inactive as an unmodified duplex (FVIIC-S52UU) was significantly more active with both the HL and the UA modification motifs. In the PTEN-targeting set, the average ED50 across all variants was 0.024 ± 0.009 mg/kg for canonical siRNAs and 0.055 ± 0.037 mg/kg for dsiRNAs. Despite the similar performance of the modified compounds on average, we did find statistically significant differences. Comparing siRNAs and dsiRNAs for each sequence and modification, we find that for HL compounds, three of four (FVIIC-S14HL, PTENC-S49HL, and PTENC-S39HL) canonical siRNAs performed better than the sequence-matched dsiRNAs. Likewise, both HA compounds performed better as siRNAs than dsiRNAs (PTENC-S49HA, PTENC-S39HA). For the UA variant, differences in general were very small, and most compounds behaved similarly, independent of their structure; however, there was an advantage of FVIIC-S52UA over its dicer-substrate counterpart.
Previous reports proposed that dsiRNAs provide enhanced duration of silencing (Kim et al. 2005); we therefore evaluated this hypothesis with our compound set in vivo. Duration is best evaluated at doses that result in linear and robust silencing at early times (Zimmermann et al. 2006; Frank-Kamenetsky et al. 2008). Thus, we considered the highest dose for FVII silencing (0.6 mg/kg) (Fig. 4A,C) and the middle dose for PTEN silencing (0.1 mg/kg) (Fig. 4B,C). Differences in target knockdown at Days 9 or 21 were considered for measures of duration.
All unmodified compounds demonstrated similar durability of silencing effect (Figs. 4, 5A). Analyses by two-way ANOVA (time × structure) indicated that FVIIC-S14UU knockdown lasted longer than the matched dsiRNA, while FVIID-S52UU showed better duration than the canonical siRNA counterpart. FVIIc-S41UU, PTENc-S49UU, and PTENc-S15UU showed similar longevity to matched dsiRNAs, although PTEND-S39UU showed somewhat greater duration than its canonical counterpart. As a simpler measure of duration, we calculated T50 values by linear regression. These represent the time at which target suppression equaled 50%. In the FVII set, active unmodified canonical siRNAs (0.6 mg/kg) yielded an average T50 of 13.6 ± 3.7 d; dsiRNA showed a similar T50 of 11.2 ± 3.9. For the PTEN set, unmodified canonical siRNAs (0.1 mg/kg) yielded an average T50 of 9.6 ± 1.3 d, compared with 11.3 ± 2.2 for dsiRNAs.
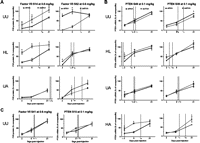
In vivo duration. (A–C) Time-course data for 0.6 mg/kg (FVII) and 0.1 mg/kg (PTEN). Duration data were compared by two-way ANOVA (structure × day). (*) Bonferoni post hoc comparisons with p < 0.05. T50 values were determined by linear regression of duration data and defined as the time at which target suppression equaled 50%. Data points represent n = 3–5, with two technical replicates. Modifications are (UU) unmodified/unmodified, (HL) heavy/light, (HA) heavy/alternating, and (UA) unmodified/alternating.
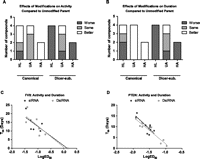
Impact of chemical modifications in vivo and activity–duration relationship. (A) Compounds demonstrating a significant impact of chemical modifications on Day 2, compared with unmodified parent, at Day 1 were categorized accordingly. Categorization is based on the direction of significant comparisons (p < 0.05) as determined by two-way ANOVA (sequence × modification) of log-transformed ED50 data. Bars are additive. (B) Compounds showing a significant difference compared with unmodified parent compound on Day 9, Day 21, or both were categorized accordingly. Categorization is based on direction of significant comparisons (p < 0.05) as determined by two-way ANOVA (time × modification) of single dose data. Bars are additive. FVII (C), and PTEN (D) T50 data from all canonical (black) and dicer-substrate (gray) compounds were plotted against corresponding LogED50 values. A linear regression was performed to describe the relationship between these measures of activity and duration. Data points represent n = 3–5, with two technical replicates. Modifications are (UU) unmodified/unmodified, (HL) heavy/light, (HA) heavy/alternating, and (UA) unmodified/alternating.
Knockdown by modified canonical siRNAs tended to be more durable than for dsiRNAs (Figs. 4, 5B). Canonical siRNAs showed greater longevity than dsiRNAs for five of 12 pairs (FVIIC-S14HL, PTENC-S39HL, PTENC-S39UA, PTENC-S49HA, and PTENC-S39HA); only FVIID-S52UA showed an improvement compared with its matched siRNA. Regarding T50 values, siRNAs demonstrated 50% silencing out to 14.6 ± 5.9 d for all FVII-targeting modified sequences (0.6 mg/kg), and 10.5 ± 4.1 d for PTEN-targeting modified sequences (0.1 mg/kg). dsiRNAs had corresponding T50 values of 16.2 ± 7.9 (FVII) and 6.3 ± 4.5 d.
siRNAs were found to tolerate chemical modifications more than dsiRNAs. Modifications led to reductions in duration for all HL- and HA-modified dsiRNAs, as well as PTEND-S39UA, compared with unmodified parent compounds; only FVIID-S14UA showed an improvement. FVIID-S52UA demonstrated mixed results, with somewhat poorer activity than the unmodified parent at day 9, and better at day 21. Conversely, all UA- and HA-modified canonical siRNAs showed improved duration; HL modification resulted in a mixed effect in which FVIIC-S52HL demonstrated improved duration and FVIIC-S14HL and PTENC-S39HL showed reduced duration. Comparing each modified compound to its unmodified parent, we found that the median effect of chemical modification across 10 modified siRNAs was a 36% lowering of ED50 and a 25% enhancement of duration (T50). Conversely, the median effect of chemical modification on dsiRNAs was a 225% increase in ED50 and a 47% reduction in duration. If we consider only UA-modified compounds, the canonical structure was still slightly favored, because we find a 31% lowering of ED50 for siRNA compared with a 24% increase in dsiRNA ED50. Upon plotting the LogED50 data against T50 for the two genes and structures, we observed a similar distribution of data across structures (Fig. 5C,D). Thus, the relationship between initial activity and duration appears similar for siRNAs and dsiRNAs, independent of modification state and structure.
As evidenced in our in vitro PBMC assay, unmodified dsiRNAs tend to be more immunostimulatory than unmodified siRNAs, but chemical modification eliminated this activity. To further explore the immunostimulatory potential of these compounds, we evaluated cytokine and chemokine stimulation in vivo. We assessed analyte levels in the blood after i.v. dosing of all LNP-formulated duplexes depicted in Figure 2. Luciferase-targeted duplexes were included as nontargeting controls. Mice were injected with 0.6 mg/kg siRNA or an equimolar amount of dsiRNA. Blood was obtained 4 h after injection, and the levels of 23 analytes were determined by Luminex assay. Of the 23 analytes measured, eight were deemed not suitable for cross-structure comparisons because their levels were below LLOQ. Two-way ANOVA analyses indicated that as a group, unmodified dsiRNAs were significantly more stimulatory for seven of the remaining 15 analytes than the sequence-matched siRNAs (Fig. 6). Furthermore, more dsiRNAs stimulated at least one analyte twofold or more above PBS (one of seven canonical vs. five of seven dsiRNAs). Data below LLOQ were also below PBS values, and thus considered nonstimulatory. While chemical modification abolished immunostimulation by canonical siRNAs for all 11 compounds, three of 11 modified dsiRNAs stimulated at least one analyte above PBS. It should be noted that due to the larger mass of dsiRNA, animals received ∼20% more lipid to achieve equimolar dosing. However, given that chemical modification reduced immunostimulation by dsiRNAs, the immunostimulation cannot be attributed solely to lipid-based effects. These data are also supported by the greater immunostimulation of INFα by dsiRNAs in vitro.
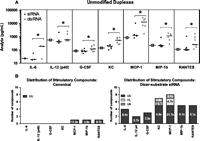
In vivo immunostimulation. Female C57/BL6 mice were injected with LNPs containing 0.6 mg/kg siRNA or dsiRNA. The dose of dsiRNA was adjusted such that the amount of 21-mer produced by Dicer cleavage in vivo (assuming 100% accurate and complete cleavage) was equal to the amount of canonical siRNA administered. Blood was collected 4 h after injection and analyzed by Luminex assay. Twenty-three cytokines were quantified. Data were log-transformed for statistical analyses. For comparison between siRNA and dsiRNA (A), ANOVA effects of structure and sequence were used (N ≥ 2 per sequence, minimum three matched sequences). Canonical siRNAs (black circles), dsiRNAs (light gray circles), and PBS (an open square in the first column for each analyte) are depicted. (*) p < 0.05. For comparison to PBS (B), two-tailed Student's t-tests were performed. Data below LLOQ were also below PBS values and thus considered nonstimulatory. The number of compounds that stimulated at least one analyte twofold or more above PBS (p < 0.05) was tallied by group and depicted in the figure. The total numbers of stimulatory compounds are organized by analyte, with subgroups indicated by shading. Median fold stimulation over PBS (non-log values) for stimulatory compounds is indicated in each bar. Data points represent n = 3–4 and technical replicates. Modifications are (UU) unmodified/unmodified, (HL) heavy/light, and (UA) unmodified/alternating.
DISCUSSION
The drive to develop RNAi-based therapeutics has led to the widespread application of chemically and structurally modified siRNAs. A variety of chemical modifications are compatible with siRNA activity, including 2′-fluoro, 2′-OMe, 2,4-difluorotoluene, phosphorothioate, and others (Bumcrot et al. 2006). Such modifications have the potential to reduce immunostimulation, enhance serum stability, increase specificity, and improve activity (Bumcrot et al. 2006; Addepalli et al. 2010). Manipulation of the naturally occurring 21-mer design has revealed that not only longer duplexes, such as dsiRNA, but also shorter duplexes and duplexes with altered overhangs participate in RNAi (Chu and Rana 2008; Chang et al. 2009).
In the present studies, we have sought to compare the target gene-silencing characteristics of canonical 21-mer duplexes and 25/27-mer duplexes characteristic of Dicer substrates. Using an unbiased gene-walk approach, we observed that the most active unmodified siRNAs and dsiRNAs show similar target knockdown in vitro as well as in vivo, with similar duration in vivo. Differences in activity, where present, were generally small in magnitude. In vitro we found that the best siRNAs and dsiRNAs differed on average by less than twofold in IC50 value. Similarly, in vivo ED50 values for active unmodified compounds differed on average less than twofold. Where present, statistically significant differences in ED50 between structures are only about twofold to threefold in magnitude. It is noteworthy that the lead compounds identified for each structural class are not sequence-matched, suggesting that sequence differentially influences the selection of the optimal targeting RNA. While the sequence-matched siRNA to the lead FVII-targeting dsiRNA was inactive unmodified, the chemically modified version showed activity similar to the dsiRNA. In the chemically modified set, one dsiRNA was rendered inactive by modification; considering the other compounds, we find comparable mean ED50 values for FVII-targeting compounds and an approximately twofold difference for PTEN-targeting compounds (favoring canonical). Statistically significant comparisons across structures indicated ED50 values for siRNAs to be twofold to fourfold lower than for dsiRNAs, with one dsiRNA ∼10-fold less active. Notably, no modified dsiRNA was more active than its matched siRNA. Chemical modification is expected to retain activity while reducing immune stimulation, compared with the unmodified parent. Differences in modification tolerability between sequence-matched siRNAs and dsiRNAs indicate an effect of structure. Therefore, we can hypothesize that for modification that reduces activity more for dsiRNAs than for siRNAs, the chemical modification interferes with Dicer cleavage or handling.
Our in vivo duration studies supported the same conclusions as the ED50 data, with similar longevity among active unmodified compounds of both structures. In the modified set, we note that for each unmodified parent siRNA, there was at least one modified variant that improved duration. The opposite was true for the dsiRNA: at least one variant in each set demonstrated reduced duration. Moreover, of the 10 modified compounds, five siRNAs showed greater longevity than the corresponding dsiRNA; only one modified dsiRNA was more durable in activity than the matched siRNA. The greater duration of modified siRNAs is particularly relevant in a therapeutic context, because a drug with shorter duration of action will require more frequent dosing. For a parenterally administered drug, such as LNP-based drugs, this could result in more frequent dose administration.
Because both unmodified siRNAs and dsiRNAs stimulated cytokine and chemokine production, it is clear that chemical modifications to abrogate immunostimulation are required for an optimal therapeutic. Unmodified dsiRNAs were more immunostimulatory than siRNAs in vitro and in vivo. This is consistent with published reports demonstrating that longer double-stranded RNAs, including dsiRNAs, are more immunostimulatory and reduce cell viability in certain cell types (Billy et al. 2001; Reynolds et al. 2006). Chemical modifications reduced immunostimulation by siRNAs and dsiRNAs in both settings, albeit to a lesser degree for dsiRNAs in vivo. Because the dsiRNA is ∼25% larger than the siRNA, more lipid is necessary for delivery and may therefore contribute to immunostimulation in vivo.
While similar in a basic research setting, dsiRNAs present certain additional challenges from a drug development point of view by virtue of their greater length. dsiRNAs are intended as a pro-drug, with the 21-mer being the active agent. For the purpose of comparing structures and matching sequences, we assumed that Dicer-mediated cleavage of the 25/27-mer would yield only the expected 21-mer. While it was beyond the scope of the present studies to empirically identify the active agent generated by Dicer, our assumptions were based on the principles outlined in Rose et al. (2005). If Dicer processing occurred at a site other than the predicted one, the active agent would then have a different target site that could result in altered activity. Alternatively, the full-length parent compound could remain unprocessed and load directly into RISC, bypassing Dicer cleavage altogether and constituting a distinct active agent (Salomon et al. 2010).
Dicer is known to be vital in a variety of biological settings, by virtue of its role in maturation of microRNAs. Dicer has been shown to be regulated, both up and down, in various cancers (Merritt et al. 2008, 2010; Catto et al. 2009; Faggad et al. 2010; Watashi et al. 2010; Wu et al. 2010), and reduced Dicer expression is associated with reduced survival in patients with ovarian cancer (Merritt et al. 2008). Additionally, cellular stresses affect Dicer levels in vitro (Wiesen and Tomasi 2009), and, as expected, dsiRNAs are less effective in diseased tissue with down-regulated Dicer (Merritt et al. 2008). RNAi therapeutics that depend on Dicer activity may thus result in a more heterogeneous effect across diverse patient populations.
In summary, we conclude that both siRNA and dsiRNA strategies are viable means of silencing genes. No class effect was observed to confer superior activity on either dsiRNAs or siRNAs, consistent with the ultimate active RNAi trigger being largely the same. Further investigation into chemical modification motifs amenable to dsiRNAs may ameliorate untoward effects on activity and reduce immunostimulation. Overall, we conclude that siRNAs are better suited for use as therapeutics than dsiRNAs due to better tolerance of chemical modifications, reduced immunostimulation, and smaller size.
MATERIALS AND METHODS
Canonical and Dicer substrate siRNA design
To facilitate a direct comparison of dsiRNA and canonical siRNA structures, we designed a large set of paired design sequences. We first constructed two sets of 21-mer siRNAs: one set targeted the conserved regions of mouse (NCBI Refseq NM_010172.3) and rat (NM_152846.1) coagulation Factor VII (FVII); the second set targets conserved regions of the PTEN gene (human, NM_000314.4; mouse NM_008960.2; rat NM_031606.1). The sense or passenger strand of the canonical 21-mer is identical to the target transcript along its entire length; the antisense or guide strand is the reverse complement of the sense strand. We selected the candidate 21-mer siRNAs using a proprietary algorithm that maximized the target transcript specificity.
Dicer-substrate compounds were designed according to the principles established by Rose et al. (2005). The antisense strand of each siRNA was aligned with the reverse complement of the mRNA transcript. dsiRNAs were then selected such that nucleotides 1–21 of the 27-nt strand are identical to the 21-mer antisense strand, and nucleotides 22–27 match the next 6 nt of the mRNA transcript. The dsiRNA sense strand is the reverse complement nucleotides 1–25 of the 27-mer. The last 2 nt at the 3′ end of the 25-mer are deoxy nucleotides, and the 5′ end is phosphorylated. Dicer cleavage of the 25/27-mer duplex should give rise to a product with the same sequence as the canonical siRNA. Sixty-seven and 63 matching duplexes were designed to target FVII (Supplemental Table S1) and PTEN (Supplemental Table S2), respectively.
Small-scale synthesis
FVII- and PTEN-targeting sequences were synthesized on a CPG solid support on a MerMade 192 synthesizer at 1-μmol scale. Amidite solutions were prepared at 0.1 M concentration, and ethyl thio tetrazole (0.6 M in acetonitrile) was used as activator. Deblock solution (3% dichloroacetic acid in dichloromethane), oxidizer solution (50 mM iodine in 9:1 pyridine–water mixture), and cap A and cap B solutions were prepared according to reported procedures (Beaucage 2008). At the end of the synthesis, solid support bound sequences were cleaved and deprotected in 96-well plates using methylamine (mixture of aqueous and ethanolic solutions in 3:1 ratio, 45°C, 90 min) in the first step and triethylamine.3HF in the second step (40°C, 90 min). Crude sequences were precipitated, and the pellet was resuspended in 20 mM sodium acetate buffer and analyzed by LC-MS to confirm the mass identity before purification.
Single strands were purified on an AKTA explorer purification system using a Source 15Q column. A salt gradient using buffer solution A (20 mM Tris-HCl, 1 mM EDTA, 10 mM NaClO4 at pH 7.5) and buffer solution B (20 mM Tris-HCl, 1 mM EDTA, 500 mM NaClO4 at pH 7.5) was used with solution B increasing from 27% to 55% over 10 min. Purification was performed using a Resource Q column (GE Life) and in-line buffer heater set at 60°C. A single peak corresponding to the full-length sequence was collected by automated fraction collection. Single strands were collected in 96-well plates and subsequently desalted on a Sephadex G25 column using an AKTA purifier instrument. Desalted FVII and PTEN single-strand sequences were analyzed for identity by LC-MS, purity by anion exchange chromatography, and concentration by UV absorbance measurement at A260 nm.
Medium-scale single-strand RNA synthesis
RNA oligonucleotides were synthesized at a scale of 1–2 μmol on an ABI 394 DNA/RNA Synthesizer or BioAutomation MerMade-12 using commercially available 5′-O-DMT phosphoramidite monomers (ChemGenes, Glen Research) following standard protocols for solid-phase synthesis and deprotection (Bernstein et al. 2001a; Fallows et al. 2001). Bis-cyanoethyl-N,N-diisopropyl phosphoramidite was used to synthesize phosphates (ChemGenes). The crude oligonucleotides were analyzed by LC-MS and purified by anion-exchange high-performance liquid chromatography (IEX-HPLC) with TSK-Gel SuperQ-5PW support (TOSOH Bioscience, Inc.) using a linear gradient (0.2%/min) of buffer B. Solutions of 0.02 M Na2HPO4 in 10%–15% CH3CN (pH 8.5) and 0.02 M Na2HPO4/1 M NaBr in 10%–15% CH3CN (pH 8.5) were used as eluents A and B, respectively. To ensure high fidelity of the data, all single strands were HPLC-purified to >85% purity, and purified oligonucleotides were desalted by size exclusion chromatography using Sephadex G25 (GE Healthcare). The purity and identity of the oligonucleotides were confirmed by ion-exchange chromatography and LC-MS, respectively.
Duplex annealing and endotoxin testing
Single-strand RNAs were verified by LC/ESI-MS using an XBridge C8 column (2.1 × 50 mm, 2.5 μm) and a linear gradient of buffers A (100 mM HFIP/16.3 mM TEA in H2O) and B (100% methanol) from 5% to 35% in 2 min and from 35% to 70% in 30.5 min. Duplexes were annealed by heating equimolar ratios of single strands in a water bath for 2 min at 95°C and slowly cooling them to room temperature. The absence of excess single strands was confirmed with an Advanced Analytics Oligo Pro 96 capillary CGE using Native Oligel matrix and buffers. A 10-kV injection and separation voltage was used for 60 min total runtime. A cutoff of 15% excess single strand was used. Endotoxin testing was performed using a QCL-1000 kit by Lonza. A maximum of 1 EU of lipopolysaccharides (LPS) per milligram of duplex was permitted for lipid nanoparticle duplexes, while 0.25 EU was allowed for unformulated duplexes.
Cell culture
The HeLa SS6 cell line was obtained from ATCC and cultured in EMEM (ATCC 30-2003) with 10% FBS (Omega Scientific, FB-02). HeLa Factor VII (FVII) cells were created by stable transfection of HeLa cells with mouse FVII and a gene providing resistance to blasticidin. Full-length mFVII CDS was cloned into pEF6/V5-His plasmid (Invitrogen) and transfected into cells. Mouse FVII cells were selected and maintained by culture in DMEM (GIBCO, 11995) with 10% FBS (Omega Scientific, FB-02) in 8 μg/mL blasticidin (InvivoGen, ANT-BL-1). Cells were maintained at 37°C and 5% CO2.
Transfection for duplex activity assessment
Duplexes and Lipofectamine RNAiMax (Invitrogen, 13778-150) were prepared in OptiMem I (GIBCO, 31985) solution. The duplexes and Lipofectamine RNAiMax solutions were mixed, and lipoplex formation proceeded for ∼20 min at room temperature before addition of cells. Ninety-six-well plates (BD Falcon, 353072) were seeded with 20,000 cells, 0.2 μL of Lipofectamine, and varying concentrations of duplex. For single-dose screens, a concentration of 0.1 nM was tested. A higher concentration (10 nM) was initially tested but was not found to be useful in distinguishing compounds by activity. IC50 values were determined for top-performing compounds from both canonical and Dicer-substrate structures using a sixfold dilution series containing eight concentrations ranging from 20 nM to 74 fM. For FVII-targeting duplexes, the media was changed 24 h following transfection. After an additional 24 h, media was recovered for FVII protein quantification, and cell lysis was performed for mRNA measurement. Samples transfected with PTEN-targeting duplexes were collected 48 h following transfection with no media change. LucC-S1HL was transfected as a control. At least two independent experimental replicates were performed, each containing a minimum of two biological replicates.
Viability assay
Canonical and sequence-matched Dicer-substrate duplexes were selected based on similar target suppression at 0.1 nM (<0.15-fold difference). Both FVII- and PTEN-targeting duplexes were tested. Duplexes were reverse-transfected at a concentration of 5 nM. As above, dilutions were prepared in OptiMem I (GIBCO, 31985). The final contents in the well of a 96-well plate were 2000 cells (HeLa SS6 or HeLa FVII), 0.2 μL of Lipofectamine RNAiMax (Invitrogen, 13778-150), and 5 nM duplex. Samples were collected on Days 2, 3, and 4 after transfection. Experiments were performed with three or six biological replicates.
Viability was assessed with the CellTiter Blue kit (Promega, G8081). Twenty microliters of CellTiter Blue was added to 100 μL of media per well and incubated for 90 min at 37°C. A Spectramax M5 (Molecular Devices) was used to measure fluorescence (560ex, 590em). Media plus CellTiter Blue, in the absence of cells, provides a measure of background fluorescence. Data are expressed as percent of same-day mock-transfected. Data were plotted in Graphpad Prism for determination of the area under curve (AUC).
Branched DNA assay (bDNA)
Quantification of mRNA was achieved by the Quantigene 2.0 branched DNA assay (Panomics, QS0011). Cell culture samples were lysed in 200 μL/well reconstituted lysis buffer containing 0.5 μg/mL proteinase K (Panomics, QS0512), followed by incubation for 60 min at 55°C. Lysates were diluted 2:3 for PTEN mRNA capture, 1:20 for mouse FVII, and 1:20 for human GAPDH. The probes used were human PTEN (Panomics, SA-10543), mouse FVII (Panomics, SB-13971), and human GAPDH (Panomics, SA-10001).
Liver samples snap-frozen in liquid nitrogen were placed in collection tubes (Nalgene, 2116-0015) for grinding. The liver tissues were processed in an SPEX GenoGrinder (250 strokes per second, 1× speed, 180 sec) with two or three stainless-steel balls per collection cup. A small amount of each tissue was transferred to Matrix Storage Tubes (ThermoFisher, 4252) on dry ice for lysis. Tissue & Cell Lysis solution (Epicentre, MTC096H) containing 0.5 μg/mL proteinase K (Panomics, QS0512) was then added to the lysates. Lysis proceeded for 30 min at 65°C with 1400 rpm agitation in an Eppendorf Thermomixer R. Samples were subjected to centrifugation at 15,000g for 10 min at room temperature, and the central 500 μL was removed to a deep-well plate. Clarified lysates were diluted 1:8 for capture by either target probes (FVII, PTEN) or the housekeeping gene (GAPDH). The probes used included mouse FVII (Panomics, SB-13971-02), mouse PTEN (Panomics, QS0051), and mouse GAPDH (Panomics, SB-10001).
The bDNA assay was performed as described by the manufacturer. Luminescence was measured on a Spectramax M5 (Molecular Devices) with an integration time of 200 msec. Background luminescence was measured by omitting sample and retaining all other assay steps; this value was subtracted from all data before data processing. Data are expressed as percentage of the nonspecific canonical control LucC-S1HL.
Chromogenic assay
Factor VII protein in cell culture media or mouse serum was quantified by chromogenic assay (Hyphen Biomed, 221304). For cell culture, the media was changed 24 h after transfection, and the media was removed after an additional 24 h for quantification of FVII protein. Media was diluted 1:400 in the supplied Tris-BSA buffer. Mouse serum was diluted 1:7000 for assay.
The assay was performed as described by the manufacturer. Absorbance was measured on a Spectramax M5 at 405 nm. Background absorbance was measured by omitting sample and retaining all other assay steps; this value was subtracted from all data. Data are expressed as percentage of the nonspecific canonical control LucC-S1HL.
Chemical modification of siRNA and dsiRNA
Chemical variants containing 2′-OMe, phosphorothioate (PS), and deoxy thymidine (dT) residues were prepared according to three motifs, termed heavy/light (HL), unmodified/alternating (UA), or heavy/alternating (HA), sense/antisense, respectively. The HL variant is composed of 2′-OMe modification of all pyrimidines on the sense strand (heavy), and 2′-OMe modification of only the uridine and cytidine residues that are followed by adenosine on the antisense strand (light). HL variant siRNAs contain two deoxythymidine residues with a phosphorothiate linkage (dTsdT) on the 3′ end; the terminal deoxythymidine bases were omitted for the HL variant of dsiRNA compounds to avoid potential disruption of Dicer-mediated cleavage. The UA variant consists of an unmodified sense strand and an antisense strand with 2′-OMe nucleotides in positions 11, 13, 15, 17, 19, 21, 23, 25, 26, and 27 from the 5′ end (alternating). FVII-targeting siRNAs contain a 3′ dTsdT on the sense strand, whereas PTEN-targeting siRNAs contain 3′ RNA (matching the expected cleavage product). The HA variant, implemented only for PTEN-targeting duplexes, was designed as a hybrid of the HL and UA motifs and used the HL sense (i.e., heavy) and the UA antisense strand designs (alternating). HA siRNAs contain a dTsdT on the 3′ end of the sense strand, which is omitted from the HA dsiRNA compounds.
PBMC isolation, culture, and assay
Whole-blood anti-coagulated with sodium heparin was obtained from healthy donors at Research Blood Components, Inc. Whole blood was diluted 1:1 in Hanks Balanced Salt solution (Invitrogen) before peripheral blood mononuclear cell (PBMC) isolation by Ficoll density centrifugation (Ficoll-Histopaque 1077; Sigma-Aldrich). Isolated PBMCs were washed three times in serum-free media and resuspended in RPMI 1640 GlutaMax Medium (Invitrogen) supplemented with 10% heat-inactivated fetal bovine serum and 1% antibiotic/antimycotic (Invitrogen). Purified PBMCs were cultured in 96-well plates at 1 × 106 cells/mL. siRNAs were complexed with 8 μg/mL DOTAP (Roche Applied Science) and transfected at a final concentration of 133 nM. Following incubation for 24 h at 37°C and 5% CO2, supernatants were harvested and stored at −80°C for future analysis. Supernatants were analyzed for IFN-α and TNF-α secretion using Instant ELISA Kits (Bender-Medsytems) according to the manufacturer's instructions.
Lipid nanoparticle (LNP) formulation
siRNAs and dsiRNAs were formulated in LNPs using the preformed vesicle process and were based on the recently described novel ionizablc cationic lipid DLin-KC2-DMA (Semple et al. Nature Biotechnology 2010). The formulation was composed of DLin-KC2-DMA, distearoylphosphatidylcholine (DSPC), cholesterol, and PEG2000-DMG (57.5/7.5/31.5/3.5 mol/mol), with a total lipid:nucleic acid ratio of ∼10:1 (w:w).
Animal studies
Female C57/BL6 mice were obtained from Charles River Laboratories at age 8 wk and used for all in vivo experiments. Animals were maintained on a 12-h light/dark cycle with ad libitum access to food and water.
LNP-formulated duplexes were diluted in PBS (GIBCO, 14190) and injected in a volume of 200 μL. Vehicle (PBS) and LNP-formulated LucC-S1HL were included as control treatments. For experiments involving PTEN or cytokine measurement, animals were deeply anesthetized at the indicated time points with an injection of Avertin. Blood was collected by abdominal bleed, followed by removal of the liver. Liver tissue was collected in jars (Nalgene, 2116-0015) and snap-frozen in liquid nitrogen. The blood was placed in serum-separator tubes (BD Microtainer, 365956) and incubated at room temperature for 30 min to 2 h before placing on ice. Animal blood was then subjected to centrifugation at 12,000 rpm for 6 min at room temperature; serum was subsequently removed to a new tube. Both serum and liver were stored at −80°C. For FVII-related experiments, blood was collected by retro-orbital bleed and handled as above.
In vivo cytokine and chemokines assay
Luminex cytokine assay serum samples were analyzed on the Bio-Plex 200 system, which uses the Luminex xMAP Technology (Luminex Corporation) for multiplex sandwich bead immunoassays. Bio-Plex Pro Mouse Cytokine 23-Plex Assay Kits were used according to the manufacturer's instructions to measure IL-1α, IL-1β, IL-2, IL-3, IL-4, IL-5, IL-6, IL-9, IL-10, IL-12(p40), IL-12(p70), IL-13, IL-17, Eotaxin, G-CSF, GM-CSF, INF-γ, KC, MCP-1, MIP-1α, MIP-1β, RANTES, and TNF-α. Serum samples were diluted 1:4 with sample diluent (provided in kit) before incubation with magnetic antibody beads. Following incubation, the plates were washed and incubated with biotinylated secondary antibodies fixing each target protein. A Streptavidin-phycoerythrin reporter complex was then added. The Bio-Plex200 instrument uses a dual-laser, flow-based reader system in which one laser identifies the specific emission signal of each bead and the second laser quantifies target protein via fluorescence intensity of bound phycoerythrin. Bio-Plex Manager Software V.5.0 specified acquisition of 50 beads per region (100 region setting), 50 μL of sample volume, and DD gates of 5000 (low) and 25,000 (high). LLOQs were determined based on BioPlex Software Manager 6.0, where LLOQ is the lowest standard above background fluorescence. Data below LLOQ were excluded from analyses.
Statistics
For activity assays, data were background-subtracted and normalized as percent of LucC-S1HL. IC50 values were calculated by XLFit (ID Business Solutions), using a four-parameter fit {fit = A + B/(1 + (x/C)D)}, where x is concentration, y is target mRNA remaining, A is the minimum of y, B is the range of y, C is the IC50 value of y, and D is slope factor. Values A, B, C, and D were pre-fit in the model. ED50 values were determined by three-parameter nonlinear regression using Graphpad Prism. Constraining the top and bottom values to 100 and zero, respectively, provided the best fit to the data. T50 values were also determined in Prism by linear regression and defined as the time at which activity equaled 50% of nonspecific control. ANOVA analysis was performed with Prism as described in the text. IC50, ED50, viability, and cytokine data were log-transformed before statistical analysis. For across-structure comparisons of cytokine data by ANOVA, a minimum N = 2 per cytokine and structure was required from each of three or more matched sequences. Comparison to PBS was performed by a two-tailed Student's t-test assuming unequal variance, with a minimum of twofold increase over PBS to be considered stimulating. Data below LLOQ were also below PBS and thus considered nonstimulating.
SUPPLEMENTAL MATERIAL
Supplemental material is available for this article.
ACKNOWLEDGMENTS
We acknowledge the contributions of Dr. Paul Peng, Haripriya Addepalli, and Boyd Conklin for duplex synthesis support; and J. Hettinger for Luminex assay performance. We also thank Dr. S. Milstein for comments on the manuscript, M. Livingston for figure preparation, and A. Capobianco for assistance in preparing the manuscript.
Footnotes
-
↵4 Corresponding author.
E-mail dfoster{at}alnylam.com.
-
Article published online ahead of print. Article and publication date are at http://www.rnajournal.org/cgi/doi/10.1261/rna.031120.111.
- Received October 25, 2011.
- Accepted December 19, 2011.
- Copyright © 2012 RNA Society








