
The C-terminal end of the Trypanosoma brucei editing deaminase plays a critical role in tRNA binding
- Frank L. Ragone1,5,
- Jessica L. Spears2,3,5,
- Jessica M. Wohlgamuth-Benedum2,3,
- Nathan Kreel1,
- F. Nina Papavasiliou4 and
- Juan D. Alfonzo1,2,3,6
- 1Ohio State Biochemistry Program, Ohio State University, Columbus, Ohio 43210, USA
- 2Department of Microbiology, Ohio State University, Columbus, Ohio 43210, USA
- 3Ohio State Center for RNA Biology, Ohio State University, Columbus, Ohio 43210, USA
- 4Laboratory of Lymphocyte Biology, Rockefeller University, New York, New York 10021, USA
-
↵5 These authors contributed equally to this work.
Abstract
Adenosine to inosine editing at the wobble position allows decoding of multiple codons by a single tRNA. This reaction is catalyzed by adenosine deaminases acting on tRNA (ADATs) and is essential for viability. In bacteria, the anticodon-specific enzyme is a homodimer that recognizes a single tRNA substrate (tRNAArgACG) and can efficiently deaminate short anticodon stem–loop mimics of this tRNA in vitro. The eukaryal enzyme is composed of two nonidentical subunits, ADAT2 and ADAT3, which upon heterodimerization, recognize seven to eight different tRNAs as substrates, depending on the organism, and require a full-length tRNA for activity. Although crystallographic data have provided clues to why the bacterial deaminase can utilize short substrates, residues that provide substrate binding and recognition with the eukaryotic enzymes are not currently known. In the present study, we have used a combination of mutagenesis, binding studies, and kinetic analysis to explore the contribution of individual residues in Trypanosoma brucei ADAT2 (TbADAT2) to tRNA recognition. We show that deletion of the last 10 amino acids at the C terminus of TbADAT2 abolishes tRNA binding. In addition, single alanine replacements of a string of positively charged amino acids (KRKRK) lead to binding defects that correlate with losses in enzyme activity. This region, which we have termed the KR-domain, provides a first glance at key residues involved in tRNA binding by eukaryotic tRNA editing deaminases.
Keywords
INTRODUCTION
Be it to affect structure or function, editing involves different post-transcriptional and post-replicative processes that change the information content of nucleic acids beyond what is encoded in genes. These sequence alterations often alter coding specificity and greatly contribute to the expansion of coding capacity. Both DNA and RNA undergo editing (Gott and Emeson 2000), which can involve nucleotide insertions and/or deletions as in trypanosomatids, Physarum and Acanthamoeba (Benne et al. 1986; Abraham et al. 1988; Simpson et al. 1989; Gott et al. 1993; Mahendran et al. 1994; Price and Gray 1999). Alternatively, editing may proceed by single chemical changes to the bases in RNA, altering base-pairing and, therefore, affecting coding specificity (Gerber and Keller 2001; Nakanishi et al. 2005).
The most widespread type of editing, in terms of substrates targeted and phylogenetic distribution, is base deamination (Gerber and Keller 2001). Two types of deamination reactions were originally discovered in RNA—adenosine to inosine (A to I) and cytidine to uridine (C to U)—which change the reading frames of mammalian mRNAs and are now known to have critical functions in cellular metabolism (Powell et al. 1987; Bass and Weintraub 1988; Wagner et al. 1989; Navaratnam et al. 1993). These initial findings have more recently led to the discovery of the deaminases that target DNA. For example, DNA deamination mediated by activation-induced deaminase (AID) plays a crucial role in antibody diversification (Muramatsu et al. 1999; Petersen-Mahrt et al. 2002). Likewise, C to U deamination of viral DNA is important in antiviral response (Mangeat et al. 2003).
To date, an ever-growing number of proven and putative targets of deamination editing have been identified, including various tRNAs, mRNAs, and 7SL RNA (Alfonzo et al. 1999; Ben-Shlomo et al. 1999; Gott and Emeson 2000; Petersen-Mahrt et al. 2002; Dickerson et al. 2003; Rubio 2005; Rubio et al. 2006). Deaminases are also responsible for free nucleotide interconversions that contribute to nucleotide pools in cells and serve as a repository for the nucleotide building blocks used in both RNA and DNA synthesis (Wolfenden 1993; Betts et al. 1994). In addition, deamination reactions have key functions in various metabolic pathways, for example, in riboflavin biosynthesis and threonine catabolism (Chen et al. 2006; Simanshu et al. 2006).
The essential A to I tRNA editing event in bacteria is orchestrated by a homodimeric adenosine deaminase acting on tRNA (ADATa) or an ADAT2/ADAT3 heterodimer in eukarya (Auxilien et al. 1996; Gerber and Keller 1999; Wolf et al. 2002). While the subunit composition varies between domains of life, the catalytic cores of these enzymes are highly conserved (Gerber and Keller 1999; Wolf et al. 2002). Interestingly, these enzymes that catalyze A to I deamination phylogenetically form a clade with the cytidine deaminase superfamily (Gerber and Keller 1999). Like all cytidine deaminases, the catalytic core of ADATs consists of conserved HAE and PCxxC (where “x” represents any amino acid) motifs. The histidine and the two cysteines coordinate a Zn2+ ion, while the glutamate residue actively shuttles a proton from an activated water molecule to the N1 position of the newly formed inosine. Through evolution, the catalytic glutamate in the HAE domain of ADAT3s has been replaced by a noncatalytic residue, for example, HPV in Trypanosoma brucei ADAT3 (TbADAT3), and this subunit is thought to be solely a structural, yet still essential, component of the enzyme (Gerber and Keller 1999).
The similarities with cytidine deaminases led Keller and coworkers to propose that, evolutionarily, all RNA deaminases are derived from an ancestral cytidine deaminase (Gerber and Keller 1999). Adding credence to this hypothesis, we recently showed that the TbADAT2/3 enzyme could perform both types of deaminations, albeit in different substrates: A to I in tRNA and C to U in DNA (Rubio et al. 2007). Therefore, TbADAT2/3 maintains some of the characteristics of Keller's ancestral deaminase, but it is still not clear how this enzyme achieves substrate recognition or catalytic flexibility. A number of bacterial ADAT structures and the recent cocrystal of the Staphylococcus aureus ADATa (SaADATa) complexed with the anticodon stem–loop (ASL) of tRNAArg has given insights into enzyme–substrate interactions by the bacterial enzyme (Losey et al. 2006). The SaADATa homodimer binds the ASL via residues within the enzyme's active site, while the tRNA backbone contributes minimally to RNA binding.
While a great deal of effort has been put forth to characterize bacterial ADATs, little is known about the eukaryotic enzymes and their interactions with tRNA substrates. Here, we show that the putative catalytic subunit of the T. brucei tRNA deaminase (TbADAT2) contains key tRNA binding residues at its C terminus. Deletion of these residues leads to defects in substrate binding and complete loss of enzymatic activity. Since a similar motif is found at the C terminus of many eukaryotic ADAT2s but is absent in most bacterial ADATs, it suggests that this represents a general tRNA recognition motif for the eukaryotic enzymes. Currently, however, nothing is known about the mode of tRNA binding by any of the eukaryotic tRNA deaminases, so it is not clear if similar sequences found at the C terminus of other ADAT2s may provide a tRNA binding function. The data presented support a previous model in which, through evolution and in order to accommodate many different substrates, a domain has been appended away from the enzyme's active site, providing critical substrate binding functions (Elias and Huang 2005).
RESULTS
Recombinant TbADAT2/3 stably binds tRNA
As a prelude to identifying regions of TbADAT2/3 important for tRNA binding, an electrophoretic mobility shift assay (EMSA) was established by incubating recombinant protein with in vitro-transcribed radioactively labeled tRNAVal (one of its natural substrates in T. brucei). Under these conditions, a band which migrated slower in the gel compared to an RNA alone control was observed only in the presence of protein (Fig. 1A, right panel), suggesting formation of a stable complex between the recombinant protein and the tRNA. Additionally, larger complexes, which did not enter the gel, were observed at the highest protein concentrations. We estimated a dissociation constant (Kd) of 1.31 +/− 0.83 μM for this tRNA. Similar values were obtained even when the larger complexes were included in the calculations. Although this Kd approximates the observed KM of the wild-type enzyme for the same tRNA (0.75 μM), the Kd was consistently higher than the KM. To ensure that the observed binding was specific for tRNA, competition assays were performed where a twofold molar excess of nonradioactive tRNAVal used as a specific competitor abrogated most of the binding (Fig. 2A, left panel). Similar experiments with an unrelated, nontRNA substrate (spliced leader RNA from T. brucei) used as a nonspecific competitor showed that even at a 64-fold molar excess over the tRNA substrate, binding still occurred (Fig. 2A, right panel).
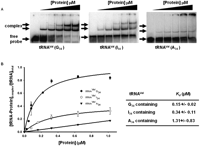
Wild-type TbADAT2/3 stably binds tRNA in vitro. Radioactively labeled tRNAVal (8 nM) from T. brucei was incubated with increasing concentrations of recombinant TbADAT2/3 expressed in E. coli and purified by Ni2+-chelate chromatography. (A) Representative electrophoretic mobility shift assay (EMSA) to determine the extent of tRNA binding. Right panel shows TbADAT2/3 binding to an A34-containing tRNA (natural substrate). Middle and left panels show a similar experiment but with either an I34- or G34-containing tRNA. In all panels, lane 1 shows a mock reaction in which the probe was incubated in binding buffer in the absence of enzyme. Lanes 2–6 show tRNA incubated with increasing concentrations of the enzyme (0.052, 0.18, 0.35, 0.65, and 1.18 μM, respectively). “Free probe” denotes the migration of the unbound tRNA, and “complex” denotes the migration of the TbADAT2/3 bound tRNA. (B) The reaction products from A were used to calculate the fraction of tRNA bound by calculating the percent of the probe shifted divided by the total (bound and unbound) probe in each reaction. These values were plotted against TbADAT2/3 concentration in μM and fitted to a single exponential curve, and the dissociation constant (Kd) was calculated by nonlinear regression using SigmaPlot software.

Wild-type TbADAT2/3 specifically binds tRNA in vitro. (A) Representative electrophoretic mobility shift assay (EMSA) to determine the extent of tRNA binding in the presence of either nonradioactive tRNA or a nontRNA (spliced leader RNA) substrate used as specific and nonspecific competitors, respectively. The left panel shows TbADAT2/3 binding to radioactive tRNAVal in the presence of the same nonlabeled tRNA. The right panel shows TbADAT2/3 binding to the same radioactively labeled tRNAVal but in the presence of splice leader RNA. Lane 1 shows a mock reaction in which the tRNA probe was incubated in binding buffer in the absence of enzyme and competitor. Lane 2 shows the tRNA probe incubated with wild-type enzyme in the absence of any competitors. Lanes 3–7 and 8–12 show tRNA incubated with an increasing excess of cold competitor (2-, 4-, 8-, 16-, and 64-fold). “Free probe” denotes the migration of the unbound tRNA, and “complex” denotes the migration of the TbADAT2/3 bound tRNA. (B) The reaction products from A were used to calculate the fraction of tRNA bound by calculating the percent of the RNA shifted divided by the total (bound and unbound) probe in each reaction. These values were plotted against competitor fold-excess and fitted to a single exponential decay curve using SigmaPlot.
With the bacterial ADATa, a stable RNA-protein complex was only obtained by replacing the target adenosine (A34) by zebularine, a purine analog that resembles the proposed transition state for the deaminase reaction. Presumably, the use of the zebularine-containing tRNA prevents turnover of the substrate, which leads to a stable enzyme–tRNA complex. This approach led to the cocrystallization of the bacterial enzyme with RNA. We decided to further explore the binding of the T. brucei enzyme to different tRNA derivatives with the hope of identifying a more stable complex that could be used to probe potential TbADAT2/3 RNA binding motifs. Since zebularine is not readily available, we tested the possibility of stable interactions with the reaction product inosine 34 (I34)-containing tRNAVal. Inosine is a guanosine analog; therefore, we also tested a tRNA where A34 was replaced by G34. We found that the recombinant enzyme formed stable complexes with these two substrates; however, binding was significantly improved in comparison to the A34-containing tRNA, with Kd values of 0.34 +/− 0.11μM and 0.15 +/− 0.02 μM for the I34- and G34-containing substrates respectively (Fig. 1). Together, the data lead to the conclusion that TbADAT2/3 can stably bind tRNA in vitro.
Single-turnover kinetics validates G34-containing tRNA substrates for binding studies
To validate our use of a product-mimicking tRNA for our binding studies and characterization of ADAT2 residues important for binding, we performed enzyme single-turnover kinetic experiments by incubation of an A34-containing tRNA with saturating concentrations of enzyme. These allowed us to calculate observed rates (kobs) by plotting the amount of inosine produced in these reactions over time. The data were fitted to the equation f = a(1−e−kt), where f and t are inosine formed and time, respectively, a represents inosine produced at the end point of the reaction, and k is kobs (Fig. 3A). The kobs values were subsequently used to calculate an apparent dissociation constant of 0.05 +/− 0.01 μM (Fig. 3B), which is only threefold better than that measured by EMSA with the G34-containing tRNA (Kd = 0.15 +/− 0.02 μM). Importantly, the kinetically determined Kd shows an ∼25-fold improvement when compared to that calculated by EMSA using the same A34-containing substrate (compare 1.3 μM to 50 nM) (Figs. 1B, 3). With these data in hand, we conclude that using a G34-containing tRNA substrate which mimics the product in EMSA studies is a fair alternative to using the natural A34-containing substrate.

Kinetic determination of the dissociation constant for TbADAT2/3. (A) Single-turnover assays were performed as described in Materials and Methods. For each protein concentration (ranging from 0.15 μM to 10 μM), the fraction of inosine produced was plotted against time and fit to the single exponential equation f = a(1 − e−kt). kobs values were calculated for each curve representing protein concentration. (B) kobs values were plotted against TbADAT2/3 concentration and fit to a single ligand-binding curve; the apparent dissociation constant (Kd app) was calculated by nonlinear regression using SigmaPlot software.
TbADAT2 has a predicted RNA binding motif at its C terminus important for tRNA binding
The relatively high affinity of the enzyme for the G34-containing tRNA allowed us to probe regions of the enzyme for RNA binding motifs. It has been known for many years that bacterial ADATs can efficiently deaminate short stem–loops corresponding to the ASL of tRNAArgACG, the only in vivo substrate for this enzyme (Grosjean et al. 1996). This minimal RNA substrate was, in fact, used in the cocrystalization experiments, which revealed a number of direct contacts between the SaADATa and RNA, including a network of hydrogen bonds that involved all five nucleotides in the anticodon loop and, to a lesser degree, contacts with specific nucleotides in the stem (Losey et al. 2006). To investigate the specific contribution of analogous residues in the ADAT2 subunit, we first aligned the sequence of SaADATa with that of the TbADAT2 protein to show the equivalent, highly conserved, active-site residues that play a role in RNA binding in the bacterial enzyme compared to the eukaryotic enzyme (Fig. 4A). This alignment showed that some of the key residues in SaADATa had already undergone nonconservative replacements in TbADAT2 during evolution. For example, the wild-type sequence of TbADAT2 already contains naturally occurring alanine and valine replacements at the equivalent positions as the conserved arginine 125 and serine 138 from bacterial deaminases (Fig. 4A). These observations indicate that active site residues important for the SaADATa enzyme to bind RNA may only have a minor contribution in the case of the T. brucei enzyme. To examine the potential contribution of these residues, we individually replaced two conserved residues by alanine: the catalytic glutamate 92 and arginine 159 of TbADAT2 (Fig. 4A). Each mutant was co-expressed with wild-type TbADAT3 in E. coli and shown to still form a heterodimer. The mutants were tested for enzymatic activity as well as binding. We found that, as expected, the catalytic glutamate mutation abolished enzymatic activity (not shown). Remarkably, however, neither mutation had any impact on tRNA binding, showing a Kd for the tRNA substrate of 0.21 +/− 0.08 μM and 0.16 +/− 0.03 μM for E92A and R159A, respectively (Table 1). These Kd values are within the range for that of the wild type (0.15 +/− 0.02 μM) (Table 1). Therefore, active-site residues involved in ADATa binding to its substrate play only minor roles (if any) in the T. brucei enzyme. This observation also suggested that, if the reason for the apparent low affinity of the wild-type enzyme for the A34-containing tRNA was due to the relatively rapid turnover of the substrate into product, then with a catalytically defective mutant (where the conserved glutamate has been replaced by alanine), one should expect binding to the A34-containing tRNA. We tested this possibility by performing EMSA with mutant E92A; as expected, this mutant formed a stable complex with the A34-containing tRNA with a Kd of 0.48 +/− 0.13 μM, which indeed approaches the KM for the reaction and is within the range for the calculated Kd for (a) the same mutant with the G34-containing tRNA (0.2 +/− 0.08 μM) (Table 1), and (b) the wild-type enzyme with the G34- and I34-containing substrates.
Binding parameters for wild type (ADAT2/3) and C terminus mutants of TbADAT2

Alignment of SaADATa and TbADAT2 suggests differences in tRNA binding. (A) Sequence comparison between the adenosine deaminases acting on tRNA of S. aureus and T. brucei shows that a number of key residues involved in substrate binding by the bacterial enzyme have naturally undergone nonconservative changes in the T. brucei enzyme. The amino acid sequences of T. brucei ADAT2 and S. aureus ADATa were aligned using Clustal W. Conservative changes are denoted by dots [(.) and (:)] placed under the sequences and identical residues by an asterisk (*). Amino acids involved in RNA interaction in the cocrystal of SaADATa and an anticodon stem–loop (ASL) representing the anticodon arm of S. aureus tRNAArg are shown in boldface letters. The amino acids in TbADAT2 shown within boxes have been mutated to alanine and are shown to play no role in tRNA binding in T. brucei. (B) The amino acid sequence of TbADAT2 was also analyzed for potential RNA binding domains by the RNAbindR program, which uses a naive Bayesian classifier for all predictions, as described by Terribilini (Terribilini et al. 2007). Potential domains are denoted by plus (+) signs under the sequence, and boldface letters denote the KR-domain which has been analyzed further in this study.
Partly due to earlier reports by Grosjean and coworkers on the requirement of a full-length tRNA for activity (Auxilien et al. 1996), our group has speculated on the basis for substrate binding by the eukaryal deaminases (Rubio and Alfonzo 2005). Most recently, Huang and coworkers suggested that eukaryotic deaminases have evolved the ability to utilize many different substrates by acquiring RNA binding motifs that are distinct from active site residues (Elias and Huang 2005). They also suggested that comparison of the bacterial deaminase sequences to those of the two subunits of the S. cerevisiae deaminase reveal an extension at the C terminus of ADAT2, which may harbor the RNA binding function. We have performed a sequence comparison between bacterial ADATa and TbADAT2 and identified a stretch of 10 amino acids (KRKRKDLSVV) at the C terminus of TbADAT2, which we term the KR-domain. This domain is found in all of the trypanosomatid ADAT2 protein sequences and generally across eukaryotic ADAT2 sequences but does not exist in most bacterial counterparts.
The TbADAT2 sequence was also further analyzed for potential RNA binding residues with the RNABindR program (Terribilini et al. 2007) (http://bindr.gdcb.iastate.edu/RNABindR/), and the KR-domain showed high probability for a potential RNA binding domain (Fig. 4B). We have constructed a series of C-terminal mutants to determine the role the KR-domain may play in substrate binding and/or deaminase activity. Initially, two mutants were generated: ADAT2 C-terΔ5, which retained all positively charged residues but bears a deletion of the last five amino acids (DLSVV), and ADAT2 C-terΔ10, in which the last 10 residues were deleted (including all five charged residues of the KR-domain). We performed binding studies with these two mutants, and indeed, the ADAT2 C-terΔ10 showed a defect in binding and yielded an eightfold increase in Kd compared to the wild type (Fig. 5A; Table 1). The ADAT2 C-terΔ5 mutant, however, showed a smaller binding defect (Kd = 0.62 +/− 0.29 μM). We then tested a mutant with the five residues in the KR-domain replaced by alanine (ADAT2 C-ter5A); this mutant showed a Kd of 3.28 +/− 0.63 μM (Fig. 5B; Table 1) and in turn could not support any detectable enzymatic activity (Table 2). This lack of activity is similar to what was observed with the 10-amino acid deletion mutant (ADAT2 C-terΔ10) (Table 2). The threefold increase in Kd of ADAT2 C-ter5A over the ADAT2 C-terΔ10 mutant, however, suggests that the string of alanine residues may affect the local structure beyond the binding defect seen with the 10-amino acid deletion. Regardless, these data indicate that the bulk of the binding may, indeed, be due to the contribution of the positively charged residues in the KR-domain.
Kinetic parameter for wild type and C-terminal mutants of TbADAT2/3
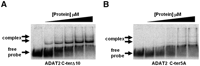
The C terminus of TbADAT2 is essential for tRNA binding. Two mutants were generated bearing either a deletion of the last 10 amino acids of TbADAT2 (ADAT2 C-terΔ10) or a replacement of the five positively charged amino acids of the KR-domain by alanines (ADAT2 C-ter5A). These TbADAT2 mutants were co-expressed with wild-type TbADAT3 in E. coli, and the resulting recombinant proteins (heterodimers) were purified by Ni2+-chelate chromatography. These mutants were used in EMSA assays. (A) Radioactive G34-containing tRNAVal was incubated with increasing concentrations of recombinant ADAT2 C-terΔ10/TbADAT3 heterodimer (Materials and Methods). (B) A similar experiment as in A but with ADAT2 C-ter5A. “Free probe” denotes the migration of the unbound tRNA, and “complex” denotes the migration of the protein-bound tRNA, also highlighted by arrows.
To study the specific contribution of individual residues further, we generated a series of mutants in which single amino acids of the KR-domain were replaced by alanine residues (Table 1). The Kd defects ranged in magnitude from three- and 4.7-fold increases for ADAT2 R217A and ADAT2 R219A and as high as sevenfold increases for ADAT2 K216A and ADAT2 K218A. Interestingly, the lysine to alanine mutations had similar and comparable effects on binding as the 10-amino acid deletion. The arginine for alanine replacements, while still affecting Kd, had smaller effects (Table 1).
Given the behavior observed with mutant ADAT2 C-ter5A and the structural argument made above, it is possible that some of the observed defects are due to more global changes in the overall structure of the enzyme. We have, thus, analyzed wild type and some of the mutants of TbADAT2/3 by circular dichroism spectroscopy. TbADAT2/3 contains a total of 15 phenylalanines, 15 tyrosines, and six tryptophans (five phenylalanines, nine tyrosines, and no tryptophan in TbADAT2, and 10 phenylalanines, six tyrosines, and six tryptophans in TbADAT3). We took measurements at wavelengths between 200 and 260 nm. Few differences were observed between the mutants described in Tables 1 and 2 and the wild type. The scans showed characteristic minima at 208 and 222 nm (α-helices) and at 218 (β-sheets) (Fig. 6). The minor differences seen at various points in the scans for the mutants compared to the wild type are within the experimental error of the machine on which the circular dichroism experiments were performed. Each protein sample was scanned at least three times, and raw data from each scan are averaged to give the data used to create each plot in Figure 6. As shown, with the exception of E92A at 224 nm, the deviations in the mutant scans from the wild type are all within the typical variability for independent scans of the wild type alone, indicating that the small differences seen are not statistically significant.
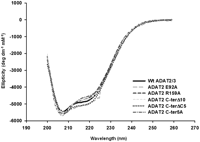
None of the mutations produced drastic effects on the overall structure of TbADAT2/3 and its variants. Both wild-type recombinant TbADAT2/3 and the various mutants from Tables 1 and 2 were analyzed by circular dichroism. The UV spectra were taken at various wavelengths between 200 and 260 nm. Molar elipticity ([θ]) was calculated as described in Materials and Methods and plotted as a function of wavelength. None of the mutants had significant spectral deviations when compared to the wild type, indicating that no major structural rearrangements had taken place.
In addition, size-exclusion chromatography showed that all of the mutants had comparable elution profiles to the wild type, and all formed heterodimers with TbADAT3, as shown by a nominal molecular weight of 68 kDa (Fig. 7; data not shown). We, therefore, conclude that the KR-domain plays a critical role in substrate binding, and the observed differences in binding behavior between the mutants and the wild type are not likely due to indirect effects from major disruptions in the global structure of the enzyme.
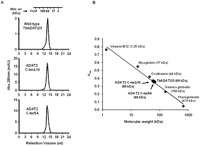
TbADAT2 C-terminal mutant still heterodimerizes with ADAT3. (A) Wild-type TbADAT2/3 and the two C-terminal mutants were fractionated by size-exclusion chromatography as indicated in the Materials and Methods section. Both mutants eluted with a size consistent with that calculated for the wild type (∼68 kDa), again suggesting that these mutations cause no major effects on the multimeric state of these mutants. (B) Kav vs. molecular weight standard curve. The standards and their respective molecular weights are as marked (gray diamonds) and labeled in gray. The wild-type and mutant ADAT2/3 enzymes are marked (black circles) and labeled in black.
The C terminus of TbADAT2 is crucial for enzyme activity
The binding studies allowed us to establish that the computer-predicted motif (KR-domain), indeed, plays a crucial role in tRNA binding. However, as explained earlier, the binding studies were performed with a G34-containing tRNA. Thus, for the binding data to be meaningful, we tested the effect of mutations of the KR-domain on enzymatic activity using the A34-containing tRNA substrate. The recombinant TbADAT2/3 enzyme was capable of 1 mole of inosine formation per 1 mole of tRNA after 40 min of incubation (data not shown). The reaction followed saturation kinetics when increasing concentration of radioactively labeled tRNA substrate was incubated with a constant amount of enzyme (Fig. 8A,B). The radioactively labeled substrate used in these reactions ranged in concentration from 0.1 μM to 1.6 μM. Plotting of the initial velocity against substrate concentration yielded a Vmax = 0.25 +/− 0.06 pmol/min, a KM value of 0.75 +/− 0.11 μM, and a kcat = 0.19 +/− 0.07 min−1 (Table 2). A similar KM value of 0.72 +/− 0.11 μM was observed for the partially purified (600-fold pure) native enzyme, suggesting that our recombinant enzyme is a fair representation of that natively expressed in T. brucei (data not shown). The observed KM is also within the range of the E. coli ADATa enzyme (KM = 0.83 μM) (Kim et al. 2006). However, the E. coli enzyme shows a 10-fold better kcat (Kim et al. 2006). The ADAT2 C-terΔ5 had a KM of 0.76 +/− 0.21 μM and a kcat of 0.14 +/− 0.05 min−1 (Table 2; Fig. 8); these values are very similar to the wild-type enzyme, and we conclude that these residues are not major contributors to enzyme activity. On the contrary, mutant ADAT2 C-terΔ10, as previously mentioned, had no detectable enzyme activity (Fig. 8; Table 2), suggesting that the binding mediated by the KR-domain may, indeed, be essential for enzyme activity.

Steady-state kinetic analysis of wild-type TbADAT2/3 and variants. (A) A representative one-dimensional thin-layer chromatography (TLC) analysis of the reaction products. pA and pI denote the migration of unlabeled 5′-AMP (pA) and 5′-IMP (pI) used as TLC markers and visualized by UV shadowing (not shown). The fraction of pA converted into pI during each reaction was calculated by dividing the amount of radioactive pI produced by the total (pA + pI); this value was then used to calculate the picomoles of 5′-IMP produced. A no-enzyme control was routinely used for background subtraction. (B) The initial velocity (Vo) is plotted as a function of substrate concentration given in μM. The data were fitted by nonlinear regression to the Michaelis–Menten equation using the SigmaPlot kinetic software. (C) Similar assays were performed with the two C terminus deletion mutants (ADAT2 C-terΔ5 and ADAT2 C-terΔ10) as indicated. A plus sign (+) refers to a reaction in which the radioactive tRNA substrate was incubated with wild-type TbADAT2/3 as a positive control. The black triangles refer to increasing, saturating concentrations of tRNA used in the assay (0.1, 0.2, 0.4, 0.8, 1.5, 2.0, 2.5, and 2.8 μM). (D) A plot of initial velocity vs. substrate concentration was used to derive the KM and kcat values as in B.
We also determined the steady-state kinetic parameters for the alanine scanning mutants. Replacement of the key catalytic glutamate in the ADAT2 by alanine, as expected, led to a complete loss of activity. Interestingly, a similar effect was observed for the ADAT2 C-ter5A mutant, indicating that the KR-domain, which by EMSA analysis was implicated in tRNA binding, is, indeed, essential for enzymatic activity. Further analysis of the individual alanine substitutions in the KR-domain showed that all the mutations led to increases in KM with negligible changes in kcat, reinforcing our proposal that the enzymatic defects observed correlate with binding defects and establishing the KR-domain as a bona fide binding determinant for this enzyme.
DISCUSSION
Mechanistically, whether acting on a riboflavin precursor, an amino acid, or nucleotides (free or within RNA and DNA polymers), all deaminases contain active sites with a tightly bound Zn2+ cofactor which generates a nucleophile by activation of water. What then separates the different members of this superfamily is their unique substrate specificity.
Free nucleotide deaminases use the 5′ and 3′ hydroxyls flanking the sugar as key recognition motifs to the exclusion of larger polymers (Navaratnam et al. 1998). On the other hand, RNA and DNA deaminases have more open active sites that allow accommodation of these larger substrates. In addition, the presence of a “flap” that closes the active site plays a critical role in what substrates can be accommodated (Xie et al. 2004). In the case of free nucleotide deaminases, this flap is short and less flexible than that of polynucleotide deaminases, as shown by Wedekind and coworkers in a comparative analysis of CDD1 (a free nucleotide deaminase) from yeast and APOBEC-1 (Xie et al. 2004).
The recently solved structure of an active deaminase domain of APOBEC3G also showed that, in addition to the active-site flap, a groove spanning the active site of the enzyme is formed by a set of both charged and aromatic amino acids (Furukawa et al. 2008; Holden et al. 2008; Shandilya et al. 2010). Groove formation for the purpose of substrate orientation is as critical for activity as direct contact of the ssDNA substrate by specific amino acids. Despite these divergences in substrate recognition, ssDNA-specific deaminases like AID or APOBEC3G still share a conserved amino acid core with tRNA deaminase (ADATa and ADAT2/3), including the presence of two core signature motifs found in all members of the deaminase superfamily: an active site glutamate (serving as a general base for catalysis), and a histidine/cysteine/cysteine triad involved in Zn2+ coordination. This evolutionary conservation has led to the proposal that all deaminases are derived from an ancestral nucleotide deaminase and through evolution a core deaminase domain has been appended to different specificity domains that will then provide binding to different substrates (i.e., riboflavin precursor vs. RNA or DNA).
In the specific case of tRNA deaminases, the process of evolution has taken the use of inosine at the wobble position in tRNAs into separate paths for bacteria and eukarya. In the eukaryal system, seven to eight different tRNAs may undergo A to I editing, depending on the organism (Gerber and Keller 1999). Searches of the T. brucei genomic database reveal the presence of eight different A34-containing tRNAs which all presumably undergo A to I editing. In bacteria, only tRNAArg undergoes A to I editing, but this single editing event is still essential for translation. Interestingly, the presence of G34-containing tRNAs in bacteria for the same codons (which are missing in eukarya) makes possible the limited use of inosine in the bacterial system, as G34 can take the place of inosine during decoding. In turn, tRNAArg is the only tRNA in bacteria for which a G34-containing counterpart is not encoded in the genome (Wolf et al. 2002). This observation has raised the question of how the eukaryal enzyme manages to accommodate the different substrates while maintaining target site specificity.
Huang and coworkers have proposed that, for recognition of seven to eight different substrates, a tRNA binding module has been appended to the eukaryal deaminases away from their active site, which in turn led to accumulation of mutations, resulting in active-site relaxation and concomitant multisubstrate specificity (Elias and Huang 2005). They asserted that likely regions in ADAT2 and ADAT3 that show little sequence conservation with the bacterial RNA deaminases might in fact harbor the appended RNA binding domain. In an attempt to test this model and elucidate some of the basis for substrate recognition, we have compared the T. brucei tRNA deaminase (TbADAT2/3) with bacterial tRNA deaminases. Given the remarkable catalytic flexibility of the TbADAT2/3 (able to perform both A to I and C to U editing in vitro) (Rubio et al. 2007), our work has concentrated on utilizing this enzyme to explore its mode of tRNA binding. We have demonstrated that, as predicted by Huang and coworkers, the C-terminal end of ADAT2 contains an essential domain for tRNA binding (Elias and Huang 2005). This domain is formed by a string of positively charged amino acids (arginines and lysines) we termed the KR-domain. Due to its overall charge, one could envision possible interactions between the KR-domain and the phosphate backbone of the tRNA, as suggested by Grosjean and coworkers for the yeast enzyme (Auxilien et al. 1996). Notably, binding of TbADAT2 alone to either the A34- or G34-containing tRNA is poor (>3.0μM) (data not shown), despite still harboring a KR-domain, indicating that the KR-domain is necessary but not sufficient to provide functional binding. This suggests that the second subunit (TbADAT3) also contributes to substrate binding.
A similar domain rich in positively charged amino acids is present in ADAR3, an orphan deaminase of unknown function (a homolog of the mRNA deaminases ADAR1 and 2) (Chen et al. 2000). It was recently shown that mutations of the analogous R-domain of ADAR3 specifically impaired ssRNA binding (Chen et al. 2000). These authors proposed that this domain might play a crucial role in recognizing loops often found near the targeted adenosine in mRNA substrates. We suggest that the KR-domain of ADAT2/3 serves an analogous binding function, but, unlike ADAR3, the targeted adenosine is distal to this binding domain.
Interestingly, in this study we have shown that the recombinant enzyme does not readily bind an A34-containing tRNA by EMSA. Admittedly this observation may appear counter to the behavior of the enzyme, which is able to efficiently catalyze inosine formation in this substrate (its natural substrate). We suggest that this may reflect on the fact that, once catalysis occurs, the enzyme turns over the product, making it difficult to catch the enzyme product complex if the assay is performed at optimal temperature (27°C). However, little binding was observed during EMSA (performed at 4°C). We suggest that it may be possible that substrate binding requires some type of conformational change in the enzyme, which may be temperature-dependent. Currently, however, we have no evidence for this, and it will thus remain an open question. Along the same lines, EMSA with the wild-type enzyme and the G34- and I34-containing substrates showed the presence of two well-defined complexes (arrows in Fig. 1A). This may again reflect either different multimeric forms of the enzyme upon substrate binding or, as mentioned above, different conformations of the protein and/or the substrate. In either case, determination of the Kd for either of the shifted bands yielded identical values (data not shown).
Kinetic measurements yielded an apparent Kd of 50 nM. This Kd is threefold better than that obtained with EMSA and the G34-containing tRNA. This is an important observation in that it partly explains how the enzyme avoids competitive inhibition due to binding naturally occurring G34-containing iso-acceptors in vivo. It is still possible that, in vivo, post-transcriptional modifications may also serve as negative determinants for nonproductive binding to other tRNAs that at the primary sequence level may resemble its natural substrate.
MATERIALS AND METHODS
Mutagenesis and recombinant expression of ADAT2/3 variants from T. brucei
ADAT2 mutants were generated by QuikChange site-directed mutagenesis (Stratagene). The coding sequences of either wild-type or mutant ADAT2s were cloned into expression vectors as previously described by us and co-expressed with wild-type TbADAT3 (Rubio et al. 2007). For expression, 10 mL of a starter culture was added to 1.0 L of 2× YT media and grown at 37°C until an OD600 of 0.8. Recombinant protein expression was then induced by addition of IPTG (0.5 mM final concentration) to the culture and grown at 25°C for 18 h or until an OD600 of 2.5 was reached. During protein purification, all procedures were conducted at 4°C and in buffers containing a protease inhibitor cocktail and phenylmethanesulphonylfluoride (PMSF) (Sigma-Aldrich). Cells were washed with lysis buffer (50 mM HEPES pH 8.0, 500mM KCl, and 50 mM imidazole) and broken by sonication with a Sonifier 450 using a microprobe at 50% output for a total of 5× 30-s intervals with a 60 s rest between sonication bursts. The resulting extract was spun at 100,000× g for 30 min, and the S100 fraction was collected and passed through a Ni2+-nitrilotriacetic acid (NTA) agarose column (1mL bed-volume). The column was washed with binding buffer and the bound protein eluted with a linear gradient of 50 mM to 1M imidazole. Peak fractions were pooled and dialyzed overnight in storage buffer (50 mM HEPES pH 8.0, 0.1 mM EDTA, 0.5 mM MgCl2, and 2mM 1,4-Dithiothreitol) and stored at −80°C in buffer containing 20% glycerol.
Electrophoretic mobility shift assay
Radio-labeled tRNAValAAC (8 nM) was incubated with reaction buffer (50 mM HEPES pH 8.0, 0.1 mM DTT, 1 mM MgCl2, and 5 mM KCl) and various concentrations of protein for 30 min at 4°C. Glycerol was added to the samples (10% v/v final concentration) and separated on a 6.5% nondenaturing poly-acrylamide gel at 100 volts for 1.5 h at 25°C. The gel was dried and exposed to a PhosphorImager screen. Products were analyzed with the Storm imaging system and quantified using ImageQuant software (GE). The binding data were fitted to a single ligand-binding curve using the SigmaPlot kinetic software.
Deamination assays
Labeled tRNA substrate was prepared as previously described by us (Rubio et al. 2006). Radio-labeled substrate was first heated to 70°C for 3 min to denature the RNA and allowed to refold at room temp for 5 min in reaction buffer (50 mM Tris-HCl pH 8.0, 2.5 mM MgSO4, 0.1mM EDTA, and 1 mM DTT). Following enzyme addition, reactions were incubated at 27°C for 45 min, phenol-extracted, and ethanol-precipitated. Pellets were washed with 70% ethanol and suspended in 10 μl of nuclease P1 buffer (MPBiomedicals) and 1 μL (0.5 units per reaction) of P1 nuclease (MPBiomedicals). The reaction was incubated overnight at 37°C and dried in a SpeedVac DNA 110 concentrator system (Savant) under high heat. Dried samples were suspended in 3 μL ddH20, and 1 μL was spotted on thin-layer chromatography (TLC) plates. Reaction products were separated by one-dimensional TLC in solvent C [0.1 M sodium phosphate (pH 6.8):ammonium sulfate:n-propyl alcohol (100:60:2, v/w/v)], visualized using the Storm imaging system (GE), and quantified using ImageQuant software. Nucleotide assignments were made using published TLC maps and cold nucleotide markers (Keith 1995). The fraction of inosine formed was calculated by dividing the inosine signal (pI) by the total signal of adenosine plus inosine (pA + pI); this value was then used to calculate the specific activities for each assay, taking into consideration the reaction time. The values obtained from a mock reaction in the absence of enzyme were used as a background control. Steady-state kinetic constants were calculated by nonlinear regression using the SigmaPlot kinetic software.
Single-turnover kinetic assay
Single-turnover deaminase assays were performed as above with the following changes. T7-transcribed tRNA was incubated with a saturating concentration of TbADAT2/3 for increasing time. The time course experiment was repeated for each different protein concentration ranging from 0.15 μM to 10 μM. At each time point, the tRNA was phenol-extracted, ethanol-precipitated, digested, and spotted on TLC plates as above. The fraction of inosine produced was plotted versus time and fitted to the equation f = a(1 − e−kt) to calculate kobs values for each protein concentration using SigmaPlot. kobs values were then plotted against each respective protein concentration. The data were fitted to a single ligand-binding curve and the Kd app calculated by linear regression using the SigmaPlot kinetic software.
Circular dichroism spectroscopy and size-exclusion chromatography
Circular dichroism (CD) spectra of wild-type and mutant ADAT2/3 (in 10 mM Tris, pH 7.5) were measured at room temperature with an AVIV 62DS Spectropolarimeter. CD data were collected at a scan rate of 1 nm/s; spectra reported represent the average of three scans. Ellipticity values collected directly from the machine were converted to molar ellipticity using the equation: [θ] = 100 × θ/cl, where [θ] refers to molar ellipticity, θ refers to ellipticity, c equals the solute molarity, and l equals the path length of the cuvette used. At least three scans were performed for each sample, and the average of those scans is represented in the plot.
For molecular weight determination, wild-type TbADAT2/3 and the C-terminal mutants (ADAT2 C-ter5A and ADAT2 C-terΔ10) were subject to size exclusion chromatography using a Superdex 200 10/300 (GE Life Sciences). Elution buffer was the same as the reaction buffer (above) without EDTA and the addition of 100mM KCl. A standard curve was first generated with protein standards (from Bio Rad) (Fig. 7). Kav values were calculated using the formula Kav = [Velution − Vvoid]/[Vcolumn − Vvoid], where V is volume (mL). Blue dextran was used to calculate a void volume for the column of 8.1 mL. The total column volume is 23.6 mL. Peak elution volumes of each ADAT2/3 sample were then converted to Kav values which were used to calculate the molecular weight in kDa.
ACKNOWLEDGMENTS
We thank all members of the Alfonzo laboratory for helpful comments and discussions. We also thank Dr. Jane Jackman (Ohio State University) for helpful suggestions. This work was supported in part by a grant GM084065 (NIGMS) to J.D.A.
Footnotes
-
↵6 Corresponding author.
E-mail alfonzo.1{at}osu.edu.
-
Article published online ahead of print. Article and publication date are at http://www.rnajournal.org/cgi/doi/10.1261/rna.2748211.
- Received March 23, 2011.
- Accepted April 8, 2011.
- Copyright © 2011 RNA Society








