
Chemical modification resolves the asymmetry of siRNA strand degradation in human blood serum
Abstract
Small interfering (si)RNAs have recently been used to therapeutically silence genes in vivo after intravenous systemic delivery. Further progress in the development of siRNA therapeutics will in part rely on tailoring site-specific chemical modifications of siRNAs to optimize their pharmacokinetic properties. Advances are particularly needed to improve the nucleolytic stability of these double-stranded RNA drugs in vivo and suppress adverse off-target gene silencing effects. Here we demonstrate that specific chemical 2′-O-methylation, which has already been shown to ameliorate the omnipresent off-target toxicity of siRNAs, selectively protects the particularly vulnerable 5′-end of the guide strand against exonucleolytic degradation in human blood serum. Specific chemical modification thus resolves the asymmetric degradation of the guide and passenger strands, which is inherent to the thermodynamic asymmetry of the siRNA termini as required for proper utilization of the guide strand in RNA interference.
Keywords
INTRODUCTION
Originally described in Caenorhabditis elegans (Fire et al. 1998), RNA interference (RNAi) is a conserved set of pathways in eukaryotes that mediates potent gene silencing triggered by the interaction of small interfering (si) and related RNA effector molecules with the RNA induced silencing complex (RISC) (for a recent review, see Rana 2007). RNAi holds promise for treatment of human disease because it is broadly, yet specifically, applicable to any undesired gene whose sequence is known. While recent studies have successfully utilized systemically administered siRNAs in nonhuman primates via intravenous injection (Zimmermann et al. 2006), at least two major obstacles remain before the therapeutic promise of RNAi can be fully realized. First, the guide strand that, together with the passenger strand, makes up a siRNA duplex must be stabilized against the ubiquitous exo- and endonucleases found in blood serum and elsewhere in the body (de Fougerolles et al. 2005). Second, adverse off-target activity due to cross-reactivity between RNAi pathways must be suppressed (Scacheri et al. 2004; Birmingham et al. 2006). We demonstrate here that the same modest chemical double modification found to decrease off-target toxicity (Jackson et al. 2006) also substantially stabilizes the siRNA guide strand against exonucleolytic attack.
A number of studies have previously examined the stability of siRNAs in blood serum (Braasch et al. 2003; Chiu and Rana 2003; Czauderna et al. 2003; Layzer et al. 2004; Allerson et al. 2005; Choung et al. 2006; Haupenthal et al. 2006; Xia et al. 2006; Turner et al. 2007). Most of this work relied on assays that could not identify the strand and/or specific site of cleavage, which is a prerequisite for designing chemical modifications to specifically protect vulnerable sites. Such information, in principle, is accessible through cleavage product analysis by either mass spectroscopy (Turner et al. 2007) or radioactive labeling combined with denaturing polyacrylamide gel electrophoresis (PAGE) (Xia et al. 2006). A robust kinetic analysis of nucleolytic cleavage products of both the guide and passenger strands at nucleotide resolution has, however, not yet been reported. We here have developed such an assay, which allows us to show that specific chemical 2′-O-methylation, already known to reduce off-target activity of siRNAs, selectively protects the vulnerable 5′-end of the guide strand against exonucleolytic degradation in human blood serum. A simple chemical modification of the guide strand thus serves dual functions essential for the efficient application of siRNA therapeutics.
RESULTS AND DISCUSSION
The guide strand is degraded more efficiently than the passenger strand
We selectively labeled each strand of a canonical siRNA against firefly luciferase (Fig. 1; Martinez et al. 2002) and followed cleavage of the full-length RNA into specific shorter products over time by gel electrophoretic separation (Fig. 2). Dephosphorylation and cleavage of both the guide and passenger strands of our siRNA is substantial after only 5 min incubation with 3% (v/v) human blood serum in a close-to-physiologic buffer (50 mM Tris-HOAc, pH 7.4, 80 mM KCl, 20 mM NaCl, and 1 mM MgCl2, at 37°C) and increases over 3 h (Fig. 2). (As expected, the degradation kinetics accelerate with the volume fraction of serum [data not shown]; 3% [v/v] serum was chosen as optimal for resolving the cleavage products by gel electrophoresis.) Strong cleavage at the 5′-terminus of the guide strand, 3′ to positions 1 and 2, causes markedly lower stability of the guide strand compared to the passenger strand (Fig. 2B,C). The kinetics are strongly biphasic so that 56% of the guide strand degrades with a half-life <3 min, while the remaining 43% degrade with a half-life of 15 min (Table 1). Thus, the guide strand is completely degraded over the course of 2–3 h while 50% of the passenger strand persists after an initial, rapid, biphasic decay (Fig. 2; Table 1). Given the strict length requirement in RNA interference (Elbashir et al. 2001), the observed loss of even a single nucleotide on the 5′-end of the guide strand is expected to be functionally detrimental. The distribution of the remaining cleavage sites on the individual strands is generally not predictable (Figs. 1, 2), except that at least one cleavage site is observed in all clusters of A–U base pairs. A significant fraction of cleavage events may be secondary hits on partially degraded strands, although the time-dependent decrease in full-length RNA as measured here quantifies only primary hits. The observed preference for cleavage 3′ to pyrimidines is consistent with earlier studies suggesting that siRNA degradation in serum is at least in part due to RNase A-type enzymes (Haupenthal et al. 2006). Human pancreatic ribonuclease (hpRNase) is the likely candidate, as it is the most competent tetrapod RNase A-type enzyme to target dsRNA substrates and accounts for 70%–80% of total ribonuclease activity in blood serum (Weickmann and Glitz 1982; Libonati and Sorrentino 2001; Sorrentino et al. 2003). Despite the biphasic nature of the observed degradation kinetics, the results are consistent with the action of a single enzyme, as we do not observe any distinct products at early and late stages of the time course (Fig. 2) and controls utilizing purified RNase A also manifest biphasic kinetics (Table 1); still, we cannot rule out the combined action of (groups of) faster and slower nucleases. Similar biphasic degradation kinetics were observed in earlier work monitoring the amount of intact siRNA duplex upon exposure to mouse plasma, suggesting that both strands survive together (Allerson et al. 2005).
Half-lives (t 1/2) describing the observed biphasic siRNA degradation kinetics in 3% (v/v) human blood serum and purified RNase A under standard conditions
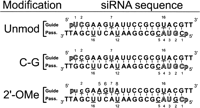
siRNAs used in this study as derived from Martinez et al. (2002), with guide and passenger strands indicated. All three constructs carry 5′-phosphates as well as two deoxythymidines on the 3′-end of each strand. In the C–G siRNA, a C–G base pair replaces the closing U–A base pair at the 5′-guide strand terminus. The 2′-O-Me siRNA harbors two 2′-O-Me modifications at positions 1 and 2 of the guide strand (depicted in lowercase letters). An outlined nucleotide indicates a nucleolytic hit 3′ to that position and represents ≥5% of the total lane intensity, while an underlined nucleotide indicates a hit 3′ to that position that is >10-fold of background, but <5% of the total lane intensity.

Comparison of the degradation of guide and passenger strands of the unmodified siRNA in the presence of 3% (v/v) human blood serum under standard conditions (50 mM Tris-HOAc, pH 7.4, 80 mM KCl, 20 mM NaCl, and 1 mM MgCl2, at 37°C). (A) Gel electrophoretic analysis of the time courses of serum induced cleavage of both the guide and passenger strands alongside RNase T1 and alkali (OH) sequencing ladders. Significant cleavage at positions 1 and 2 of the guide strand leads to a marked instability of the guide strand compared to the passenger strand. (B,C) Quantification of the gels in A where the fraction of intact siRNA, position 1, position 2, and PO4 3− band intensity are plotted as a function of time. Both the half-life (t 1/2) of <3 min and the 63% fraction cleaved at position 1 of the guide strand illustrate the particular lability of this position (Fig. 2B; Table 1). The guide strand is completely degraded over the course of 3 h, whereas the passenger strand persists at ∼50% over the full course of the assay.
Thermodynamic asymmetry is defined by weaker terminal U–A base pairing
The activity of RNase A-type nucleases on double-stranded RNAs depends on the ability of the enzyme to invade the duplex and thus access the in-line attack conformation required for RNA cleavage (Raines 1998; Libonati and Sorrentino 2001). Our observation of a cleavage preference 3′ to U over C is consistent with this requirement for strand invasion, as the weaker base pairing of U–A compared to C–G would overcome the reported preference of RNase A-type enzymes for poly-C over poly-U (Raines 1998; Sorrentino et al. 2003). We therefore hypothesized that the same thermodynamic asymmetry that leads to the siRNA end fraying used by RISC to select a 5′-phosphate and thus define the guide strand (Tomari et al. 2004) predisposes this strand to ribonuclease exposure.
To test this hypothesis, we mutated the 5′-terminal U–A base pair of our guide strand to the more stable C–G. This U-to-C transition may be expected to enhance reactivity toward hpRNase due to the enzyme's C-over-U preference (Raines 1998; Sorrentino et al. 2003). In contrast, yet consistent with our hypothesis, we observe protection of the 5′-terminus of the C–G modified guide strand compared to that of the unmodified siRNA (Fig. 3A,B; Table 1). A control experiment using a mismatched duplex between the U-to-C mutated guide strand and the unmodified passenger strand exhibits degradation profiles that are practically identical to those of the unmodified duplex (data not shown). Passenger strand cleavage in the C–G modified siRNA remains essentially unchanged (Table 1). Given the increased stability of the C–G modified siRNA (Fig. 3C, ΔG°37C = −29.0 ± 1.5 kcal/mol) relative to the unmodified siRNA (Fig. 3C, ΔG°37C = −25.7 ± 0.6 kcal/mol), we therefore suggest that thermodynamic stabilization of the duplex terminus protects it from RNase degradation.

Stabilization of the guide strand by mutation and chemical modification in the presence of 3% (v/v) human blood serum under standard conditions (50 mM Tris-HOAc, pH 7.4, 80 mM KCl, 20 mM NaCl, and 1 mM MgCl2, at 37°C). (A) Gel electrophoretic analysis of the time course of serum induced cleavage of the guide strand of our three different siRNA constructs, as indicated, alongside RNase T1 and alkali (OH) sequencing ladders. Both terminal C–G mutation and 2′-O-Me modification substantially stabilize the guide strand. (B) Quantification of the gels in A where the fraction of intact guide strand is plotted as a function of time. (C) Thermal denaturation of our three siRNA constructs as monitored by UV melting. The derived melting temperatures (Tm ) and ΔG°37C values are indicated on the plot in °C and kcal/mol, respectively.
2′-O-Me modification of positions 1 and 2 selectively stabilizes the guide strand
Selection rules for effective siRNA design dictate full complementarity to the target sequence and asymmetric melting of the termini for proper selection of the guide strand by RISC (Tomari et al. 2004; Yuan et al. 2004; Pei and Tuschl 2006; Patzel 2007), typically ruling out a terminal U–A to C–G mutation as employed above. We therefore sought to introduce protective chemical modifications into the ribonucleolytically most vulnerable positions 1 and 2 at the 5′-end of the guide strand. A number of modifications, especially of the RNA backbone, have been previously explored for broadly stabilizing siRNAs in gene therapy applications (Dorsett and Tuschl 2004; Manoharan 2004; Chen et al. 2005; Eckstein 2005; Zhang et al. 2006; Rana 2007). In addition, recent studies have found that 2′-O-methyl (2′-O-Me) modification of positions 1 and 2 at the 5′-ends reduces the widespread problem of off-target silencing effects induced by siRNAs (Jackson et al. 2006; Snove and Rossi 2006). We thus set out to test whether this same double modification may serve the added purpose of extending the lifetime of a siRNA in human blood serum by applying our quantitative degradation assay. We find that this modest chemical double modification completely suppresses ribonucleolytic cleavage 3′ to position 1. It also significantly reduces, but does not completely block cleavage 3′ to position 2 (Fig. 3A). The residual cleavage upon 2′-O-Me modification of position 2 suggests either that hpRNase uses an alternate cleavage mechanism not requiring the 2′-OH moiety or that less abundant nucleases partake in degradation (that also do not require the 2′-OH). The latter possibility is supported by the unusual cleavage sites 3′ to purine residues that are induced upon 2′-O-Me modification of the guide strand (Fig. 3A). It is possible that interference with preferred cleavage sites by chemical modification enhances slower, less optimal ribonucleolytic activity at purine residues. The overall degradation kinetics of the doubly 2′-O-Me protected guide strand in human blood serum is monophasic (at the time resolution of our data) such that 52% of the RNA is still rapidly degraded with a half-life of <3 min, but the remaining RNA persists for long periods of time (Fig. 3B; Table 1). Serving as a control, passenger strand degradation is again largely unaffected by modification of the guide strand (Table 1). Thermal denaturation experiments indicate only a slight increase in stability of the 2′-O-Me modified siRNA duplex relative to the unmodified duplex (Fig. 3C, ΔG°37C = −26.8 ± 0.2 kcal/mol versus ΔG°37C = −25.7 ± 0.6 kcal/mol), which is a lesser increase in stability than afforded by the terminal U–A to C–G mutation (Fig. 3C, ΔG°37C = −29.0 ± 1.5 kcal/mol). These observations suggest that both thermodynamic and chemical stabilization likely contribute to the observed prolonged half-life of our 2′-O-Me modified siRNA duplex.
Interestingly, we observe reduced target RNA cleavage by RISC (Martinez et al. 2002) upon introduction of our 2′-O-Me modifications into the siRNA guide strand, as we do for the C–G mutant (Fig. 4). To investigate the possibility that having 2′-O-Me modifications only on the guide strand perturbs RISC function, we conducted target cleavage assays with an siRNA carrying 2′-O-Me modifications in the 1 and 2 positions of both the guide and passenger strands [Fig. 4, denoted as OMe(2)], better reflecting the experiments described in the study identifying the off-target amelioration of 2′-O-methylation (Jackson et al. 2006). Interestingly, this symmetrically modified siRNA does not rescue activity in our target cleavage assay. Since the mechanism by which 2′-O-Me modification reduces off-target activity is not well understood (and target RNA cleavage was not described in the earlier study) (Jackson et al. 2006), our findings raise the intriguing possibility that RNAi mechanisms not involving target RNA cleavage, such as target sequestration, may be involved in enhancing siRNA specificity.
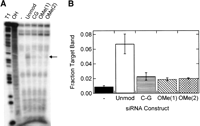
RISC-mediated, siRNA-induced cleavage of pp-luc target RNA by the siRNAs examined in this study. (A) Gel electrophoretic analysis of a representative target cleavage assay, where the “-” lane is target RNA incubated with cell extract in the absence of siRNA, “Unmod” is incubated in the presence of our unmodified siRNA, “C–G” is incubated in the presence of the C–G modified siRNA, “OMe(1)” is incubated in the presence of the siRNA with only the guide strand double 2′-OMe modified, and “OMe(2)” is incubated in the presence of the siRNA with double 2′-O-Me modification (in positions 1 and 2 from the 5′-end) of both the guide and passenger strands. “T1” and “OH” indicate RNase T1 and alkali sequencing ladders, respectively. The arrow indicates the siRNA-induced cleavage site on the target RNA. (B) Quantification of the target band intensity from A relative to the sum of all bands in the respective lane.
CONCLUSIONS
In this report we have found that a relatively modest double 2′-O-Me modification at the 5′-end significantly prolongs the half-life of a large fraction of the siRNA guide strand when exposed to human blood serum. The promise of siRNA drugs that can be specifically tailored to downregulate previously “undruggable” gene products via the RNAi pathway may usher in a new era of pharmaceuticals. Still, several problems need to be overcome, including off-target effects, nucleolytic degradation, cell-specific delivery, and cellular uptake of the siRNA drug before RNAi therapies are widely applicable. Showing here that a simple double 2′-O-Me modification, which previously has been shown to enhance siRNA specificity, selectively protects the vulnerable 5′-end of the guide strand against exonucleolytic degradation in human blood serum may help simultaneously address two of the barriers to successful implementation of siRNA therapeutics for human disease.
MATERIALS AND METHODS
RNA synthesis
All RNAs were synthesized by the HHMI Biopolymer/Keck Foundation Biotechnology Resource Laboratory at the Yale University School of Medicine, and were deprotected and purified as previously reported (Hoerter et al. 2004). Sequences are derived from the firefly luciferase gene (Martinez et al. 2002) and are shown in Figure 1.
RNA labeling
RNAs were first treated with calf intestine alkaline phosphatase to remove the synthetic 5′-phosphate and prepare the 5′-OH necessary for labeling. Strands were 5′-32P labeled with T4 polynucleotide kinase (PNK) and γ-32P-ATP at an RNA concentration of ∼800 nM. PNK was inactivated by heating to 90°C for 10 min. The appropriate complementary RNA strand was then added to the reaction at a final concentration of 2 μM and annealed by heating to 70°C for 2 min, followed by cooling to room temperature over 10 min. Cold nondenaturing loading buffer was added to a final concentration of 1 × TBE, 0.025% bromophenol blue, and 10% glycerol, and the annealed, labeled siRNA purified at 4°C by electrophoresis on a nondenaturing, 1 × TBE, 12% (w/v) polyacrylamide gel run at ∼20 V/cm, excision of the radiolabeled duplex band, diffusion elution into 1 mM EDTA overnight at 4°C and ethanol precipitation. The purified siRNA was dissolved in 25 mM Tris-HCl, pH 7.5, and 25 mM NaCl. Single-stranded 5′-32P labeled RNA for the T1 and alkali ladders was prepared similarly and dissolved in water.
siRNA degradation assays
A 5′-32P labeled, purified siRNA duplex was prepared in a close-to-physiologic standard buffer of 50 mM Tris-HOAc, pH 7.4, 80 mM KCl, 20 mM NaCl, and 1 mM MgCl2. Degradation was initiated by the addition of 3% (v/v) human blood serum (Innovative Research; single healthy donor) at 37°C. Time points were taken after 5, 10, 20, 40, 80, 120, and 180 min and quenched by mixing with a final concentration of 10% (v/v) Contrad 70 at pH 9.3 (Decon Labs) (Uhler et al. 2003). Loading buffer was added to a final concentration of 1 × TBE, 0.025% bromophenol blue, 0.025% xylene cyanol, and 40% formamide, and the degradation products (50,000 cpm per lane) separated on a denaturing, 8 M urea, 20% (w/v) polyacrylamide gel. Individual bands were identified by comparison with sequencing ladders from partial digestion with G-specific RNase T1 and alkali as described (Harris et al. 2004), quantified and normalized to the sum of all bands in a lane using a PhosphorImager Storm 840 with Image Quant software (Molecular Dynamics). Error bars were derived from at least two independent determinations. Time traces of the loss of the full-length RNA were fit with the double-exponential first-order rate equation y = y 0 + A 1 e −tk1 + A 2 e −tk2, employing Marquardt–Levenberg nonlinear least-squares regression (Igor Pro 5.03), where A 1 and A 2 are the fractions cleaved and ln 2/k 1,2 are the reported half-lives (t 1/2) of the fast and slow phases, respectively. The t 1/2 values for the fast phase of the reaction were estimated to be <3 min based on an interpolated lower estimate for the cleavage extent at a hypothetical 2.5-min time point. The 2′-O-Me guide strand data were fit with the above equation with only a single exponential. RNase induced siRNA degradation assays were identical to the serum assays except that purified RNase A was added to a final concentration of 290 nM, yielding the data shown in Table 1.
Thermal denaturation
siRNA duplexes at a concentration of 1 μM per strand were annealed in standard buffer (50 mM Tris-OAc pH 7.4, 80 mM KCl, 20 mM NaCl, and 1 mM MgCl2) by heating to 70°C for 2 min and cooled at room temperature over 10 min. Solutions were filtered through a 0.45 μM centrifugal filter device (Millipore Ultrafree-MC) and degassed by spinning under vacuum in an Eppendorf Vacufuge for 5 min. Samples were placed in the Micro Auto 6 Tm cell holder of a Beckman DU640B UV-Vis spectrophotometer. Samples were equilibrated at 30°C for 15 min, and then heated to 95°C at a rate of 0.5°C/min while taking 1-sec absorbance readings at 260 nm every min, which were corrected against a 320-nm background measured in standard buffer at the corresponding temperature. Each melting curve was measured in duplicate and independently analyzed to extract melting temperatures (Tm ) and thermodynamic parameters as described (Plum et al. 1999). Reported ΔG°37C values are the average±error from the two melting curves. Tm s are reported as the average of the two extracted values; the error in all cases was <±0.1°C.
Target cleavage
RISC-mediated, siRNA-induced target cleavage of cap labeled pp-luc RNA was performed essentially as described (Martinez et al. 2002). Purified Pp-luciferase RNA was 5′-cap 32P labeled using α-32P-GTP and guanylyltransferase enzyme (Ambion) as recommended by the supplier. Cap labeled RNA was purified on a denaturing, 8 M urea, 6% (w/v) polyacrylamide gel run at 27 V/cm. The RNA was excised, eluted into 1 mM EDTA at 4°C overnight, ethanol precipitated, and dissolved in water.
We used each of our three siRNAs (Unmodified, C–G, 2′-O-Me) as well as an siRNA that was 2′-O-Me modified at positions 1 and 2 of both the guide and passenger strands [termed OMe(2) for distinction from the guide strand only modified OMe(1)]. siRNA strands were annealed at 1:1 ratio such that the final concentration of RNA was 100 nM in the target cleavage reaction and the final MgCl2 concentration was 1.1 mM. After annealing, GTP, ATP, RNasin, creatine phosphate, creatine kinase, and HeLa S100 cytoplasmic extract (Jena Bioscience) were added as specified previously (Martinez et al. 2002), and incubated at 37°C for 15 min, at which point 100,000 cpm of cap-32P labeled pp-luc target RNA was added. Incubation was continued at 37°C for 2.5 h, when proteinase K and the corresponding buffer were added (Martinez et al. 2002). The solution was incubated at 65°C for 15 min, phenol/chloroform extracted, chloroform extracted, ethanol precipitated overnight, and the RNA dissolved in formamide loading buffer and loaded onto a 37-cm-long denaturing, 8 M urea, 10% (w/v) polyacrylamide gel run at 25 V/cm. The gel was wrapped in plastic foil and exposed to a phosphor screen, which was recorded on a Storm PhosphorImager Storm 840, quantified using Image Quant software (Molecular Dynamics), and analyzed in IgorPro. Data reported results from two to three independent experiments.
ACKNOWLEDGMENTS
The authors are grateful to Aimee Jackson for helpful discussions. This work was supported in part by a Camille Dreyfus Teacher-Scholar award to N.G.W., while J.A.H.H. was supported by an Eli Lilly Predoctoral Fellowship.
Footnotes
-
Reprint requests to: Nils G. Walter, Department of Chemistry, 930 N. University Avenue, University of Michigan, Ann Arbor, MI 48109-1055, USA; e-mail: nwalter{at}umich.edu; fax: (734) 647-4865.
-
Article published online ahead of print. Article and publication date are at http://www.rnajournal.org/cgi/doi/10.1261/rna.602307.
-
- Received April 19, 2007.
- Accepted July 23, 2007.
- Copyright © 2007 RNA Society








