
PTBP1 controls miRNA loading on target RNAs: lessons from the CyCoNP lncRNA
- 1Center for Life Nano- & Neuro-Science of Istituto Italiano di Tecnologia (IIT), Rome 00161, Italy
- 2Department of Biology and Biotechnologies “Charles Darwin,” Sapienza University of Rome, Rome 00185, Italy
- Corresponding authors: irene.bozzoni{at}uniroma1.it; fabio.desideri{at}cbm.csic.es
-
Handling editor: Eric Phizicky
Abstract
The concerted action of regulatory RNA and RNA binding proteins (RBPs) provides cells with highly versatile and transient tools to fine-tune gene expression in a broad variety of cellular systems (Unfried and Ulitsky, Nat Cell Biol 24: 608–615 [2022]; Hentze et al., Nat Rev Mol Cell Biol 19: 327–341 [2018]; Suzuki et al., Nat Genet 50: 657–661 [2018]). In this work, we explore the function of a specific interaction between PTBP1 and the cytoplasmic long noncoding RNA (lncRNA) CyCoNP, highly expressed in neural progenitors (Desideri et al., NAR 52: 9936–9952 [2024]), in which the RBP regulates the abundance of the lncRNA by a miRNA-mediated mechanism. PTBP1 is a well-known splicing regulator, although limited and peculiar examples of its involvement in other cellular processes, such as IRES-dependent translation and miRNA recognition of target RNAs, have been described (Dorn et al., Cell Death Dis 14: 6429 [2023]; Kim et al., Nat Commun 12: 5057 [2021]). We have recently characterized CyCoNP lncRNA as a regulator of NCAM1, which acts through a mechanism that involves direct RNA–RNA interaction with NCAM1 mRNA, balancing the availability and the localization of miR-4492 in its vicinity (Desideri et al., NAR 52: 9936–9952 [2024]). Here we expand the repertoire of molecular players acting in this circuitry by describing a direct interaction between PTBP1 and CyCoNP lncRNA. Through endogenous RNA purification, protein immunoprecipitation, and exploiting CyCoNP mutant constructs, we found that PTBP1, when interacting with CyCoNP, hampers miR-4492 binding to the lncRNA and in turn impedes its regulation on NCAM1 mRNA. This work aims to expand the biochemical characterization of regulatory networks relying on RBPs and their cognate target RNAs, highlighting the relevance of the analysis of the subcellular environment for each case of study.
Keywords
INTRODUCTION
RNA-binding proteins (RBPs) are a large class of molecules involved in nearly all aspects of RNA metabolism, such as splicing, transport, stability, and translation. These proteins form ribonucleoparticles (RNPs) with their target RNA, ultimately exerting a powerful regulation of gene expression at transcriptional and posttranscriptional levels (Hentze et al. 2018; Choi et al. 2024). RBP can recognize their RNA targets through RNA recognition motifs (RRMs), which are able to bind to specific nucleotide sequences or RNA secondary structures (Ray et al. 2017; Kuret et al. 2022; Dorn et al. 2023).
RBP's activity is strictly dependent on subcellular localization and cell maturation stage at which they are expressed. Human polypyrimidine tract–binding protein 1 (PTBP1), also known as hnRNP I (Ghetti et al. 1992), has been predominantly studied as a nuclear splicing factor in several cell types (Han et al. 2014; Desideri et al. 2020; Iannone et al. 2023; Liu et al. 2023). Additionally, its cytoplasmic functions in regulating RNA translation, through IRES recognition (Kafasla et al. 2009; Dorn et al. 2023), and localization (Sawicka et al. 2008) have been extensively characterized. In the neural context, PTBP1 represses the alternative splicing of key determinants of various developmental and physiological traits, to the extent of regulating the specification of the neuronal fate (Makeyev et al. 2007; Qian et al. 2020; Iannone et al. 2023; Liu et al. 2023). Thus, its expression is reduced during neural differentiation to allow the correct translation of differentiation drivers, such as the homologous RNA-binding protein PTBP2 (also named nPTB, given its prevalent neural expression) and elements of the REST complex, in the early phases of neurogenesis (Xue et al. 2013, 2016).
PTBP1 and PTBP2 are both targets and regulators of microRNA (miRNA) activity in the progression of early neuronal differentiation (Xue et al. 2016). PTBP1 has recently been found to broadly influence miRNA targeting in the neural context, through the modulation of RNA secondary structure and possibly other mechanisms, such as steric hindrance on the target RNA (Xue et al. 2013; Kim et al. 2021). On the other hand, miRNA-based regulatory mechanisms include their interaction with competing endogenous RNAs (ceRNAs), which act as molecular sponges for specific miRNAs, preventing them from binding to their target transcripts. This mechanism has been extensively described for lncRNA (Cesana et al. 2011; Bosson et al. 2014; Denzler et al. 2014; Carvelli et al. 2022; Unfried and Ulitsky 2022). Such molecules could act as platforms where protein- and RNA-based mechanisms of gene expression regulation occur simultaneously. A significant description of this mode of action involves the human lncRNA MACC1-AS1, which acts both as a miRNA sponge and binds to PTBP1 in breast cancer cells (Zhang et al. 2019).
We recently described the mechanism of action of CyCoNP, a human lncRNA that regulates SK-N-BE cells motility and hiPSC-derived motor neuron branching by acting as a strong regulator of NCAM1, a key protein involved in many aspects of neuron physiology (Desideri et al. 2024). CyCoNP acts in the cytoplasm of neural progenitors as a miR-4492 ceRNA, and at the same time, through direct interactions with the NCAM1 mRNA, it regulates the activity of miR-4492 on this mRNA.
Here we extended the characterization of CyCoNP, by detailing PTBP1 as a main protein interactor of the lncRNA in the cytoplasm. Using different approaches, we demonstrate that PTBP1, by regulating the amount of miR-4492 loaded on CyCoNP, controls the levels of the lncRNA and consequently those of NCAM1. These data shed light on a mechanism exerted by PTBP1 in the cytoplasm as a miRNA regulator and add insights into the multiple modes of action of RBPs on target RNAs.
RESULTS
PTBP1 interacts with CyCoNP in the cytoplasm of neuroblastoma cells
The RNA interactome of CyCoNP lncRNA was recently resolved through an endogenous RNA pull-down strategy followed by direct Nanopore RNA sequencing. This analysis led to the identification of NCAM1 mRNA as one of the main CyCoNP interactors and was crucial to resolve the complex mechanism of action exerted by the lncRNA in the cytoplasm of neuronal cells (Desideri et al. 2024). With the aim to go deeper into the regulatory mechanisms revolving around CyCoNP, beyond its RNA partners, we investigated whether RNA binding proteins (RBPs) could also control and participate in this regulatory circuitry.
We first looked for candidate interacting proteins of the CyCoNP RNA sequence by performing an in silico analysis running the catRAPID algorithm (Armaos et al. 2021) and scanning for interactions occurring between CyCoNP and the whole human proteome. The catRAPID algorithm predicted the presence in CyCoNP of numerous RNA motifs possibly recognized by PTBP1 (Fig. 1A), a human RBP mainly known to act as a splicing regulator in the nucleus but also described to act in the cytoplasm (Liu et al. 2023; Xue et al. 2013).
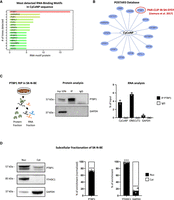
PTBP1 protein and CyCoNP lncRNA interact in the cytoplasm of SK-N-BE cells. (A) catRAPID analysis showing the proteins with detected RNA-binding motifs in CyCoNP transcript sequence among those available in the human proteome. The protein with the highest number of detected RNA-binding motifs, PTBP1, is highlighted in red. (B) Schematic representation of the list of proteins interacting with CyCoNP transcript according to the data retrieved from POSTAR3 database. The only interacting protein detected in a neuroblastoma cell line (SH-SY5Y) is demarcated in red. (C, left) Schematic representation of PTBP1 RNA immunoprecipitation (RIP) assay workflow in SK-N-BE cells (D 1.5). (Middle) Representative western blot analysis of the retrieved protein fractions in PTBP1 IP and IgG samples. GAPDH protein serves as a loading control. Input (Inp) samples represent 10% of the total protein extracts. (Right) RT-qPCR quantification of CyCoNP transcript recovery in PTBP1 IP and IgG samples. ONECUT2 transcript serves as positive control, while GAPDH transcript serves as a negative one. Values represent means ± SD of two biological replicates and are expressed as percentage (%) of input. (D, left) Representative western blot analysis of the nuclear and cytoplasmic fractions of PTBP1, YTHDC1 (nuclear control), and GAPDH (cytoplasmic control) in SK-N-BE cells (D 1.5). Quantification of PTBP1 (middle), YTHDC1, and GAPDH (right) signal intensities in each subcellular compartment. PTBP1 quantification was normalized over the enrichment of GAPDH in the cytoplasmic fraction. Data represent mean ± SEM of three biological replicates. Data information: (***) P < 0.001, unpaired Student's t-test.
We then queried the POSTAR3 database (Zhao et al. 2022) searching for the interaction between CyCoNP and PTBP1 and found that CyCoNP (named linc-02381 on the database) interacts with PTBP1 in the SH-SY5Y human neuroblastoma cell line (Fig. 1B; Uemura et al. 2017). Specifically, one PTBP1 binding site (bs) was detected on the CyCoNP sequence in that study. To experimentally confirm this, we performed PTBP1 RNA immunoprecipitation (RIP) experiments on extracts from SK-N-BE cells at 1.5 days of differentiation, when CyCoNP reach its highest expression levels (Fig. 1C, left panel). The successful precipitation of PTBP1 (Fig. 1C, middle panel) was paralleled by the detection of CyCoNP only in the precipitated fraction, along with ONECUT2 mRNA, a known interactor of PTBP1 (Uemura et al. 2017). Instead, no enrichment was detected for the GAPDH mRNA, used as a negative control (Fig. 1C, right panel; Uemura et al. 2017). These results were confirmed and strengthened by PTBP1 cross-linking and immunoprecipitation (CLIP) analysis followed by RT-qPCR performed on SK-N-BE cells that highlighted a direct and specific in vivo interaction between CyCoNP and PTBP1 (Supplemental Fig. 1A).
Since PTBP1 has been described as a splicing factor mainly localized in the nucleus, we performed nuclear/cytoplasmic fractionation in SK-N-BE cells at 1.5 days of differentiation in order to assess its presence and abundance in the compartment where CyCoNP is localized. Western blot analysis of the retrieved fractions showed that 27% of PTBP1 is present in the cytoplasm, whereas 73% resides in the nucleus (Fig. 1D). We then reanalyzed proteomic data generated by Zhang et al. (2021) during a differentiation experiment in SH-SY5Y cells. Examination of PTBP1 expression revealed that its levels peak at the onset of differentiation (Supplemental Fig. 1B) and that, compared with all the proteins identified in the data set, it falls within the upper tier of the distribution, classifying it as a medium to highly expressed protein (Supplemental Fig. 1C). Since CyCoNP expression corresponds to that of a medium abundant transcript (15 FPKM in SK-N-BE cells, equivalent to approximately five to seven molecules per cell; Desideri et al. 2024), and given that its expression trend parallels that of PTBP1 (Desideri et al. 2024), these data well endorse the hypothesis that PTBP1 is in a large excess with respect to CyCoNP, also in the cytoplasm, thus supporting the experimental results.
PTBP1 influences CyCoNP expression and activity by modulating its interaction with miR-4492
PTBP1 is known to influence RNA abundance in the cytoplasm by modulating miRNA-mediated RNA interference (RNAi; Kim et al. 2021). To test if PTBP1 is able to modulate the expression of CyCoNP, we depleted PTBP1 in SK-N-BE cells using a pool of siRNAs targeting PTBP1 mRNA compared to a scrambled control (si-PTBP1 and si-SCR, respectively). Upon 80% PTBP1 depletion (Fig. 2A,B), we observed a significant reduction in the abundance of CyCoNP that was paralleled by a slighter but significant decrease in NCAM1 expression, the main target of the lncRNA (Fig. 2A,B). Interestingly, PTBP1 depletion had no effect on the expression of the other linc-02381 isoforms (linc-02381-203 and -204), which remained undetectable as in the scrambled condition (Supplemental Fig. 2A; Desideri et al. 2024). This observation rules out the existence of a PTBP1-mediated splicing regulation affecting the different CyCoNP lncRNA isoforms. These results suggested that PTBP1 could affect CyCoNP stability through the modulation of miR-4492 activity.
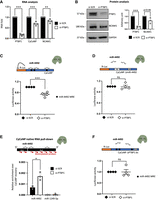
PTBP1 binds to CyCoNP to control the loading of miR-4492 on the lncRNA. (A) RT-qPCR quantification of PTBP1, CyCoNP, and NCAM1 transcripts in SK-N-BE cells (D 1.5) treated with si-SCR or si-PTBP1. Data were normalized to GAPDH mRNA and represent means ± SEM of four biological replicates. (B, left) Representative western blot analysis for PTBP1 and NCAM1 in SK-N-BE cells (D 1.5) treated with si-SCR or si-PTBP1. GAPDH was used as a loading control. (Right) Quantification of PTBP1 and NCAM1 signal intensities relative to GAPDH. Data represent means ± SEM of three biological replicates. (C, upper panel) Schematic representation of the CyCoNP luciferase-based reporter construct. The entire sequence of CyCoNP lncRNA was cloned downstream from the Renilla luciferase ORF (orange). The construct was cotransfected in SK-N-BE cells with a control siRNA (si-SCR) or siRNAs targeting PTBP1 (si-PTBP1). (Lower panel) Quantification of Renilla luciferase activity in SK-N-BE cells cotransfected with si-SCR or si-PTBP1. Data represent the mean luciferase activities ± SEM of four biological replicates. The relative position of miR-4492 MREs on the CyCoNP sequence is shown. (D, upper panel) Schematic representation of the CyCoNP ΔmiR-4492 luciferase-based reporter construct. (Lower panel) Quantification of Renilla luciferase activity in SK-N-BE cells cotransfected with the CyCoNP ΔmiR-4492 luciferase construct and a control siRNA (si-SCR) or siRNAs targeting PTBP1 (si-PTBP1). Data represent the mean luciferase activities ± SEM of four biological replicates. (E, upper panel) Schematic representation of CyCoNP endogenous RNA pull-down (PD). (Lower panel) RT-qPCR quantification of miR-4492 in CyCoNP PD RNA samples from SK-N-BE cells (D 1.5) treated with si-SCR or si-PTBP1. miR-1249-5p transcript serves as a negative control. Values are expressed as relative enrichment normalized over the % of CyCoNP transcript precipitated in each condition and represent means ± SEM of three biological replicates. (F, upper panel) Schematic representation of the CyCoNP ΔPTBP1 bs luciferase-based reporter construct. (Lower panel) Quantification of Renilla luciferase activity in SK-N-BE cells cotransfected with the CyCoNP ΔPTBP1 bs luciferase construct and a control siRNA (si-SCR) or siRNAs targeting PTBP1 (si-PTBP1). Data represent the mean luciferase activities ± SEM of four biological replicates. miR-4492 MREs relative position on CyCoNP sequence is shown. Data information: ns (nonsignificant) P > 0.05, (*) P < 0.05, (**) P < 0.01, (***) P < 0.001, unpaired Student's t-test.
Our previous work demonstrated the existence of a functional interplay between CyCoNP and its RNA interactors: miRNA-4492 and NCAM1 mRNA (Desideri et al. 2024). To further expand the molecular circuitry controlling CyCoNP expression and to investigate the role of PTBP1 in this context, we performed luciferase assays using a construct bearing the entire CyCoNP sequence cloned downstream from the Renilla luciferase gene (Rluc). We cotransfected this luciferase construct with siRNAs against the PTBP1 mRNA (si-PTBP1) or a scrambled control (si-SCR), in SK-N-BE cells, observing a significant reduction in the Renilla luciferase signal upon PTBP1 depletion, compared to the control (Fig. 2C). This reduction could be due to either a destabilization of the Renilla luciferase transcript conjugated to CyCoNP sequence, or to the increased binding of miR-4492 to CyCoNP, a process that could be modulated by PTBP1, as previously described (Kim et al. 2021). To distinguish between the two possibilities, we performed luciferase assays using a CyCoNP construct lacking the miR-4492 binding sites (validated in Desideri et al. 2024) cloned downstream from the Renilla luciferase ORF (CyCoNP ΔmiR-4492). Interestingly, we observed that without the miR-4492 binding sites, CyCoNP expression was insensitive to PTBP1 depletion (Fig. 2D).
These data suggest that the decrease of CyCoNP observed upon PTBP1 depletion could be linked to an increased binding of miR-4492 to its sequence. To verify this hypothesis, we performed CyCoNP endogenous RNA pull-down (PD) using antisense biotinylated probes (Desideri et al. 2024), from extracts of SK-N-BE cells that were either treated with si-SCR or si-PTBP1. Notably, RT-qPCR analysis on the retrieved RNA highlighted a twofold increase of miR-4492 enrichment in CyCoNP pull-down fractions depleted for PTBP1 compared to the scrambled control (Fig. 2E), despite having obtained a comparable CyCoNP PD efficiency in the two conditions (Supplemental Fig. 2B). miR-1249-5p, used as negative nonbinding control, was not detected in either condition (Fig. 2E). Notably, these results allowed us to confirm that miR-4492 binding to CyCoNP increases in absence of PTBP1 and showed that PTBP1 can modulate the binding and activity of miR-4492 on CyCoNP. Importantly, the total amount of miR-4492 in cells did not vary upon PTBP1 depletion (Supplemental Fig. 2C).
To exclude putative indirect effects that could originate from the overall PTBP1 depletion, we mapped the PTBP1 binding site identified by PAR-CLIP in SH-SY5Y cells (Uemura et al. 2017) onto the CyCoNP sequence. We identified one binding site in the 3′ portion of CyCoNP and then generated a CyCoNP construct lacking this region of interaction (CyCoNP ΔPTBP1 bs) to be tested in a luciferase assay (Fig. 2F).
Notably, we found that this construct, which still bears miR-4492 MREs and only lacks the region corresponding to the PTBP1 bs, is insensitive to PTBP1 depletion (Fig. 2F), reinforcing the data that the direct interaction between the lncRNA and PTBP1 influences miR-4492 regulation.
Finally, we wondered whether this effect could be achieved through the binding of PTBP1 to AGO2—hinting at a broader role for RBP in modulating miRNA targeting by interacting with AGO2, a mechanism previously shown for FUS (Zhang et al. 2018). To test this, we performed complementary PTBP1 and AGO2 co-IP experiments, checking for the possible enrichment of each protein in the immunoprecipitated fraction of the other. Western blot analysis of the precipitated protein fractions allowed us to exclude the existence of a direct binding between PTBP1 and AGO2 in SK-N-BE cells (Supplemental Fig. 2D,E). These data are in accordance with a previous study that did not observe such interaction in another human cellular system (Cui and Placzek 2018).
DISCUSSION
Ever-growing evidence is confirming the tight interplay between lncRNAs and RBPs as a critical mechanism of gene expression regulation. In this work, we have expanded the molecular regulatory circuitry related to a human cytoplasmic lncRNA, CyCoNP, which is known to interact with miRNA-4492 and NCAM1 mRNA and thus regulate the specification of several neurophysiological traits both in hiPSC-derived motor neurons and SK-N-BE cells (Desideri et al. 2024). To fully understand CyCoNP's role in the cytoplasmic environment, it is important to consider the possible interactions with RBPs, which could have a crucial regulatory function on the lncRNA. Therefore, by employing in silico predictions and experimental validations, we have studied the effect of a RBP, PTBP1, on CyCoNP expression. The modulation exerted by PTBP1 on the interaction between CyCoNP and miR-4492 underscores the importance of considering both RNA-binding proteins and RNA partners to fully define the mechanism of action of a target RNA molecule. In our case of study, PTBP1 acts as a negative regulator of miRNA-4492 binding to CyCoNP, thus appearing as a modulator of CyCoNP stability and in turn of its regulated targets, such as the NCAM1 mRNA.
Ample experimental evidence has shown that the gene expression perturbations induced by RBP and miRNA are strongly context-specific and highly dependent on structural and sequence features of target RNAs, especially in their 3′ UTR (van Kouwenhove et al. 2011; Jens and Rajewsky 2015; Kim et al. 2021; West et al. 2025). In fact, RBP show opposing effects on miRISC targeting efficiency in different cellular contexts, with phenotypical outcomes that greatly vary depending on what transcripts are affected (Kedde et al. 2010; Xue et al. 2013). The coregulation of target RNA by RNA-binding proteins and the miRISC is shaped globally by several factors, including the stoichiometric balance of specific RBP and miRNA around their targets, the physical distance between their binding sites, which may generate a competition between RBP and miRISC (Jiang et al. 2013; Preusse et al. 2015; Cottrell et al. 2018; Suzuki et al. 2018; Unfried and Ulitsky 2022), and the binding dynamics of RBP, including the miRISC, during target RNA processing (Choi et al. 2024).
Systematically investigating the combinatorial regulation of RNA-binding proteins and miRNA on target RNA in vivo could illustrate the importance of their coordinated actions in shaping cellular metabolism, identity, and fate, even when studying single regulatory RNAs.
MATERIALS AND METHODS
Cell culture
SK-N-BE (SK) cells were cultured in growth medium with RPMI-1640 (Sigma-Aldrich R0883), 10% fetal bovine serum (FBS; Sigma-Aldrich), 1% GlutaMAX (Thermo Fisher Scientific 35050061), 1% Pen/Strep (Thermo Fisher Scientific 15070063), 1% sodium pyruvate (Thermo Fisher Scientific 11360070). Differentiation medium: RPMI-1640 (Sigma-Aldrich), 2.5% heat-inactivated FBS (Sigma-Aldrich), 1% GlutaMAX (Thermo Fisher Scientific), 1% Pen/Strep (Thermo Fisher Scientific), 1% sodium pyruvate (Thermo Fisher Scientific), and 10 μM retinoic acid (RA; Sigma-Aldrich R2625).
RNA preparation and analysis
Total RNA from SK cells was extracted with the Direct-zol RNA Purification Kit (Zymo Research) and reverse transcribed with PrimerScript RT reagent Kit (Takara-Clontech), SuperScript VILO cDNA Synthesis Kit (Thermo Fisher Scientific), or miRCURY LNA RT Kit (QIAGEN). For mRNAs, RT-qPCR analysis was performed with SYBR Green Power-UP (Life Technologies), using the housekeeping glyceraldehyde-3-phosphate dehydrogenase (GAPDH) gene as an internal control. For miRNAs, RT-qPCR analysis was performed with SYBR Green PCR Master Mix (QIAGEN) for microRNAs. Each reaction was performed in three technical replicates and according to the manufacturer's protocol.
Protein analysis
For western blot analysis, proteins were collected in RIPA protein buffer completed with Proteinase Inhibitor Complex (PIC) 100×, loaded on 4%–12% bis-tris-acrylamide gel (Thermo Fisher Scientific), and transferred to a nitrocellulose membrane (Millipore). The membrane was blocked in 10% milk and hybridized with the specific antibodies overnight at 4°C at the appropriate dilutions, according to the manufacturer's instructions. After three washes in TBST buffer, the filter was hybridized with the corresponding secondary antibody for 1 h at room temperature. Protein detection was carried out with the Long-Lasting Chemiluminescent Substrate (EuroClone) using ChemiDoc MP System. Images were analyzed using Image Lab Software (Bio-Rad). See Supplemental Table S1 for antibody details.
Subcellular fractionation
Subcellular fractionation was performed on SK-N-BE cell extracts at 1.5 days of differentiation. Cells were harvested in cold buffer A (Tris-HCl pH 8.0 20 mM, NaCl 10 mM, MgCl2 3 mM, NP40 0.1%, glycerol 10%, EDTA 0.2 mM, 1× PIC, and RNase inhibitors), incubated on ice for 10–15 min, and centrifuged at 500g for 5 min at 4°C. The supernatant was collected as cytoplasmic protein fraction and used for subsequent western blot analysis. The nuclear pellet was resuspended in cold buffer C (Tris-HCl pH 8.0 20 mM, NaCl 400 mM, glycerol 20%, DTT 1 mM, 1× PIC, and RNase inhibitors) in a volume corresponding to 25% of the cytoplasmic volume. Nuclei were sonicated at low intensity to promote nuclear membrane disruption and centrifuged at 16,000g for 15 min at 4°C. The supernatant was collected as nuclear protein fraction and used for subsequent western blot analysis.
Plasmid construction
The detailed procedure used to generate the full-length CyCoNP and CyCoNP ΔmiR-4492 luciferase plasmids is described in Desideri et al. (2024).
Briefly, the full-length CyCoNP sequence was amplified with CloneAMP PCR HiFi (Takara-Clontech) and cloned downstream from the Renilla luciferase stop codon in the psiCHECK-2 plasmid (Promega), previously linearized with NotI (NEB) and XhoI (NEB) enzymatic digestion, by using T4 DNA ligase (Thermo Fisher Scientific). The CyCoNP ΔmiR-4492 plasmid was obtained by removing the miR-4492 MREs from the full-length CyCoNP luciferase plasmid. The plasmid lacking PTBP1 binding site on CyCoNP sequence (CyCoNP ΔPTBP1 bs) was obtained from the CyCoNP WT luciferase plasmid by performing a deletion of the sequence bearing the PTBP1 binding site, retrieved from the POSTAR3 database. To achieve this, the plasmid was linearized by inverse PCR and then ligated using the T4 DNA ligase.
Cell transfection and dual-luciferase reporter assay
SK cells were plated (100,000 cells/well of a 12 well plate) in growth medium (FBS 10%, GlutaMAX 1%, Pen/Strept 1%, sodium pyruvate 1%, and RPMI-1640) and transfected 24 h later with 75 nM of siRNA pool targeting PTBP1 mRNA (si-PTBP1) or the respective scrambled siRNA (si-SCR) using Lipofectamine RNAiMAX Transfection Reagent (Thermo Fisher Scientific) according to the manufacturer's specifications. Eighteen hours after transfections, cells were exposed to differentiation medium (2.5% heat-inactivated FBS, GlutaMAX 1%, Pen/Strept 1%, sodium pyruvate 1%, RA 10 μM, and RPMI-1640) for additional 36 h prior to cell collection. For the luciferase assays, cells were cotransfected with 100 ng of psiCHECK-2 luciferase plasmid containing the wild-type CyCoNP sequence, or CyCoNP ΔmiR-4492 sequence, or CyCoNP ΔPTBP1 bs sequence in combination with 75 nM of siRNA pool targeting PTBP1 mRNA (si-PTBP1, see Supplemental Table S1) or a siRNA negative control (si-SCR). Transfections were performed using Lipofectamine 2000 (Thermo Fisher Scientific) according to the manufacturer's specifications.
Luciferase activity was measured in GloMax-Multi+ Detection System (Promega), using the Dual-Luciferase Reporter Assay System (Promega).
RNA pull-down assay
Native RNA pull-down on total extract from SK cells was performed according to Desideri et al. (2022). Briefly, cells were harvested in lysis buffer (Tris-HCl pH 7.5 50 mM, NaCl 150 mM, MgCl2 3 mM, NP40 0.5%, EDTA 2 mM, DTT 1 mM, 1× PIC, and RNase inhibitors) and incubated on ice for 10–15 min, prior to centrifugation at 15,000g for 15 min. After lysis and clearing by centrifugation, 1 mg of extract was diluted in a 1:2 ratio with hybridization buffer containing Tris-HCl pH 7.5 100 mM, NaCl 300 mM, MgCl2 1 mM, SDS 0.2%, formamide 15%, NP40 0.5%, EDTA 10 mM, DTT 1 mM, 1× PIC, and RNase inhibitors. Ten percentage of the total extract was collected for input (Inp). One hundred picomoles of previously heat-denatured biotinylated probes were added (see Supplemental Table S1). To enhance RNA recovery, 2.5% dextran sulfate was then added to the PD and control samples (LacZ). After a 4 h incubation at 4°C, 0.1 mL of Streptavidin MagnaSphere Paramagnetic Beads (Promega) was added to pull down the complex, and the mixture was incubated for 1 h at room temperature. Beads were then washed four times with hybridization buffer, and RNA was extracted and DNase treated for further analyses. Pull-down (PD) RT-qPCR results were represented as a percentage of PD/input signal (% of input) normalized over the % of CyCoNP transcript precipitated in each condition.
Cross-linking immunoprecipitation (CLIP) assay
Plated SK cells (D 1.5) were UV cross-linked with 4000 μJ/cm2 energy using a Stratalinker and harvested in NP40 lysis buffer pH 7.5 (50 mM Hepes-KOH, 150 mM KCl, 2 mM EDTA, 1 mM NaF, 0.5% NP40, 0.5 mM DTT, 1× PIC, and RNase inhibitors) and incubated on ice for 10–15 min followed by centrifugation at 18,000g for 10 min at 4°C. Resulting cellular lysates were incubated (overnight on a rotating wheel, at 4°C) with 30 μL of Dynabeads Protein G magnetic particles (Invitrogen) preincubated with either 8 μg of PTBP1 Antibody (Invitrogen 32-4800) or mouse IgG (Santa Cruz sc-2025, see Supplemental Table S1). After incubation, beads were washed with a high-salt buffer (50 mM Hepes-KO, 500 mM KCl, 0.5 mM DTT, and 0.05% NP40). Before RNA extraction, 1/4 of the cell lysate was heated for 5 min at 95°C, and the supernatant was collected and resuspended in protein elution buffer (4× Laemmli sample buffer [Bio-Rad]) with DTT 50 mM and analyzed by western blot. RNA fraction was treated with Proteinase K (Thermo Fisher Scientific AM2546) for 30 min at 50°C; the samples were then placed for 10 min at 95°C, and finally, the RNA was extracted using Direct-zol RNA Purification Kit (Zymo Research) with on-column DNase treatment, according to the manufacturer's instructions.
RNA immunoprecipitation (RIP) assay
Plated SK cells (D 1.5) were harvested in RIP buffer pH 7.5 (150 mM NaCl, 25 mM Tris-HCl, 5 mM EDTA;,0.5% NP40, 0.5 mM DTT, 1× PIC, and RNase inhibitors). The lysate was incubated on ice for 10–15 min followed by centrifugation at 15,000g for 15 min at 4°C. Lysates for IP reactions were diluted with RIP buffer and precleared with 10 μL of Dynabeads Protein G magnetic particles (Invitrogen) for 45 min on a rotating wheel at 4°C.
Precleared cellular lysates were incubated (overnight on a rotating wheel, at 4°C) with 30 μL of Dynabeads Protein G magnetic particles (Invitrogen) preincubated with either 5 μg of PTBP1 Antibody (Invitrogen 32-4800) or mouse IgG (Santa Cruz sc-2025, see Supplemental Table S1). After incubation, beads were washed four times with RIP buffer. Before RNA extraction, 1/4 of the cell lysate was heated for 5 min at 95°C, and the supernatant was collected and resuspended in protein elution buffer (4× Laemmli sample buffer [Bio-Rad]) with DTT 50 mM and analyzed by western blot. The remaining RNA fraction was extracted using Direct-zol RNA Purification Kit (Zymo Research) with on-column DNase treatment, according to the manufacturer's instructions.
Co-immunoprecipitation (Co-IP) assay
Plated SK cells (D 1.5) were harvested in co-IP lysis buffer pH 7.5 (150 mM NaCl, 50 mM Tris-HCl, 1 mM EDTA, 1% NP40, 0.1% SDS, 1× PIC, and 1 mM DTT). The lysate was incubated on ice for 10 min followed by centrifugation at 15,000g for 15 min at 4°C. One milligram of lysate per reaction was diluted 1:4 with co-IP buffer pH 7.5 (150 mM NaCl, 50 mM Tris-HCl, 1 mM EDTA, 0.25% NP40, 5% glycerol, 1× PIC, and 1 mM DTT), and 5% of the volume was collected as input sample. IP and IgG samples were incubated (O.N. on rotating wheel at 4°C) with 5 μg of specific antibodies against PTBP1 or AGO2 (Invitrogen MA5-23515, see Supplemental Table S1), or IgG.
The following day, lysates were incubated for 4 h on rotating wheel at 4°C, with 30 μL of Dynabeads Protein G magnetic particles (Invitrogen) previously washed with PBS with Ca2+/Mg2+ and Tween 0.02% (Sigma-Aldrich P9416). After incubation, beads were washed four times with co-IP buffer and then resuspended in the same volume as the input. All samples were resuspended in protein elution buffer (4× Laemmli sample buffer [Bio-Rad]) with DTT 50 mM and heated for 5 min at 95°C. The supernatant was collected and analyzed by western blot.
Statistical analysis
Data are expressed as mean values, and error bars represent SEM or SD. Statistical differences were analyzed by a two-tailed unpaired Student's t-test. A P-value <0.05 was considered statistically significant.
All the oligonucleotides and antibodies used in this work are listed in Supplemental Table S1.
SUPPLEMENTAL MATERIAL
Supplemental material is available for this article.
ACKNOWLEDGMENTS
This work was supported by grants from ERC-2019-SyG 855923-ASTRA and “National Center for Gene Therapy and Drugs based on RNA Technology” (CN00000041) and NextGenerationEU PNRR MUR.
Footnotes
-
Article is online at http://www.rnajournal.org/cgi/doi/10.1261/rna.080705.125.
-
Freely available online through the RNA Open Access option.
- Received July 29, 2025.
- Accepted January 6, 2026.
This article, published in RNA, is available under a Creative Commons License (Attribution-NonCommercial 4.0 International), as described at http://creativecommons.org/licenses/by-nc/4.0/.
REFERENCES
MEET THE FIRST AUTHOR

Meet the First Author(s) is an editorial feature within RNA, in which the first author(s) of research-based papers in each issue have the opportunity to introduce themselves and their work to readers of RNA and the RNA research community. Alessandro Grazzi is the first author of this paper, “PTBP1 controls miRNA loading on target RNAs: lessons from the CyCoNP lncRNA.” Alessandro is a PhD student at Sapienza University of Rome, Italy, in the laboratory of Professor Irene Bozzoni. The focus of the laboratory is to understand the involvement of noncoding RNAs and RNA–protein interactions in molecular networks related to neural cellular physiology and pathology.
What are the major results described in your paper, and how do they impact this branch of the field?
In a previous study, we showed that CyCoNP lncRNA controls the expression of NCAM1 mRNA and other targets through RNA–RNA interactions, which include sequestering the miRNA-4492. In the present work, we find that CyCoNP is also regulated by the RNA-binding protein PTBP1. This RBP, when interacting with CyCoNP, hampers miR-4492 binding to the lncRNA and affects its regulation of NCAM1. This work addresses aspects of the complexity of regulatory networks that rely on RBPs and their cognate target RNAs, illustrating how the molecular characterization of interactions within heterogeneous subcellular environments can contribute to our understanding of cellular physiology.
What led you to study RNA or this aspect of RNA science?
My interest in noncoding RNA sparked from a course in genomics which I attended during my Bachelor's degree, where we were first taught about the noncoding genome. The fascination for noncoding RNAs and their regulatory potential led me to work in Irene Bozzoni's lab, where I could delve deeper into this incredible field.
During the course of these experiments, were there any surprising results or particular difficulties that altered your thinking and subsequent focus?
Our initial approach was to identify the most prominent RBP interactors of our lncRNA of interest through in silico predictions in the least biased way. The key challenge was to characterize how PTBP1, a protein mainly known as a nuclear splicing regulator, could influence CyCoNP lncRNA expression in the cytoplasm, where the lncRNA is localized. This question led us to explore alternative ways in which PTBP1 could be acting on CyCoNP homeostasis, and to investigate the involvement of miRNAs in this regulatory mechanism. The approach we employed to study PTBP1 regulation of CyCoNP could prove useful for understanding other regulatory mechanisms that may occur in a highly context-specific manner but nevertheless have a relevant effect on gene expression.
If you were able to give one piece of advice to your younger self, what would that be?
I would tell my younger self to be patient with uncertainty and to never lose sight of the bigger picture. Curiosity, technical flexibility, and learning new approaches can take time, but it is always worth it. Also, rest can really help reset thoughts and spark new ideas!








