
Widespread RNA 3′-end oligouridylation in mammals
- 1Biomedical Sciences Graduate Program, University of California, San Francisco, San Francisco, California 94143, USA
- 2Department of Microbiology and Immunology, Diabetes Center, University of California, San Francisco, San Francisco, California 94143, USA
- 3Department of Laboratory Medicine, University of California, San Francisco, San Francisco, California 94143, USA
Abstract
Nontemplated 3′-end oligouridylation of RNA occurs in many species, including humans. Unlike the familiar phenomenon of polyadenylation, nontemplated addition of uridines to RNA is poorly characterized in higher eukaryotes. Recent studies have reported nontemplated 3′-end oligouridylation of small RNAs and mRNAs. Oligouridylation is involved in many aspects of microRNA biology from biogenesis to turnover of the mature species, and it may also mark long mRNAs for degradation by promoting decapping of the protective 5′-cap structure. To determine the prevalence of oligouridylation in higher eukaryotes, we used next-generation sequencing technology to deeply examine the population of small RNAs in human cells. Our data revealed widespread nontemplated nucleotide addition to the 3′ ends of many classes of RNA, with short stretches of uridine being the most frequently added nucleotide.
Keywords
INTRODUCTION
RNA can undergo several modifications between transcription and eventual degradation. Well-known examples of post-transcriptional RNA processing such as 5′-cap addition and polyadenylation of mRNAs and 3′-CCA addition to tRNAs illustrate the critical importance of RNA modifications in gene expression (Martin and Keller 2007; Moore and Proudfoot 2009). Another form of processing is the generation of a mature RNA from a longer precursor transcript. Many pre-mRNAs require removal of introns, and primary transcripts of rRNAs and tRNAs are processed by endo- and exonucleases to release the mature RNA (Granneman and Baserga 2005; Phizicky and Hopper 2010). Maturation of other noncoding RNAs such as miRNAs and snoRNAs also requires processing by endo- and exonucleases (Filipowicz and Pogacic 2002; Olena and Patton 2010).
Altering RNA 3′ ends can help stabilize or degrade RNA. The 3′ end of U6 snRNA is stabilized after the addition of nontemplated uridines by 2′-3′ cyclization of the final nucleotide (Lund and Dahlberg 1992). The destabilizing effect of uridine addition to mRNAs has been observed in yeast and mammalian cells and has been proposed to function by stimulating removal of the 5′-cap and degradation of mRNA (Song and Kiledjian 2007; Rissland and Norbury 2009). After DNA replication is complete, degradation of histone mRNA is initiated by nontemplated oligouridylation to coordinate histone expression with DNA abundance (Mullen and Marzluff 2008).
Oligouridylation is associated with miRNA biogenesis, function, and turnover. The addition of uridines to let-7 precursors has been reported to regulate the expression of let-7 in embryonic stem cells (Heo et al. 2009). Mature miRNAs are often sequenced with nontemplated uridines or adenines (Landgraf et al. 2007). These nontemplated bases may mark miRNAs to promote their decay (Ameres et al. 2010). Furthermore, small RNAs with extensive sequence complementarity to a target RNA can guide AGO2 to cleave, or slice, the target RNA (Liu et al. 2004; Meister et al. 2004). Sequencing the 5′ fragment of the sliced RNA often shows nontemplated uridines added to the cleavage site (Shen and Goodman 2004).
In vitro studies suggest that uridylyltransferase may act on many RNAs in human cells (Sinha et al. 1998). To determine the extent of nontemplated 3′-end oligouridylation in mammals, we used next-generation sequencing with strand-specific RNA linker-ligation to directly examine the 3′ ends of small RNAs (<200 nt) in human cells. Examples of previously identified RNA processing steps and nontemplated 3′-end nucleotide addition are present. We also find many novel cases of nontemplated 3′-oligouridylation on several classes of RNA including transcriptional start site-associated RNAs (TSSa-RNAs) and spliced introns, suggesting that oligouridylation may play a larger role in RNA metabolism in mammals.
RESULTS
Identification of nontemplated 3′-end nucleotide addition to RNA
Because of the growing body of literature showing that nontemplated 3′-end oligouridylation can regulate RNA metabolism in mammals, we used next-generation sequencing to better define the scope of this RNA modification. Small RNAs (<200 nt) of the optimal size for deep sequencing were purified without shearing to maintain native 3′ ends. While this method excludes long RNAs, it also avoids ribosomal RNAs, the major fraction of total RNA. Sequential ligation of adaptors maintains RNA strand information and is specific for a subset of the total RNA population: small RNAs with 5′-phosphate and 3′-OH, the functional groups that result from many endo- and exonucleases.
Illumina paired-end sequencing was used to directly examine the 3′ end of RNA. Because the paired-end 2 (PE2) primer begins sequencing 3′ → 5′ relative to the sense orientation, this data set provides high quality reads starting at the 3′ end of captured RNA. Post-transcriptional processing of RNA poses a challenge for aligning RNA-seq data because nontemplated nucleotides are likely to disagree with the genomic sequence at the aligned loci. Mismatches between the read and reference genome are penalized with some alignment programs having a maximum cutoff for number of mismatches allowed before rejecting a read. To compensate for homopolymeric, nontemplated nucleotide addition to RNA 3′ ends, all initial homopolymer clusters for all four nucleotides, regardless of length, were removed from the start of sequences in the PE2 data set. These initial clusters represent the 3′-end terminal homopolymer (THP). Alignment after removal of the THP resulted in more aligned reads and more usable reads that aligned to unique loci (Supplemental Table 1). The mismatch rate in the THP was more than 10-fold greater than the mismatch rate in the following 20 nucleotides (Supplemental Table 2). Stripping the THP allowed read mapping using bases that were more likely to match the reference genome.
A broad overview shows that, out of 16.3 million reads that aligned to unique genomic loci, nearly 20% (3.2 million) had at least one mismatch in their 3′ end (Fig. 1A). Partitioning reads by gene class found nontemplated nucleotides on many classes of RNA, often at high frequency (Fig. 1B–F). The gene family that made up the largest fraction of reads (73%) with nontemplated 3′-end nucleotides was miRNAs (Fig. 1A). The mir-302–367 cluster produces an embryonic stem cell-specific family of miRNAs identified by traditional and next-generation sequencing of small RNAs (Suh et al. 2004; Morin et al. 2008). One member of this cluster, hsa-mir-302c, accounted for the vast majority of miRNA reads with nontemplated 3′ ends. Sequencing H9 genomic DNA of the mir-302-367 cluster confirmed that the cell line agrees with the reference genome and that the high frequency and abundance of miR-302c +U was not encoded in the DNA (data not shown). Examination of other miRNAs showed that our data is consistent with previous studies that have reported frequent uridine or adenine addition to miRNAs (Fig. 1B; Landgraf et al. 2007).

The nontemplated 3′ ends of small RNAs in human ES cells. (A) The distribution of paired-end-2 sequencing primer reads by RNA gene class. The distribution for all uniquely aligned reads (left) and those reads with nontemplated 3′-end THP (right) are shown. (B–F) The frequency at which each nucleotide was found as the nontemplated THP was calculated for each RNA gene class. The first column is the percent of reads for a specific gene or gene class that had nontemplated 3′ ends. The numbers in the (MM) column are the number of reads with mismatches between the THP and the reference genome. (C) The overall frequency of nontemplated 3′ ends for several classes of RNA genes. Shown are the most highly sequenced genes of miRNAs (B), tRNA anti-codons (D), snRNAs (E), and snoRNAs (F).
Uridines and adenines accounted for nearly all cases of nontemplated nucleotides for most gene classes (Fig. 1C). Relatively few tRNA reads were captured in our library, presumably due to 3′- or 2′-aminoacylation or other modifications that would inhibit adapter ligation. We identified known examples of nontemplated RNA 3′-end addition, such as the nearly universal post-transcriptional 3′-CCA addition to tRNAs (Fig. 1D). Essentially all reads with unambiguous, nontemplated 3′-CCA or 3′-CA aligned to tRNA genes. Also noted were rare cases of nontemplated uridine or adenine addition to mitochondrial tRNAs and U or UU addition to nuclear encoded tRNAs following the 3′-CCA modification. Reads with more extensive nontemplated adenylation (>10 nt) were usually found on mitochondrial tRNAs with much fewer cases of polyadenylated nuclear-encoded tRNAs.
Another example of previously identified RNA 3′-end nucleotide addition found in our data set was the oligouridylation of U6 snRNA. The low read counts for U6 and for snRNAs as a group (Fig. 1E) likely demonstrates the selection of the cloning protocol. Before snRNAs can be captured, they must be processed to remove their 5′-cap or 5′-tri-phosphate. U6 needs to be doubly processed to remove the 2′-3′ cyclic UMP found on the mature form of U6. By contrast, snoRNA genes had the most overall reads but were least likely to have nontemplated nucleotide addition (Fig. 1F). Unlike snRNAs, mature snoRNAs have been processed by nucleases (Filipowicz and Pogacic 2002) and already possess the functional groups selected for by the cloning protocol.
Transcriptional start-site-associated RNAs are oligouridylated
After confirming the presence of known cases of nontemplated 3′-end nucleotide addition in our data set, we searched for new examples of this modification, particularly from RNAs derived from long transcripts. Although the RNA purification protocol enriched for small RNAs, ∼400,000 reads mapped to long coding and noncoding genes. This relatively small population contributed significantly to the diversity of unique sequences with nontemplated nucleotides. The overall population of 3.2 million reads with nontemplated RNA 3′ ends (Fig. 1A) collapses to 82,176 unique sequences with 24,079 derived from long RNA transcripts. The cumulative frequency of 3′-end THP lengths shows nontemplated uridines were the most frequent base for tails up to five nucleotides (Fig. 2). Because different types of RNA processing occurs on RNAPol-II transcripts, reads that aligned to the UCSC refGene database were grouped on the basis of where the 3′ end aligned with respect to the gene annotation (Table 1).
Alignment of refGene annotated genes
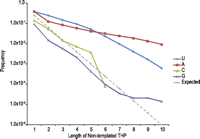
Distribution of nontemplated THP lengths. The THP lengths were counted for reads with nontemplated 3′-end nucleotides. Overall, 3.2 million reads covering 11,616 genes were collapsed to 82,176 unique sequences. The cumulative frequency of THP lengths of this collapsed data set is shown; that is, if an RNA had a THP of five uridines, it was included in the totals for lengths of one through five. The expected line is the probability of finding a homopolymer of a given length at random, based on equal representation of all nucleotides.
Recent studies in mouse and human embryonic stem cells have described TSSa-RNAs, short RNAs that map near transcriptional start-sites (Guenther et al. 2007; Seila et al. 2008). These short RNAs are derived from transcripts that have aborted or paused after initiation and have been postulated to result from regulation of RNAPol-II elongation and escape from the promoter. TSSa-RNAs are present at low copy numbers per cell per gene (Guenther et al. 2007; Seila et al. 2008); 84,298 sense and 35,284 antisense TSSa-RNAs (Table 1) were identified in our data set. The read length distribution of TSSa-RNAs in our RNA-seq library made from H9 human embryonic stem cells shows a broad peak from 22–50 nt, with 6.9% of sense and 5.4% of antisense TSSa-RNAs extending the full 76 cycles of sequencing (Fig. 3A). The distribution of reads with respect to their distance from the TSS is similar to earlier reports describing this class of RNA (Fig. 3B). Most of the sense TSSa-RNAs that aligned upstream of the TSS are due to incomplete annotations that lack the 5′ UTR. Nontemplated nucleotides were found on 20% of TSSa-RNAs, with uridines being sixfold more abundant than adenine homopolymers for both sense (Fig. 3C) and antisense orientations (Fig. 3D). The position of reads with nontemplated uridines is similar to the distribution for all sense TSSa-RNAs (Fig. 3B). It should be noted that not all TSSa-RNAs would be detected by the cloning method used to prepare our RNA-seq library. The initial 5′-tri-phosphate or added 5′-cap would have to be removed, an activity which is promoted by the addition of nontemplated uridines (Song and Kiledjian 2007).
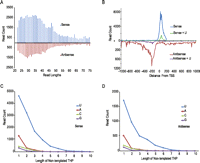
Characteristics of TSSa-RNA. (A) The length distribution of TSSa-RNAs. (B) The distribution of distances of TSSa-RNAs measured from the TSS to the 3′ end of the read for all TSSa-RNAs and TSSa-RNAs with nontemplated uridine addition. (*) Note the different scales for sense and antisense. The distribution of nontemplated 3′-end THP lengths for sense (C) and antisense (D) TSSa-RNAs.
Spliced introns are oligouridylated
Of all categories of sequences that aligned to long genes in the UCSC refGene database, splice site sequences were the RNA most likely to have nontemplated 3′-end nucleotides (Table 1). They were also the rarest population with 5600 total reads. Most genes had fewer than five reads, with only two intron splice sites covered by more than 100 reads (Supplemental Fig. 1). In contrast, the much larger population of other intronic reads had the lowest frequency of nontemplated 3′-end nucleotide addition (Table 1). After intron splicing, the lariat must be processed before it can be captured for sequencing. Analysis of the lengths of splice site reads reveals a peak for short reads, ∼25% were ≤26 nt, and a broad population that extends past the branch point, with 23% of all splice site reads and 38% of splice site reads with nontemplated THPs spanning the full 76 cycles of PE2 sequencing (Fig. 4A). Uridines were the most frequently found nontemplated nucleotide in this population of processed RNA, three- to 15-fold more abundant than nontemplated adenines depending on the length of the homopolymer (Fig. 4B).
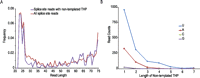
Characteristics of spliced introns. (A) The distribution of lengths of all splice site reads and splice site reads with nontemplated 3′-end THPs plotted as a fraction of their respective populations. (B) The distribution of nontemplated 3′-end THP lengths sequenced on splice site reads.
The fraction of spliced introns with nontemplated nucleotides may be larger than what our analysis reports. Sequence analysis can only identify mismatches between the read and the reference genome. It cannot distinguish between templated transcription and nontemplated nucleotide addition after RNA processing if the nontemplated base happens to match the genome. Alignment of sequences to splice sites with relatively high read coverage illustrates this problem. Ninety-six reads that mapped to the 3′-splice site of intron 7 of TATDN1 included an extra adenine that could be templated and the result of mis-splicing, or it could be due to nontemplated nucleotide addition after correct splicing (Supplemental Fig. 1). Similarly, the extra uridine found on nine reads of an intron of HSPG2 could either be due to templated transcription or added following splicing. Nontemplated RNA 3′-end modification would be underestimated on other classes of RNAs as well by the requirement for unambiguous mismatches in the alignment to the reference genome.
DISCUSSION
Deep sequencing of small RNA from human embryonic stem cells captured many previously identified examples of post-transcriptional RNA 3′-end processing. We also found many new examples of nontemplated nucleotide addition, with oligouridylation being the most frequently observed modification. The abundance of nontemplated uridines in our data set cannot be simply explained by the enzymes used in preparing the RNA-seq library. The nontemplated activity of Taq DNA polymerase adds to the end of the DNA template, and there are no reports of this enzyme being prone to insert multiple nucleotides in the middle of a template. The error rate of DNA polymerase also does not account for the clearly higher rate of mismatches to the reference genome in the THP compared to the adjacent 20 nucleotides, even after excluding the first PE2 cluster (Supplemental Table 2). T4 RNA ligase 2-truncated, the enzyme used to add the 3′ adaptor, is catalytically impaired and requires an activated, preadenylated DNA or RNA oligomer as a substrate (Ho et al. 2004). It is unlikely to act as a polymerase to add free nucleotides to RNA. Extra uridines are not likely introduced by the 3′ adaptor which was PAGE- purified and begins: 5′-AGATCG. RNA ligases are known to have different ligation efficiencies depending on the sequence of the donor and acceptor molecules (England and Uhlenbeck 1978). Even if this enzyme was particularly efficient at ligating RNA with 3′-uridines, that would not explain why the uridines are so often mismatched to the reference genome.
Our RNA-seq preparation and analysis method may explain the high rate of 3′-end mismatches we observed in our data set and why other methods may overlook this RNA modification. The common practice of excluding reads with mismatches to the reference genome, especially when aligning short sequences, would be blind to nontemplated nucleotides. Previous studies of TSSa-RNAs used DNA microarrays (Guenther et al. 2007), which do not detect sequence variants of the probe. Others used short sequencing in the sense orientation (Seila et al. 2008) that excluded reads with mismatches and would not have reached the 3′ ends of most TSSa-RNAs (Fig. 3A). RNA-seq studies of long coding transcripts often use polyA selection to enrich for mRNAs from total RNA, thereby excluding spliced introns.
The RNA-seq library preparation method that we used has limitations that must be considered when interpreting the data. The low total count of reads that aligned to 3′ splice sites may be caused in part by the size fractionation of RNA during purification. Half of splice site reads aligned to short introns <300 nt, with the other half originating from introns up to 129-kb- long. The scarcity of reads is also seen in the low read coverage of most introns (<5 reads). It was rare for more than one intron of a single transcript to be detected, usually in genes with several short introns. While it would be premature to draw conclusions about specific splice sites with such low read coverage, the scarcity of splice site reads does not address why these processed RNAs as a whole were so often sequenced with nontemplated uridines.
Though we highlighted the nontemplated oligouridylation of TSSa-RNAs and splice site-derived RNAs, it should be noted that sequences derived from longer mRNA or pre-mRNA transcripts were in the “other” category of refGene reads (Table 1). These reads included fragments of 5′ UTR, exon, or exon with upstream intron. As a group, these reads had a similar rate of nontemplated nucleotide addition as TSSa-RNAs, which may allude to prior nuclease processing of these small RNA fragments necessary to be in our library.
The low rate of nontemplated nucleotide addition to snoRNAs may provide a clue as to the relative abundance of modified RNAs compared to the mature form. Unlike RNAs that are protected at the 5′ end, mature snoRNAs can be captured without further processing, which could explain why modified snoRNAs were rarely seen against the background of mature transcripts. Further refinements of RNA-seq methods may be required to identify more examples of RNA 3′-end oligouridylation.
Though most human snoRNAs are processed from introns and have 5′-phosphates (Dieci et al. 2009), there are a few snoRNAs that are known to be 5′-capped. Many studies on U3, U8, and U14 have examined the changes in methylation status of their caps and subcellular localization of these RNAs (Reddy et al. 1985; Tyc and Steitz 1989; Terns et al. 1995; Jacobson and Pederson 1998; Speckmann et al. 2000). The low read counts for U3 (18) and U14 (2270 U14A and 13 U14B) are consistent with the requirement for RNAs to have 5′-phosphates in order to be captured by the RNA-seq cloning method used to prepare our library. However, U8 is the most frequently sequenced gene in our library, with 2,768,660 reads aligned to that locus. Reads to U8 also had a low rate of nontemplated nucleotide addition (0.93%). Because almost all reads to U8 (98.4%) in the PE2 library extended the full 76 cycles of sequencing, we searched the paired-end 1 data set to identify the 5′ ends of U8. Over 80% of reads had the annotated 5′-end sequence of U8 and were not cleaved fragments, indicating that there was a significant population of U8 snoRNAs with 5′-phosphates. This poses several questions about the processing of U8 in human embryonic stem cells. Examination of the U8 locus shows that this snoRNA is not intronic, but it overlaps the 3′ UTR of TMEM107. One possibility is that, in some cases, the cap may be added after processing the snoRNA from the 3′ UTR of TMEM107 and not due to independent transcription of U8. In support of this, there were 47 reads to TMEM107 in our data set: three TSSa-RNAs, one mRNA fragment of intron 2 and exon 3, with the remainder in the 3′ UTR downstream from U8. Other questions about the rates of capping and decapping of snoRNAs are beyond the scope of this study, but we have made available the PE1 sequencing primer data set for those interested in human small RNAs.
While we must be cautious about inferring the in vivo abundance of an RNA from sequencing read counts (Linsen et al. 2009), the different rates at which nontemplated uridines are observed on different classes of RNAs may hint at a broader functional role for nontemplated uridine addition in RNA metabolism in mammals. There is precedence for the addition of nontemplated nucleotides to mark RNA for degradation. In contrast to the stabilizing effect of long polyadenylation of eukaryotic mRNA, polyadenylation in prokaryotes destabilizes RNA (Arraiano et al. 2010). Polyadenylation can destabilize eukaryotic RNA as well (West et al. 2006). The TRAMP complex (Trf4/Air2/Mtr4p) adds poly-A tails to improperly processed nuclear RNAs to promote their degradation by the nuclear exosome (LaCava et al. 2005; Vanacova et al. 2005). Studies have identified and characterized a conserved family of nucleotidyl transferases present in many eukaryotes, including yeast, plants, and humans (Kwak and Wickens 2007; Rissland et al. 2007). These enzymes, called poly(U) polymerases (PUPs) because they have a strong preference in vitro to incorporate uridines on the 3′ ends of RNA, are related to the Trf4 and Trf5 components of the TRAMP complex. It is has already been shown that oligouridylation can regulate diverse RNA species. Studies in plant and animal models show that small noncoding RNAs can be regulated by oligouridylation (Li et al. 2005; Heo et al. 2009; Ameres et al. 2010). Longer mRNAs can also be marked for degradation by the addition of uridines (Mullen and Marzluff 2008). We find it interesting that the highest frequency of nontemplated 3′-end oligouridylation is found on RNAs that required processing by nucleases to be captured by our cloning method. Our analysis shows that post-transcriptional oligouridylation of RNA is widespread and found on many classes of RNA genes. These observations suggest that oligouridylation may be involved in several aspects of RNA metabolism and opens new questions for further study.
MATERIALS AND METHODS
Cell culture
The hESC line H9 (NIH designation, WA09), obtained from the University of Wisconsin Alumni Research Foundation, was cultured as described (Gaur et al. 2006). In brief, H9 cells were maintained as undifferentiated colonies by coculture on irradiated CF1 MEFs (Bodnar et al. 2004), plated on 0.1% gelatin-coated 6-well dishes in DMEM high glucose supplemented with 2 mM L-glutamine, 1× penicillin/streptomycin (Gibco BRL), and 10% fetal bovine serum (Hyclone). One day after plating, the media was removed, and the MEFs were washed once with phosphate-buffered saline without calcium or magnesium (PBS-CMF; Gibco BRL) immediately prior to plating the hESCs. The hESCs were grown on MEFs in KSR media: Knockout-DMEM medium containing 20% Knockout Serum Replacement, 2 mM L-glutamine, 0.1 mM (1×) nonessential amino acids, 4 ng/mL human basic fibroblast growth factor (Gibco BRL), and 0.1 mM β-mercaptoethanol (Sigma). To selectively release hESC colonies from the MEFS, the cells were incubated with a combination of collagenase (Type IV)/dispase (Gibco BRL) for 20 min at 37°C. Released hESC colonies were gently transferred to a 15-mL conical tube and allowed to settle by gravity for 5 min. Supernatant was removed, and hESC colonies were gently washed once with KSR media and once with PBS. Clumps were again allowed to gravity-settle, supernatant was removed, and clumps disrupted by P-1000 pipet. Cells were >99% free of MEFs and were used for RNA isolation.
RNA and DNA purification
Small RNAs (<200 nt) were purified from ∼3 million cells using the mirVana miRNA Isolation Kit (Ambion) according to the manufacturer's directions. DNA from H9 cells was purified using the PureLink Genomic DNA Mini Kit (Invitrogen).
Strand-specific RNA linkering and cDNA library preparation
DNA and RNA oligos for RNA sequencing were synthesized by IDT. The 3′-linker (5′-rApp-AGATCGGAAGAGCGGTTCAGCAGGAATGCCGAG/3InvdT/) was adenylated and purified as described in Pfeffer et al. (2005). RNA linker ligation was done as previously described (Neilson et al. 2007) but briefly, preadenylated 3′-linker was ligated to small RNAs using T4 RNL2truc (NEB) without ATP in the reaction buffer. After ethanol precipitation, the reaction products were used for 5′-linker ligation (/5Cy3/rArCrA rCrUrCrUrUrUrCrCrCrUrArCrArCrGrArCrGrCrUrCrUrUrCrCrGrArUrCrU) without gel purification with T4 RNL1 (NEB) with ATP. RNA was reverse-transcribed (5′-GCTGAACCGCTCTTCCGATCT) with SuperScript III (Invitrogen) and amplified with 25 cycles of PCR with Platinum Taq polymerase (Invitrogen) and Illumina PE PCR primers (Illumina). PCR products >140 bp were agarose gel-purified (Qiagen QIAquick Gel Extraction Kit), ethanol-precipitated, and resuspended in elution buffer. Taking into account the length of Illumina PE linkers, this corresponds to RNA inserts > ∼21 nt. Library fragment size and concentration were measured with the Bioanalyzer High Sensitivity DNA chip assay (Agilent).
Paired-end next-generation sequencing and analysis
The prepared library was submitted for one lane of Illumina GAIIx pair-end sequencing (76 × 76 cycles) at the Michael Smith Genome Sciences Center, Vancouver, Canada. The paired-end 2 (PE2) sequencing primer library was used to analyze the 3′ end of RNAs. Using custom Python programs, reads were trimmed of the initial homopolymer, regardless of length, for each nucleotide. Trimmed nucleotides were noted and the remaining sequence was aligned to the full hg18 genome (UCSC Genome Browser) with Novoalign (version 2.07 Novocraft Technologies). Only reads that aligned to unique loci were further analyzed. Reads were also compared to the mm9 mouse genome. Reads that had fewer mismatches when mapped to mm9 were considered contamination from mouse feeder cells and removed from further analysis. The previously removed homopolymer portion of each read was compared to the expected genomic sequence at the aligned locus to identify RNAs with mismatches in the 3′ end.
Read annotation
Reads were annotated by checking for any overlap of coordinates on the same strand when searched for in annotation databases in the following order: mirBase 16, UCSC (hg18) rnaGene.txt, UCSC tRNAs.txt, UCSC refGene.txt, and UCSC knownGene.txt. For reads that did not match, the search was repeated after adding 50 bp to the coordinates of mirBase, rnaGene, and tRNA genes and adding 1000 bp to refGene and knownGene coordinates. Reads matching these relaxed coordinates were labeled “Gene Name–flank”. For remaining reads that failed to match the expanded coordinates, the search was repeated to check for anti-sense strand origin and reads matching those conditions were labeled “Gene Name–anti” or “Gene Name–anti–flank”. Sequences that included “SNORD” or “SCARN” in the annotation to refGene were removed and included as snoRNAs genes.
SEQUENCING DATA
The data discussed in this publication have been deposited in NCBI's Gene Expression Omnibus (GEO) (Edgar et al. 2002) and are accessible through GEO Series accession number GSE31051 (http://www.ncbi.nlm.nih.gov/geo/query/acc.cgi?acc=GSE31051).
SUPPLEMENTAL MATERIAL
Supplemental material is available for this article.
ACKNOWLEDGMENTS
We thank Shaohui Wang for her help with ES cell culture. This work was supported by a National Science Foundation Graduate Research Fellowship (Y.S.C.), a grant from the California Institute for Regenerative Medicine (RC1-00347) (A.D.L., M.T.M.), and a National Institutes of Health grant (5R01DA026065) (M.T.M.). Contributions to this research were made through NIH P30 DK063720. We are grateful to members of the McManus lab, especially Matt Hangauer, Greg Ku, and Nikki Shariat for their help in drafting this manuscript.
Footnotes
-
↵5 Corresponding author.
E-mail michael.mcmanus{at}ucsf.edu.
-
Article published online ahead of print. Article and publication date are at http://www.rnajournal.org/cgi/doi/10.1261/rna.029306.111.
- Received July 19, 2011.
- Accepted November 29, 2011.
- Copyright © 2012 RNA Society








