
Aptamer redesigned tRNA is nonfunctional and degraded in cells
Abstract
An RNA aptamer derived from tRNAGln isolated in vitro and a rationally redesigned tRNAGln were used to address the relationship between structure and function of tRNAGln aminoacylation in Escherichia coli. Two mutant tRNAGln sequences were studied: an aptamer that binds 26-fold tighter to glutaminyl-tRNA synthetase than wild-type tRNAGln in vitro, redesigned in the variable loop, and a mutant with near-normal aminoacylation kinetics for glutamine, redesigned to contain a long variable arm. Both mutants were tested in a tRNAGln knockout strain of E. coli, but neither supported knockout cell growth. It was later found that both mutant tRNAs were present in very low amounts in the cell. These results reveal the difference between in vitro and in vivo studies, demonstrating the complexities of in vivo systems that have not been replicated in vitro.
Keywords
INTRODUCTION
The in vitro study of tRNA aminoacylation has revealed many intricacies of the complex process of tRNA recognition by aminoacyl tRNA synthetase (aaRS). However, in vitro studies still fail to account for many observations made from in vivo studies. For example, the aminoacylation levels of certain tRNA mutants are 104-fold higher in vivo than in vitro (McClain et al. 1999). This observation alludes to the complexity of in vivo tRNA aminoacylation. In vitro studies do not account for the complex interactions between all of the cellular machinery; generally, they are designed to simplify a complex problem. In vitro techniques are therefore valuable in initiating the study of a complex system like tRNA aminoacylation. In fact, the isolation of tRNA mutants through in vitro selections of RNA aptamers would provide a basis for many mutant studies of tRNA aminoacylation.
The in vitro study of RNA aptamers is a fruitful field of scientific research. Aptamers are small nucleic acid ligands that are isolated through in vitro combinatory library selection methods (Gold et al. 1995). Aptamers have been used in a variety of applications from use as molecular sensors to use in protein inhibitors for drug discovery, and use in membrane permeability studies (Khvorova et al. 1999; Famulok et al. 2000; Cerchia et al. 2002). RNA aptamers have also presented an opportunity to study RNA–protein interactions, an important aspect in the study of the structure–function relationships in tRNA aminoacylation. The careful in vitro study of RNA aptamers coupled with in vivo analyses would reveal many workings of the tRNA aminoacylation system in the context of the living cell.
A tight binding tRNAGln aptamer derived from wild-type tRNAGln was isolated through an in vitro library selection method (Stubenrauch 1996), and it was found that the majority of the tight binding phenotype was due to the AGGU sequence in the variable loop (Bullock et al. 2000). A mutant tRNAGln containing four mutant bases in the variable loop (GlnAGGU tRNAGln mutant; Fig. 1B) was therefore synthesized through in vitro transcription and found to have a Kd for binding to glutaminyl-tRNA synthetase (GlnRS) that was 26-fold lower than that of wild-type tRNAGln in vitro (Bullock et al. 2000). The substantially tighter binding was attributed to a rearrangement in the complex core of tRNAGln. The crystal structure of the GlnRS–GlnAGGU complex (Bullock et al. 2000) showed that the bases (AGGU) in the tRNAGln mutant are stacked more efficiently than in wild-type tRNAGln in which the variable loop bases U47 and U46 are flipped out of the complex core base stacking. The GlnAGGU mutant tRNAGln suggests that stabilization of the tRNA tertiary structure as opposed to optimization of RNA–protein contacts can strengthen GlnRS–tRNAGln binding (Bullock et al. 2000). In another mutant, a tRNAGln with a serine tRNA long variable loop (Fig. 1C), and a cytosine at position 44 (GlnQSerC44) was synthesized. GlnQSerC44 was shown to be folded properly and aminoacylated in vitro, although the kcat/Km was reduced slightly (fourfold) relative to wild-type tRNAGln (Nissan et al. 1999). The aminoacylation kinetics show the capacity of GlnRS to tolerate large differences in the tertiary core of the tRNA while still retaining near wild-type substrate properties in vitro.
Below, we investigate the behavior of the mutants GlnAGGU and GlnQserC44 in the GlnΔ114 tRNAGln knockout strain of E. coli (W.H. McClain, unpubl.). The GlnΔ114 knockout strain lacks all chromosomal copies of the tRNAGln genes. Because tRNAGln is required for cell survival, the knockout strain is supported by a maintenance plasmid containing a wild-type tRNAGln gene, whose expression is under the control of the araCpBAD promoter, which is induced by the presence of arabinose and turned off by the presence of glucose. The properties of these maintenance plasmids have been described (Gabriel and McClain 2001). The two mutant tRNA genes were constructed and constitutively expressed from a high copy test plasmid pGFIB (Masson and Miller 1986) in the GlnΔ114 knockout strain. The phenotypes of the two mutants were tested by growing the knockout cells on glucose-containing media, so growth of the cells would be solely dependent on the activity of the tRNA genes expressed from the constitutive test plasmid, given that expression of the wild-type tRNAGln gene is turned off in the presence of glucose.
RESULTS AND DISCUSSION
The function of two tRNA mutants was studied: GlnAGGU (Bullock et al. 2000), a mutant tRNAGln containing four (AGGU) instead of the wild-type five (CAUUC) nucleotides in its variable loop (Fig. 1B); and GlnQSerC44 (Nissan et al. 1999), a tRNAGln rationally redesigned to contain a tRNASer variable loop, a cytosine at position 44, and changes at positions 9, 13, 20, and 20b in the D stem and loop to accommodate the large variable region (Fig. 1C). The synthetically assembled tRNA genes were ligated into a high-copy pGFIB plasmid. The plasmids expressing the mutant tRNAGln genes were electroporated separately into LB liquid cultures of GlnΔ114 tRNAGln knockout cells where expression of wild-type tRNAGln from the regulatable maintenance plasmid is repressed; neither mutant supported growth after 72 h of incubation at 37°C (data not shown). While the GlnΔ114 knockout cells did contain the maintenance plasmid, growth as a result of leakiness of the araCpBAD promoter is not a concern because no growth was detected.
The growth phenotype of GlnΔ114 knockout cells containing the mutant tRNA genes GlnAGGU and GlnQSerC44 was also determined in a glucose strip plate assay (Gabriel and McClain 2001; Moulinier et al. 2001). Cells transformed with test plasmid pSU81 constitutively expressing each mutant tRNA gene separately were streaked across a plate containing arabinose, which induces expression of the wild-type tRNAGln gene from the maintenance plasmid. A strip of sterile paper was then saturated with glucose, which inhibits expression of wild-type tRNAGln from the maintenance plasmid, and placed on the surface of the plate. The positive control streak of wild-type tRNAGln constitutively expressed from pSU81 showed growth through the entire streak, while the GlnAGGU, GlnQSerC44, and a negative control only grew at the edges of the plates, as the tRNA genes that were constitutively expressed were inactive and expression of the wild-type tRNAGln from the maintenance plasmid was inhibited by the glucose strip (Fig. 2). Again, growth as a result of leaky expression of wild-type tRNAGln from the maintenance plasmid is not a concern because it would result in growth of the test streaks and negative control near the glucose strip. This was not the case.
To further investigate the no-growth phenotype of the two mutant tRNAs, a Northern analysis was performed ascertaining the amount of the mutant tRNAs GlnAGGU and GlnQSerC44 in transformed E. coli XAC/A16 wild-type cells containing all chromosomal tRNAGln gene copies. Extracted RNAs were hybridized to radioactive probes complementary to the anticodon and part of the respective variable loop regions of wild-type tRNAGln, GlnQSerC44, and GlnAGGU in a Northern blot. A preliminary film showed neither mutant tRNAGln was present. Upon prolonged exposure, the probe for the GlnAGGU tRNA mutant revealed a tRNA-size band in both the GlnAGGU and the pGFIB negative control lane (Fig. 3), suggesting the probe was crosshybridizing to wild-type tRNAGln derived from the chromosome. Even so, the presence of the GlnAGGU mutant tRNA would have been detectable as an increase in band intensity because GlnAGGU was expressed from a high-copy plasmid. The comparable intensities of the negative control band and the GlnAGGU band indicate the mature form GlnAGGU mutant tRNA is either not present or is present in small amounts.
Northern signals from GlnQSerC44 also appeared after prolonged exposure, showing two bands that ran slower than wild-type tRNAGln (Fig. 3), one in the position of tRNAGln with a longer variable loop, and the other typical of precursor tRNA. The slower migrating band in the GlnQSerC44 lane may reflect a defect in processing of the GlnQSerC44 precursor tRNA. The faster migrating band does indicate that there is some mature GlnQSerC44 tRNA in the cell. However, the considerably reduced intensity of the GlnQSerC44 tRNA band relative to the intensity of plasmid expressed wild-type tRNAGln suggests that there is not enough mature GlnQSerC44 tRNA to support GlnΔ114 cell growth.
The apparent absence of the GlnAGGU mutant and the low intensity bands shown in the GlnQSerC44 lane suggests that both tRNAGln mutants and their precursor tRNAs are degraded by cellular RNases. The tertiary core of GlnAGGU is stacked more efficiently than wild-type tRNAGln when bound to GlnRS. However, it is unclear that free, unbound GlnAGGU tRNA, which is recognized in the cell by cellular RNA processing, tRNA synthesis, and degradation machinery, has the same efficient stacking in its tertiary core relative to that in the GlnRS–tRNAGln complex. Furthermore, the accumulation of precursor molecules of the mutant tRNA GlnQSerC44 points to the difficulties the cell has in processing this peculiar molecule (Altman 1971). Although thermal melting analyses have shown that the GlnQSerC44 mutant tRNA is well folded as a class II-like fold in vitro (Nissan et al. 1999), the structure may still be unusual with respect to the in vivo tRNA synthesis and/or stability processes. Processing of tRNAs by RNase P is exquisitely sensitive to the structure of substrate precursor tRNAGln; the tRNAGln portion of the precursor is folded into its canonical tertiary L-shaped form (Seidman et al. 1974; McClain and Seidman 1975). The sensitivity of RNase P to GlnQSerC44 is indicative of the high level of specificity in in vivo processing and degradation of tRNAGln. Indeed, in vivo studies of 10 separate mutants have shown that single replacements of Watson–Crick G-C base pairs with A–C or G–U mispairs throughout tRNAGln can cause the cell to degrade the tRNA, while subsequent changes of A–C or G–U mispairs to Watson–Crick base pairs prevents tRNA degradation (McClain and Seidman 1987).
All samples showed tRNA size bands when probed by the wild-type tRNAGln probe, as expected, because wild-type tRNAGln is expressed from the chromosomal tRNAGln genes of the wild-type XAC/A16 strain of E. coli used in the Northern analysis.
In an attempt to isolate a stable and functional tRNAGln mutant with four nucleotides in the variable loop, a library was constructed by randomizing the four variable loop nucleotides of the GlnAGGU mutant tRNA. The randomized tRNAs were assembled using a one-step synthesis method (Choi et al. 2002), ligated into the pGFIB plasmid, and introduced into GlnΔ114 tRNAGln knockout cells by electroporation. The experiment did not yield any growth-supporting mutants among 5 × 1010 independent clones tested. Thus, the library, which contained 256 possible sequences, was saturated and did not contain an active tRNA.
CONCLUDING REMARKS
The absence of both GlnAGGU and GlnQSerC44 in growing cells reveals the distinctions between in vitro and in vivo studies. Although T7 phage tRNA transcripts of both GlnAGGU and GlnQSerC44 were aminoacylated in vitro with purified GlnRS, neither tRNA was present in sufficient amounts in vivo to support knockout cell growth. In vitro studies can give valuable insights into the aminoacylation reaction that are difficult to obtain in vivo. Complicated systems can be broken down to individual parts and studied separately for deeper understanding. Nonetheless, the ultimate goal is to understand how biological systems work in vivo. It is therefore imperative that results from in vitro studies are tested in vivo before conclusions about a system are solidified. The complexities of in vivo systems sometimes makes them difficult to study; however, in vitro systems may excessively simplify a system, leaving out key points. It is therefore essential that both in vivo and in vitro studies are employed to understand the aminoacylation system.
MATERIALS AND METHODS
Bacterial strains and plasmids
The following strains and plasmids have been described. E. coli XAC/A16 (Δlacpro, nalA, rif, argEam/F′lacproB, IQA16am-Z fusion; KnR; (Normanly et al. 1986), GlnΔ114 tRNAGln knockout strain is a derivative of the MG1655 E. coli strain and lacks the four tRNAGln isoacceptors (ΔglnXVWU; W.H. McClain, unpubl.). The pGFIB plasmid is a high copy, ampicillin-resistant vector with efficient constitutive promoter and terminator (lpp and rrnB, respectively) for tRNA gene expression (Masson and Miller 1986); the pSAD plasmid is a medium copy, chloramphenicol-resistant vector with an AraCpBAD promoter that is induced by arabinose and repressed by glucose (Gabriel and McClain 2001); pGAD2 is a high-copy, ampicillin-resistant vector that contains the same AraCpBAD promoter as pSAD (Gabriel and McClain 2001); pSU81 is a medium-copy, chloramphenicol-resistant vector that contains a constitutive lpp promoter (Gabriel and McClain 2001).
Construction of tRNA genes
The construction of tRNA genes was performed in two ways, both employing the use of synthetic oligonucleotides. The six-piece construction method separates the tRNA into three regions: the 5′ end, core region, and 3′ end. The regions of the tRNA were separated as follows: 5′ end: 1–13 (upper strand), 1–16 (lower strand); core region: 14–63 (upper strand), 17–58 (lower strand); 3′ end: 64–76 (upper strand), 59–76 (lower strand). The restriction sites EcoRI and PstI were added to the 5′ and 3′ ends of the tRNA construct, respectively, as sticky ends. The oligonucleotides were obtained from either IDT or the UW-Biotech Center, and were resuspended in TE to a concentration of 20 pmole/μL. All oligonucleotides with the exception of the 5′ ends were phosphorylated, using PNK (New England Biolabs). Oligonucleotide (100 pmole) was combined with the phosphorylation reaction mixture and was incubated for 1 h at 37°C. The PNK was inactivated at 65°C for 20 min. The DNA was then extracted with Bonner, centrifuged at maximum speed in a microfuge for 5 min, and precipitated. The precipitated, phosphorylated DNA oligonucleotides were combined with the 5′ oligonucleotides and annealed by heating the mixture to 80°C for 5 min and slow cooling the mixture for 30 min. Once annealed, the tRNA gene construct was ligated into EcoRI and PstI digested vector. The primary structures of tRNA genes were confirmed by DNA sequencing.
The one-step synthesis method has been described (Choi et al. 2002). The tRNA gene library for the GlnAGGU library experiment was constructed using this method. The synthetic oligonucleotides 5′-AAGCTTGAATCCNNATTGGGGTATCGCCAAG CGGTAAGGCACCGGTTTTTGATA-3′ and 5′-GGATCCTGCA GNNNGATGGCTGGGGTACCTGGATTCGAACCAGGNNNNC CGGTATCAAAAACCGGT were obtained from IDT, and were used to construct a GlnAGGU tRNA gene, where N indicates a 25% mix in the four base variable loop. The oligonucleotides were then used in a one-cycle synthesis reaction, digested with EcoRI and PstI, and were ligated into the pGFIB plasmid. The tRNA gene library was tested in GlnΔ114 knockout cells in a plasmid switch as described below. The total number of cells examined was 5 × 1010 based on an appropriate dilution of a control sample, which gave 500 colonies per plate.
Plasmid switches
To prepare cells for a plasmid switch, an overnight culture of GlnΔ114 knockout cells supported by a wild-type tRNAGln gene carried on either the pSAD or pGAD2 maintenance plasmid was grown in MinA arabinose media (Gabriel and McClain 2001). The cells were then pelleted, washed three times in equal volume sterile dH2O, and resuspended in 1/20 volume sterile distilled H2O. The cells were then separated into 20-μL aliquots, and electroporated without added plasmid; this treatment reduces the intracellular pool of maintenance plasmid. The cells were allowed to recover for 20 min in SOC broth at 37°C. The cells were then pelleted and washed as above. The appropriate test plasmid was added to the GlnΔ114 cells, which were then electroporated a second time. The cells were allowed to recover in SOC broth for 1 h at 37°C. The cells were then subcultured into LB plus the appropriate antibiotic. Cells that lost the maintenance plasmid were found by picking and stabbing onto plates with the appropriate antibiotic. Loss of resistance to chloramphenicol (for pSAD) or ampicillin (for pGAD2) is evidence that the maintenance plasmid has been physically eliminated from the cell.
Glucose strip test
The glucose strip test has been described (Gabriel and McClain 2001; Moulinier et al. 2001). The plasmid pSU81 containing the appropriate test genes was transformed into GlnΔ114 knockout cells. The cells were plated on MinA plates plus supplements, ampicillin and chloramphenicol, and were incubated at 37°C overnight. An individual colony was picked into 75 μL of MinA without supplements, and was left to stand for 10 min. A 0.8-μL aliqot was then spotted onto a MinA plate plus supplements, arabinose, ampicillin, and chloramphenicol. The spot was streaked left to right across the plate in one continuous motion using a glass spreader. Once the streak was dry, a sterile paper strip saturated with 20% (w/v) glucose was laid in the center of the plate, perpendicular to the streaks. The plate was incubated at 37°C overnight. Cells at the edge of the plate were supported by wild-type tRNAGln expressed from the maintenance plasmid and possibly tRNA expressed from the test plasmid. Cells near the glucose strip will only be supported by the constitutively expressed test plasmid because maintenance plasmid gene expression is repressed by glucose.
Northern blot analysis
The Northern blot analysis method has been described (Gabriel and McClain 2001). The tRNA for Northern blot analysis was isolated by growing cells expressing the appropriate tRNA gene from a plasmid to an OD650 of 0.4. Phenol was added directly to the growing culture, and left to shake for 10 min. The culture was then centrifuged and the aqueous phase was precipitated with ethanol. The samples were dried and resuspended in TE. A 2-μL aliquot was taken from the sample and heated at 65°C for 5 min. The sample was then loaded and electrophoresed on a 10% polyacrylamide gel containing 7 M urea. The samples were electroblotted onto Nytran SuperCharge membrane for 45 min. The membrane was hybridized to (5′-32P)-labeled probe at a hybridization temperature of 50°C. The probes for most tRNAs were complementary to tRNAGln positions 29–47; that for GlnQSerC44 covered residues 29–48.
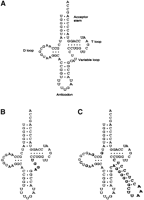
Cloverleaf structures of (A) wild-type tRNAGln; (B) the GlnAGGU tRNA mutant (Bullock et al. 2000); and (C) the GlnQSerC44 tRNA mutant (Nissan et al. 1999). Nucleotides that differ from the wild-type tRNAGln sequence are shown as bold letters on the tRNA mutant structures.
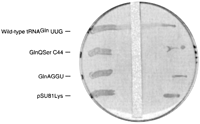
Glucose strip test of GlnAGGU and GlnQSerC44 growth properties in GlnΔ114 tRNAGln knockout cells. GlnΔ114 cells are supported by pSAD plasmid whose expression of the wild-type tRNAGln gene is repressed by glucose, and pSU81 plasmid constitutively expressing wild-type tRNAGln, GlnAGGU, GlnQSerC44, and amber tRNALys.
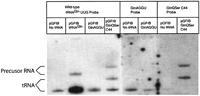
Northern analysis of GlnAGGU and GlnQSerC44 mutant tRNAs. Low molecular weight RNAs were prepared from XAC/A16 cells expressing the plasmids pGFIB, pGFIB tRNAGln, pGFIB GlnAGGU, or pGFIB GlnQSerC44. The high background on the films on the right reflects prolonged exposure. Identification of tRNA and precursor tRNA bands were based on gel mobility (Comer et al. 1974; Seidman et al. 1974; McClain and Seidman 1987). The probes for GlnQSerC44 showed precursor tRNAs, while the probes for GlnAGGU crosshybridized with wild-type tRNAGln transcribed from the XAC/A16 chromosome, and showed no signal of overexpressed GlnAGGU tRNA.
Acknowledgments
This work was supported by NIH Grant GM42123.
The publication costs of this article were defrayed in part by payment of page charges. This article must therefore be hereby marked “advertisement” in accordance with 18 USC section 1734 solely to indicate this fact.
Footnotes
-
Article and publication are at http://www.rnajournal.org/cgi/doi/10.1261/rna.5165804.
-
- Accepted September 22, 2003.
- Received August 22, 2003.
- Copyright 2004 by RNA Society








