
Identification of the enzyme responsible for N1-methylation of pseudouridine 54 in archaeal tRNAs
- Jan Philip Wurm1,
- Marco Griese1,
- Ute Bahr2,
- Martin Held2,3,
- Alexander Heckel2,3,4,
- Michael Karas2,4,
- Jörg Soppa1 and
- Jens Wöhnert1,4,5,6
- 1Institut für Molekulare Biowissenschaften, Johann-Wolfgang-Goethe-Universität, 60438 Frankfurt/M., Germany
- 2Institut für Pharmazeutische Chemie, Johann-Wolfgang-Goethe-Universität, 60438 Frankfurt/M., Germany
- 3Institut für Organische Chemie und Chemische Biologie, Johann-Wolfgang-Goethe-Universität, 60438 Frankfurt/M., Germany
- 4Cluster of Excellence “Macromolecular complexes,” Johann-Wolfgang-Goethe-Universität, 60438 Frankfurt/M., Germany
- 5Center for Biomolecular Magnetic Resonance (BMRZ), Johann-Wolfgang-Goethe-Universität, 60438 Frankfurt/M., Germany
Abstract
tRNAs from all three kingdoms of life contain a variety of modified nucleotides required for their stability, proper folding, and accurate decoding. One prominent example is the eponymous ribothymidine (rT) modification at position 54 in the T-arm of eukaryotic and bacterial tRNAs. In contrast, in most archaea this position is occupied by another hypermodified nucleotide: the isosteric N1-methylated pseudouridine. While the enzyme catalyzing pseudouridine formation at this position is known, the pseudouridine N1-specific methyltransferase responsible for this modification has not yet been experimentally identified. Here, we present biochemical and genetic evidence that the two homologous proteins, Mja_1640 (COG 1901, Pfam DUF358) and Hvo_1989 (Pfam DUF358) from Methanocaldococcus jannaschii and Haloferax volcanii, respectively, are representatives of the methyltransferase responsible for this modification. However, the in-frame deletion of the pseudouridine N1-methyltransferase gene in H. volcanii did not result in a discernable phenotype in line with similar observations for knockouts of other T-arm methylating enzymes.
Keywords
INTRODUCTION
Modified nucleotides are common in rRNAs and tRNAs from all branches of life. The chemical nature of these modifications ranges from the simple introduction of methyl groups on one hand to hypermodified nucleosides such as wybutosine (Blobstein et al. 1973) or agmatidine (Mandal et al. 2010) on the other hand. To date, more than 100 chemically different modifications have been described for tRNA nucleotides (Rozenski et al. 1999). Modified nucleotides in tRNAs can be functionally important and help to stabilize the tertiary structure of the tRNA or prevent misfolding and premature tRNA-turnover (Motorin and Helm 2010). For example, modifications at position 34 in the anticodon loop often enhance the precision of decoding by stabilizing codon–anticodon interactions at the “wobble-position” (e.g., Murphy et al. 2004). A thiolated nucleotide at position 54 was found to stabilize the interactions between the T-arm and the D-arm of tRNAs from thermophilic bacteria (Yokoyama et al. 1987), whereas the introduction of a single N1-methylation at position A9 is sufficient to prevent a human mitochondrial tRNA transcript from misfolding (Helm et al. 1999). Some modifications, such as the pseudouridine (Ψ) at position 55, are very common in tRNAs, while others are restricted to specific tRNA species of certain organisms. The so-called D- and T-arms in the classical tRNA-cloverleaf secondary structure actually received their names due to the common occurrence of specific modifications in their loops. In most eubacterial and eukaryotic tRNAs dihydrouridine (D) is present at position 15 or 16 in the D-loop, whereas ribothymidine (rT), pseudouridine (Ψ), and cytidine (C) are typically encountered at positions 54, 55, and 56, respectively, in the T-loop (or TΨC-loop).
Archaeal tRNAs not only contain some unique types of modified nucleotides such as archaeosine (Edmonds et al. 1991; Gregson et al. 1993), but also deviate with regard to their modification patterns from bacterial and eukaryal tRNAs. One prominent example is the above-mentioned modification at position 54 of the T-arm. Instead of ribothymidine (rT), most branches of archaea, except for members of the Thermococcales and Nanoarchaea (Urbonavicius et al. 2008), contain the isosteric N1-methyl pseudouridine at this position (Pang et al. 1982; Gupta 1984). The biosynthetic pathway for N1-methyl pseudouridine at position 54 starts with the U to Ψ transformation catalyzed by the pseudouridine synthase Pus10 (Gurha and Gupta 2008), which also modifies U at position 55 to pseudouridine. While the enzymes responsible for the conversion of the uridine into ribothymidine at position 54 have been described in eukaryotes (Nordlund et al. 2000), bacteria (Ny and Björk 1980; Urbonavicius et al. 2005) and archaea (Urbonavicius et al. 2008); the specific pseudouridine N1 methyltransferase responsible for the methylation of pseudouridine 54 has not yet been identified. However, bioinformatic approaches suggested a possible role for members of the COG1901 protein family (Tkaczuk et al. 2007) in pseudouridine methylation, since their genes sometimes colocalize with the genes for Pus10 homologs. This COG includes Mja_1640 from Methanocaldococcus jannaschii and other proteins with similarities to Hvo_1989 from Haloferax volcanii (Pfam DUF358). However, some members of COG1901 are proteins from archaea that have a ribothymidine modification at position 54, e.g., Pyrococcus abyssi (Urbonavicius et al. 2008). Furthermore, no biochemical or genetic data have been presented so far in the literature that support a pseudouridine N1-methyltransferase activity of COG1901 (Pfam DUF358) family members.
To date, only two enzymes have been described that are able to further modify pseudouridines by methylation. One is the S-adenosylmethionine (SAM)-dependent pseudouridine N3-methyltransferase YbeA found in eubacteria that acts on 23S rRNA (Ero et al. 2008; Purta et al. 2008). The other is the N1-specific pseudouridine methyltransferase Nep1 found in archaea and eukaryotes (Wurm et al. 2010). Nep1 is a member of the so-called SPOUT-class of RNA-methyltransferases (Tkaczuk et al. 2007; Leulliot et al. 2008; Taylor et al. 2008; Wurm et al. 2009) and is responsible for the N1-specific pseudouridine methylation of position 1191 (Saccharomyces cerevisiae numbering, nucleotide 913 in M. jannaschii) in the small ribosomal subunit RNA (Wurm et al. 2010; Meyer et al. 2011). A mutation in human Nep1 is the cause for the Bowen-Conradi syndrome, a deadly developmental disorder (Armistead et al. 2009). A costructure with an RNA inhibitor that contains a U at the position of the pseudouridine reveals how the enzyme achieves its specificity for the N1 nitrogen of the pseudouridine (Thomas et al. 2011). Interestingly, the rRNA sequence that is modified by Nep1 has notable similarities to the T-arm of archaeal tRNAs (Fig. 1).
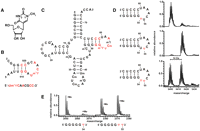
MjNep1 mis-methylates pseudouridine containing substrates corresponding to tRNA T-arms at position 55. (A) Chemical structure of N1-methyl pseudouridine. (B) Secondary structure and sequences of minimal substrates for MjNep1 derived from the small subunit rRNA helix 35 (Wurm et al. 2010). Nucleotides that are similar between these substrates and T-arm sequences of archaeal tRNAs are highlighted in red. (C) Secondary structure of H. volcanii tRNATrp with the modified nucleotide positions indicated and sequence similarities to MjNep1 substrates highlighted in red. The T-arm sequences and secondary structures of H. volcanii and M. jannaschii tRNATrp are virtually identical. (D) Methylation activity of MjNep1 toward 17 mer RNA substrates resembling the T-arm of M. jannaschii tRNATrp with pseudoruridines at position 54 (top), 55 (middle), or both (bottom) analyzed by MALDI mass spectrometry. Only the RNAs with pseudouridine at positions 55 or 54 and 55 are methylated after 5-min reaction time at 65°C. (E) Identification of the methylation site in the RNA substrate containing two pseudouridine residues at positions corresponding to Ψ54 and Ψ55 in full-length tRNAs by partial acidic hydrolysis coupled to MALDI mass spectrometry of the reaction product. Shown are the sections of the spectrum for two indicated 5′ fragments.
Here, we show by biochemical means that Nep1 from Methanocaldococcus jannaschii (MjNep1) itself is not the methyl transferase responsible for the N1-methyl pseudouridine modification at position 54 of the T-arm in archaeal tRNAs. However, another SPOUT-class methyltransferase with a highly similar spatial arrangement of amino acids in its active center—Mja_1640—is capable of methylating pseudouridine containing RNAs resembling the T-arm or complete transcripts of archaeal tRNAs in vitro with the proper sequence specificity. Furthermore, a H. volcanii mutant with a knockout of the gene coding for the Hvo_1989 protein—the H. volcanii homolog of Mja_1640—is no longer able to introduce the N1-methylation at position 54 in its tRNAs in vivo.
RESULTS AND DISCUSSION
M. jannaschii Nep1 mismethylates T-arm resembling RNAs
The sequence of the Nep1 target site of the small ribosomal subunit rRNA shows notable sequence similarities to the T-arms of M. jannaschii tRNAs (Fig. 1). Thus, using synthetic 17 mer RNAs resembling the T-arm of M. jannaschii tRNATrp that contained a pseudouridine at either position 54, 55, or both, we tested the capability of MjNep1 to methylate these substrates. To this end, the RNAs were incubated with the purified protein in the presence of SAM at 65°C for 5 min and analyzed by MALDI-mass spectrometry. MjNep1 methylated the substrate with a pseudouridine at position 55 with reasonable efficiency, but markedly slower than its native small ribosomal subunit rRNA-derived substrate (Wurm et al. 2010) as seen from the increase in mass by 14 Da corresponding to a methyl group (Fig. 1D). However, no methylation was observed for the substrate with pseudouridine at position 54 (Fig. 1D). Furthermore, the substrate with pseudouridines at both positions 54 and 55 is methylated only once, since only a reaction product with a mass shift of 14 Da is observable (Fig. 1D), but with reduced efficiency compared with the RNA with pseudouridine at position 55. HPLC-analysis of nucleosides after RNA digestion and dephosphorylation showed that, based on its retention time, the modified nucleoside is N1-methyl pseudouridine (Supplemental Fig. 1) as expected for a Nep1-catalyzed methylation reaction. In order to also verify that in the substrate with pseudouridines at the positions corresponding to 54 and 55 in tRNA only pseudouridine 55 is modified, we sequenced the reaction product using partial acidic hydrolysis in conjunction with MALDI mass spectrometry. Mass analysis of the resulting RNA fragments showed that the fragment 5′-GGGGGΨ-3′ is only present in the nonmethylated form, whereas for the fragment 5-GGGGGΨΨ-3′ signals for both the methylated and the unmethylated form are observable with similar intensities (Fig. 1E; Supplemental Table 1) as expected for a methylation reaction involving exclusively Ψ55. Methylation at pseudouridine 55 has not yet been observed in nature. Furthermore, we did not observe any modification of bacterial tRNAs that contain an unmodified Ψ at position 55 in vivo after MjNep1 overexpression in E. coli (Supplemental Fig. 2), nor was MjNep1 capable of modifying mature H. volcanii tRNAs (either from a wild-type strain with N1-methyl-Ψ and Ψ at positions 54 and 55, respectively, or from the mutant strain described below with two unmodified Ψ's at these positions) in vitro (Supplemental Fig. 3). Thus, the enzymatic activity of MjNep1 with regard to Ψ55 modification in small T-arm resembling RNA hairpins is most likely an in vitro artefact. The latter observations also could explain why there is no mismethylation of full-length tRNAs by MjNep1 in vivo. Apparently, either their stable tertiary structure, which involves interactions between the D- and T-arms, or the other post-transcriptional modifications of the T-arm that are not present in the 17 mer synthetic RNAs prevent accidental Ψ55 methylation by MjNep1 in full-length tRNAs.
Because of its different sequence specificity, MjNep1 is apparently not the pseudouridine-N1 methyltransferase responsible for the tRNA-modification at position 54 in archaeal tRNAs. This is further supported by the observation that some archaea such as H. volcanii, which do not have a Nep1 gene, still harbor the N1-methylpseudouridine modification at position 54.
Mja_1640 methylates T-arm resembling tRNAs in vitro
Recently, Chen and Yuan (2010) solved the structure of protein Mja_1640 from the hyperthermophilic M. jannaschii, revealing another member of the SPOUT-class family of RNA methyltransferases. Interestingly, Mja_1640 has an overall structure very similar to MjNep1 (Fig. 2). However, a structure-based sequence alignment with the MUSTANG algorithm (Konagurthu et al. 2010) yields only 12% identity and 24% similarity. Notably, the structural similarities include the exact spatial arrangement of amino acid side chains in the putative active centers of the two proteins (Fig. 2B). Most importantly, an Arg-Xaa-Asp motif at the beginning of α-helix 2 absolutely conserved in Nep1-homologs, is also present in Mja_1640 at the same position. This motif is required to fix the position of the arginine side chain by strong hydrogen bonds to the aspartate side chain of the same motif in such a way that it allows specific hydrogen-bonding interactions with the N3-face of the substrate pseudouridine (Fig. 2B). This, in turn, leads to an orientation of the bound pseudouridine base with the N1 nitrogen pointing toward the methyl group of the SAM-cofactor (Fig. 2B). The motif is conserved in all members of the DUF358 family either as Arg-Xaa-Asp (archaeal sequences) or as His-Xaa-Glu (eubacterial sequences), both of which might be able to orient a pseudouridine base similar to Nep1. Furthermore, Chen and Yuan (2010) showed that Mja_1640 binds the same small ribosomal subunit rRNA-derived substrates as MjNep1. However, they failed to detect RNA-methyl transferase activity with these rRNA-derived substrates. Thus, we speculated that Mja_1640 might, instead, be able to act on the archaeal tRNA T-arms.
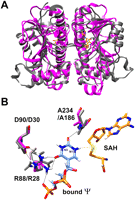
Structural similarity between Nep1 and Mja_1640. (A) Overlay of the structures of the MjNep1 (pdb 3bbe) and Mja_1640 (pdb 3aia) dimers revealing the similar overall fold typical for SPOUT-class RNA methyltransferases. (B) Model of pseudouridine bound to the active center of Nep1 derived from the structure of yeast Nep1 bound to S-adenosylhomocysteine and an inhibitor RNA containing uridine instead of pseudouridine at the reactive position (pdb 3oin). The amino acids R88, D90, and A234, which form hydrogen bonds to the pseudouridine carbonyl groups and orient the N1 nitrogen toward the sulfur atom of the cofactor, are colored by atom types. The equivalent amino acids in Mja_1640 are shown in gray and numbered according to the Mja_1640 numbering scheme.
As expected for a hypothetical pseudouridine methyltransferase, no reaction product with an increased mass was observed when an RNA-substrate with U at positions 54 and 55 was incubated with Mja_1640 and SAM (Fig. 3A). However, Mja_1640 methylated the substrate containing pseudouridine at position 54, as well as the substrate with pseudouridines at positions 54 and 55 very efficiently, as seen in the MALDI-mass spectra of the RNA after a 5-min reaction time. In contrast, the substrate with pseudouridine only at position 55 was not methylated at all (Fig. 3A). In the case of the doubly pseudouridinilated substrate, only the signal corresponding to a single methylation with a mass increase of 14 Da was observable. Analysis of the modified nucleoside after RNA digestion and dephosphorylation by HPLC revealed that the modified nucleotide has the same retention time as commercially available N1-methyl pseudouridine (Fig. 3B; Supplemental Fig. 4). To establish the sequence specificity of Mja_1640 in the reaction with the substrate containing pseudouridines at positions 54 and 55, we again subjected the reaction product to sequencing by MALDI-mass spectrometry after partial acidic hydrolysis (Bahr et al. 2009). The mass analysis of the resulting fragments showed in this case that the fragment 5′-GGGGGΨ-3′ is present almost exclusively in its methylated form (Fig. 3C; Table 1) in contrast to what was observed for the reaction with MjNep1, where this fragment was observed only in its unmethylated form. Thus, based on the MALDI sequencing results (Table 1), Mja_1640 preferentially modifies the Ψ in the T-arm hairpins that correspond to Ψ54, in agreement with the results obtained for the RNA substrates containing only one pseudouridine.
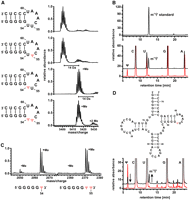
RNA-methyltransferase activity of Mja_1640. (A) Methylation activity of Mja_1640 toward T-arm substrates containing no pseudouridine (top), pseudouridine at position 55 (second from top), pseudouridine at position 54 (second from bottom), or pseudouridine at positions 54 and 55 (bottom). In the last two cases a complete methylation is observed after a 5-min reaction time as indicated by the shift in the mass spectrum by 14 Da. (B) Identity of the modified nucleotide. HPLC chromatograms of nucleoside mixtures after digestion and dephosphorylation of the 17 mer RNA substrate containing pseudouridine at position 54. The HPLC chromatogram of the unreacted substrate RNA contains only peaks corresponding to Ψ, C, U, G, and A (red trace), whereas upon methylation by Mja_1640 the Ψ peak disappears, and a novel peak appears with a retention time typical for N1-methyl pseudouridine (black trace). A reference for commercially obtained N1-methyl pseudouridine is shown on top. (C) Mass spectra of the T-arm substrate containing two pseudouridines after methylation with Mja_1640 and limited acidic hydrolysis. Shown are the sections of the spectrum for two indicated 5′- fragments indicative of methylation at the pseudouridine corresponding to position 54 in full-length tRNAs. (D) Secondary structure for a 74 mer RNA substrate resembling unmodified M. jannaschii tRNATrp with a single pseudoruridine at position 54. HPLC traces for unreacted (red) and Mja_1640-treated 74 mer RNA after digestion and dephosphorylation and after a 1-h reaction time (black). A novel peak corresponding to N1-methyl pseudouridine appears following incubation with Mja_1640 and SAM.
Masses of the T-arm analog fragments determined by MALDI mass spectrometry after methylation with Mja_1640 and partial acidic hydrolysis
Thus, in vitro Mja_1640 displays the same sequence specificity as expected for the putative pseudouridine 54 modifying methyltransferase, at least on substrates resembling the isolated T-arm of archaeal tRNAs.
To test the in vitro activity of Mj1640 on a substrate more similar to native tRNAs, we used a synthetic 74 mer RNA corresponding to nucleotides 1–74 of the unmodified M. jannaschii tRNATrp that contained a single pseudouridine at position 54. HPLC analysis of the nucleoside digest of this RNA showed that this substrate is methylated as well at the only pseudouridine in the molecule. Furthermore, the retention time of the modified pseudouridine corresponds to N1-methyl pseudouridine, as observed previously for the modified nucleotide of the T-arm substrates (Fig. 3D). The reaction was observed to proceed both in the absence and in the presence of Mg2+-ions, indicating that the fully folded tertiary structure of the tRNA is not necessary for recognition by this enzyme.
A Hvo_1989 knockout strain of H. volcanii lacks N1-methyl pseudouridine modifications at position 54 of its tRNAs
While Mja_1640 is very well suited for in vitro studies of its enzymatic activity, its activity in vivo is hard to address, since M. jannaschii is not easily genetically manipulated. However, knockout mutants can be created in H. volcanii, a halophile archaeon that contains the gene hvo_1989 coding for a close homolog of Mja_1640 (42% identity, 60% similarity).
Both proteins are members of the Pfam DUF358 family. A sequence alignment for the two proteins and other members of DUF358 is given in Figure 4A. Position 54 of all H. volcanii tRNAs contains the N1-methyl pseudouridine modification (Gupta 1984). Importantly, there is no N1-methyl pseudouridine modification present in any other position of the H. volcanii tRNAs. To enable the characterization of the biological role of Hvo_1989 in vivo an in-frame deletion mutant of the gene was constructed using the so-called Pop-In-Pop-Out strategy (Allers et al. 2004; Hammelmann and Soppa 2008). The genomic organization of the mutant at the Hvo_1989 locus was verified by Southern blot analysis (Supplemental Fig. 5).
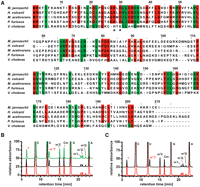
Hvo_1989 activity in vivo. (A) Hvo_1989 and Mja_1640 are highly homologous proteins, as seen from the sequence alignment of members of the DUF 358 family. Identical amino acids of the archaeal members of this family are colored in red, similar amino acids are shown in green. The arginine and the aspartate important for pseudouridine recognition and orientation are indicated by a star. Only in the bacterial member of this family these amino acids are a histidine and a glutamate, respectively. (B) Nucleoside analysis of unfractionated tRNAs isolated from wild-type (black) and the in-frame deletion mutant cells (red) and mutant cells complemented with the pSD1/M2-18 plasmid (green). For the tRNAs from wild-type and complemented strains the N1-methyl pseudouridine signal (indicated in red) is clearly present, while the signal is absent in the case of the in-frame deletion mutant. The putative assignments for the signals corresponding to other modified nucleosides are also indicated (Edmonds et al. 1991). (C) Nucleoside analysis of tRNAs isolated from in-frame deletion mutant cells (red) and the same tRNAs after incubation with SAM and Mja_1640 (black). The N1-methyl pseudouridine signal only appears after incubation with Mja_1640.
Native tRNAs were isolated from both the wild-type and the knockout mutant, and their nucleoside modification patterns were analyzed by HPLC. Whereas in tRNAs from the wild-type cells N1-methylpseudouridine was clearly present, the peak corresponding to N1-methyl pseudouridine was totally absent for the tRNAs derived from the knockout strain. At the same time, the peak corresponding to unmodified pseudouridine increased in intensity (Fig. 4B), while the signals corresponding to the other modified nucleosides in the HPLC chromatogram do not differ between wild-type and knock-out cells. Thus, the knockout of Hvo_1989 leads to a specific loss of the N1-methyl modification of pseudouridine 54 in vivo in agreement with its suspected role as the methyltransferase responsible for this specific modification.
We then amplified the hvo_1989 gene and cloned it into the plasmid pSD1/M2-18 (Danner and Soppa 1996), thereby placing it under the control of a synthetic promoter of intermediate strength. The H. volcanii deletion strain described above was transformed with the resulting plasmid and successful complementation was verified. tRNAs were isolated from the wild-type, the deletion mutant, and the complemented deletion mutant and analyzed for the presence of the N1-methyl pseudouridine modification. As expected, Hvo_1989 expression from the plasmid indeed restored the modification of tRNAs in H. volcanii cells carrying the hvo_1989 in-frame deletion (Fig. 4B).
Unfortunately, the function of Hvo_1989 cannot be analyzed in vitro using purified protein, since it is expressed mainly in insoluble form in inclusion bodies in E. coli, and attempts to refold it failed in our hands. However, to strengthen the link between the biochemical observations described for Mja_1640 and the knockout phenotype observed for Hvo_1989, we analyzed whether purified Mja_1640 is able to modify native tRNAs isolated from the H. volcanii knockout mutant that lack the N1-methylpseudouridine at position 54, but are otherwise fully modified. In agreement with the expected similar function of the two proteins, Mja_1640 is able to introduce the N1-methylpseudouridine modification into these tRNAs in vitro (Fig. 4C).
To test the importance of the N1-methyl modification of pseudouridine 54 in vivo we compared growth curves of the wild-type and the hvo_1989 in-frame deletion strain at different temperatures. Wild type and mutant grew indistinguishable at the optimal growth temperature of 42°C, at 37°C, and even at the maximal growth temperature of H. volcanii, 51°C.
As the magnesium concentration can have an effect on RNA folding, wild-type and the in- frame deletion mutant of Hvo_1989 were also grown at two different magnesium concentrations (110 mM, 55 mM) and in the absence of magnesium. However, all growth curves were identical (data not shown), indicating either that the magnesium concentration does not differentially influence folding of methylated and unmethylated tRNAs or—more likely—that H. volcanii accumulates magnesium and it is impossible to generate cells that have a magnesium-depleted cytoplasm.
The absence of a clear phenotype for the hvo_1989 in-frame deletion mutant indicates that the methyl modification at the N1 position of Ψ54 is, at best, only marginally important for the proper biological function of H. volcanii tRNAs, even under extreme conditions. The addition of the methyl group to the N1 of the pseudouridine renders this nucleotide isosteric to the ribothymidine found in the equivalent position of eubacterial and eukaryotic tRNAs. In vitro studies by biophysical methods (e.g., Davanloo et al. 1979) suggested a stabilizing role for this modification in maintaining the three-dimensional structures of tRNA. However, mutant or knockout strains missing the ribothymidine modification at position 54 in yeast (Trm2) (Nordlund et al. 2000), Bacillus subtilis (TrmFO) (Urbonavicius et al. 2005), and E. coli (Björk and Neidhardt 1975) show no or only very slight growth impairments, even under extreme conditions. Only the simultaneous lack of modifications at positions 54 and 55 in the T-arm and position 18 in the D-arm resulted in reduced growth and translational defects in E. coli (Urbonavicius et al. 2002). Thus, in line with these observations the structural effects of the absent modification in the in-frame deletion mutant of Hvo_1989 are apparently too subtle to lead to a detectable phenotype.
Phylogeny of the methyltransferase family
A similarity search in the nonredundant UniProt protein sequence database revealed the existence of nearly 200 proteins with meaningful similarity to HVO_1989. About 70 sequences with the highest similarity were all from euryarchaea, the remaining sequences were from γ proteobacteria. Fourteen sequences stemmed from Halobacteriales and exhibited a degree of identity of at least 60% to the H. volcanii HVO_1989 protein. Thirteen proteins exemplifying many euryarchaeal genera and three bacterial proteins (from Vibrio cholerae M66-2, Photobacterium profundum, and Shewanella baltica OS223) were selected, and a multiple sequence alignment was generated (Supplemental Fig. 6). It was used to construct phylogenetic trees using the parsimony and the distance matrix program of the PHYLIP program package (Felsenstein 1996). Notably, the obtained trees were very similar to the 16 S rRNA tree (Supplemental Fig. 7). In particular, the haloarchaea, as well as the euryarchaea, were monophyletic groups supported by high bootstrap values (100 of 100). This indicates that the lateral gene transfer between the euryarchaea and the γ proteobacteria was an ancient event, and the broad distribution in euryarchaea indicates that the methyltransferase gene arose in the euryarchaea and was later transferred to some proteobacterial species.
CONCLUSIONS
In summary, we have shown that the two closely related archaeal members of the COG1901 and Pfam DUF358 families, Mja_1640 and Hvo_1989, are able to modify pseudouridine 54 in the T-arm of archaeal tRNAs by methylation of its N1 position in vitro and in vivo. Thus, these proteins are only the second example for enzymes with a specific pseudouridine N1-methyltransferase activity. Structural similarities in the active centers of these proteins with the Nep1-protein family rationalize their preference for modification of the N1-face of pseudouridine and will be helpful to identify other methyltransferases with a similar specificity based on their three-dimensional structures.
In line with findings for the isosteric ribothymidine modification at the equivalent position in eubacterial and eukaryotic tRNAs, the in-frame deletion mutants for hvo_1989 are viable and show no growth impairment even under extreme temperature conditions. Thus, the presence of the N1-methylpseudouridine modification contributes only marginally to the stability and function of H. volcanii tRNAs in vivo.
MATERIALS AND METHODS
Protein production and purification
Full-length MjNep1 (aa 1–205) was overexpressed in BL21(DE3)-RIPL Escherichia coli cells (Stratagene) and purified as described previously (Taylor et al. 2008). For the expression of Mja_1640, a synthetic gene with the appropriate sequence including an N-terminal His-tag optimized for overexpression in E. coli was cloned into the pET11a overexpression vector. Mja_1640 was expressed in BL21(DE3)-RIPL E. coli cells (Stratagene) for 3 h at 37°C in LB medium after induction with 1 mM IPTG at an OD600 of 0.8. Cells were harvested by centrifugation and resuspended in 500 mM NaCl, 10 mM imidazole, 5 mM β-mercaptoethanol, 50 mM sodium phosphate (pH 7.5). Complete protease inhibitor (Roche), RNase, and DNase were added, and cells were lysed by sonication. Cleared lysate was applied to a Ni-NTA column (GE healthcare) equilibrated in 500 mM NaCl, 10 mM imidazole, 2 mM β-mercaptoethanol, 50 mM sodium phosphate (pH 7.5). Mja_1640 was eluted with a linear imidazole gradient from 10 mM to 500 mM and further purified by gel filtration (Sephacryl S-100 16/60 column [GE healthcare]).
Methyltransferase assays
RNAs were purchased (Dharmacon) and deprotected according to the manufacturer's instructions. Methyltransferase reactions with 17 mer RNAs and tRNAs extracted from H. volcanii cells were performed in 100 mM KCl, 15 mM sodium phosphate (pH 7.5) with 20 μM RNA, 500 μM SAM, and 20 μM protein at 65°C for 1 h (tRNAs) or 5 min (17 mer RNAs). Methylation of the 74 mer was carried out in 100 mM KCl, 25 mM Tris/HCl (pH 7.5) in the presence or absence of 1 mM MgCl2 at 50 μM RNA concentration for 1 h at 65°C. Samples for HPLC and MALDI mass analysis were phenol–chloroform extracted, ethanol precipitated, lyophilized, and resuspended in water.
For mass analysis, 0.5 μL of RNA solution were mixed with 2 μL of matrix solution directly on the MALDI target and dried in a stream of air. 3-hydroxypicolinic acid (40 mg/mL in water) containing 5 mg/mL of diammonium hydrogen citrate was used as matrix. MALDI mass spectrometry was carried out on a MALDI TOF mass spectrometer (Voyager STR, Applied Biosystems) in the reflector positive ion mode. The following settings were used: sample target 20 kV, grid voltage 76%, delay time 600 nsec. Spectra were accumulated over 100 laser shots. Sequence analysis was performed by partial acidic hydrolysis coupled to MALDI mass spectrometry according to the procedure published recently (Bahr et al. 2009).
Construction of a knockout strain for Hvo_1989 in H. volcanii
The vector for the construction of an in-frame deletion of gene hvo_1989 was generated using a recently described method (Hammelmann and Soppa 2008). In short, the upstream and the downstream regions of the gene were amplified by PCR using the following oligonucleotides:
-
Hvo_1989-H1-For: 5′-GACGAGGTCGTGTTCCGCGACGC-3′,
-
Hvo_1989-H1-Rev: 5′-GCCCGCCGTGTC GGTGACGATGAATTGGCGCATGCG-3′
-
Hvo_1989-H2-For: 5′-CAATTCATCGTCACC GACACGGCGGGCTATTCGCGGTAC-3′
-
Hvo_1989_H2-Rev: 5′-GTCGGAGACACCGTGGATTCGTCGG-3′.
The nucleotides shown in bold led to an overlap of the two PCR fragments, which could be “fused” into one fragment via a third PCR reaction with the primers Hvo_1989-H1-For and Hvo_1989-H2-Rev. The fragment was cloned into the vector pMH101 using restriction selection cloning (Hammelmann and Soppa 2008). The resulting plasmid was used to transform H. volcanii H26 and select for the in-frame deletion mutant using the so-called Pop-In-Pop-Out method (Allers et al. 2004). The mutant was identified by colony PCR and the deletion was verified by Southern blot analysis.
Complementation of the deletion mutant
The hvo_1989 gene was amplified with genomic DNA from H. volcanii as the template using the following primers:
-
Kompl.1989-for “caaactCCATGGCGATGCGCCAATTCATCGTCACCGGC” and
-
Kompl.1989-rev “caaactGGTACCTCAGTACCGCGAATAGCCCGCCGTG.”
The primers introduced a NcoI and a KpnI site, respectively (shown in bold). The PCR fragment was cut with both enzymes and cloned into the vector pSD1/M2-18 (Danner and Soppa 1996) that had been cut with the same enzymes. The hvo_1989 gene thereby replaced the dhfr gene present in pSD1-M2-18 and is put under the control of a synthetic constitutive promoter of intermediate strength. Error-free integration of the gene was verified by sequencing. The resulting plasmid was used to transform H. volcanii, and the complemented strain was characterized by comparative growth experiments with the deletion mutant and the wild type and by analyzing the methylation pattern of its tRNAs (see below).
tRNA-isolation from H. volcanii and E. coli
E.coli BL21(DE3)-RIPL (Stratagene) cells were transformed with the plasmids used for protein production and grown in LB medium supplemented with 100 mg/L of ampicillin and 50 mg/L of chloramphenicol. As a control, untransformed cells were grown in LB medium supplemented with 50 mg/L of chloramphenicol. Protein expression was induced at an OD600 of 0.8 by addition of IPTG (final concentration 1 mM), and cells were harvested after 3 h at 37°C. Total RNA from E. coli, H. volcanii H26, and HVO_1989 cells was extracted according to previously described protocols (Chomczynski and Sacchi 1987). tRNAs were purified by preparative gel electrophoresis on a 12% denaturing polyacrylamide gel, eluted from gel slices in 300 mM sodium acetate (pH 6.5) overnight at 4°C ethanol precipitated and dissolved in water.
HPLC-analysis of modified nucleosides
RNAs were hydrolyzed and dephosphorylated using P1 nuclease (Sigma Aldrich) and bacterial alkaline phosphatase (Sigma Aldrich) following the protocols of Gehrke and Kuo (1989). Analytical HPLC of the resulting nucleoside mixture was performed on a Supelcosil LC-18-S HPLC column (Sigma Aldrich) equipped with a pre-column on an Agilent 1200 HPLC system (Agilent) at 30°C using buffers and gradients according to Pomerantz and McCloskey (1990). Elution of nucleosides was monitored at 280 nm. Chemically synthesized N1-methylpseudouridine used as an external reference was purchased from Berry & Associates.
Software and construction of phylogenetic trees
Molecular graphics images were produced using the UCSF Chimera package from the Resource for Biocomputing, Visualization, and Informatics at the University of California, San Francisco (Pettersen et al. 2004).
The databases and softwares of the European Bioinformatics Institute were used to construct a multiple sequence alignment of HVO_1989 and 12 archaeal and three bacterial homologs (Blast and ClustalW at www.ebi.ac.uk). The alignment was formatted for phylogenetic analyses using Microsoft Word. Construction of phylogenetic trees was performed as described previously (Wanner and Soppa 2002). The programs ProtPars, ProtDist, Fitch, and CONSENSE of the PHYLIP program package (Felsenstein 1996) were used at the Server of the Institute Pasteur (http://bioweb.pasteur.fr/seqanal/phylogeny/phylip-uk.html). A total of 100 bootstrap replications were performed and a consensus tree was calculated.
SUPPLEMENTAL MATERIAL
Supplemental material is available for this article.
ACKNOWLEDGMENTS
We thank Ramesh Gupta (University of Southern Illinois, Carbondale) for bringing the N1-methyl pseudouridine modification at position 54 of archaeal tRNAs to our attention and for noticing the similarities to the Nep1 target sequence. We are grateful to M. Görlach, U.A. Hellmich, H. Keller, O. Mirus, and E. Duchardt-Ferner for helpful discussions and their critical reading of the manuscript. This work was supported by an Aventis Foundation Endowed Professorship (to J.W.), the Center for Biomolecular Magnetic Resonance (BMRZ), the Cluster of Excellence, Macromolecular complexes, Johann-Wolfgang-Goethe-University Frankfurt, and the Deutsche Forschungsgemeinschaft (DFG) through the SFB 902 “Molecular Mechanisms of RNA based regulation” (to J.W. and J.S.) and grant So264/14 (to J.S.).
Footnotes
-
↵6 Corresponding author.
E-mail woehnert{at}bio.uni-frankfurt.de.
-
Article published online ahead of print. Article and publication date are at http://www.rnajournal.org/cgi/doi/10.1261/rna.028498.111.
- Received May 31, 2011.
- Accepted November 23, 2011.
- Copyright © 2012 RNA Society








