
How tailored Ribo-seq methods probe unique translation events
- James Marks1 and
- Sezen Meydan1,2,3
- 1Department of Biochemistry, Vanderbilt University School of Medicine, Nashville, Tennessee 37232, USA
- 2Vanderbilt Brain Institute, Vanderbilt University, Nashville, Tennessee 37235, USA
- 3Vanderbilt-Ingram Cancer Center, Vanderbilt University Medical Center, Nashville, Tennessee 37232, USA
- Corresponding author: sezen.meydan{at}vanderbilt.edu
Abstract
Ribosome profiling (Ribo-seq) is a next-generation, high-resolution sequencing technique that captures ribosome-protected mRNA fragments to map ribosome positions across the transcriptome. This method serves as a powerful proxy for global translational activity by revealing where ribosomes engage with mRNAs. Recent advances have expanded the utility of Ribo-seq to resolve distinct ribosome populations, including initiating ribosomes, small subunits, collided ribosomes, mitochondrial ribosomes, and those associated with specific translation factors or localized to subcellular compartments. These methodological advances have significantly broadened the scope of Ribo-seq, enabling new insights into the molecular mechanisms that govern translation across diverse eukaryotic systems. In this mini-review, we highlight key innovations in Ribo-seq technology and discuss how they have deepened our understanding of the spatial, temporal, and regulatory dimensions of translational control.
Keywords
INTRODUCTION
Ribosome profiling (also referred to as “Ribo-seq”) builds on classical ribosome footprinting approaches (Steitz 1969; Wolin and Walter 1988), which use nucleases to digest unprotected regions of mRNA, leaving behind fragments that are protected by ribosomes (Fig. 1A). These ribosome-protected mRNA fragments (“ribosome footprints”) are then subjected to next-generation sequencing (Ingolia et al. 2009). Once sequenced, the footprints are computationally mapped to the genome of the organism of interest, revealing the in vivo positions of translating ribosomes at single-nucleotide resolution. Since its development over a decade ago, Ribo-seq has become a powerful tool for investigating diverse aspects of protein synthesis. It has been widely used to uncover mechanisms of translational regulation, quantify translation efficiency (defined as the ratio of Ribo-seq signal relative to RNA-seq expression), and examine how these processes respond to various cellular conditions. Ribo-seq has also emerged as a genomic tool for systematically annotating open reading frames (ORFs) and for identifying previously unrecognized ORFs across a broad range of model organisms.
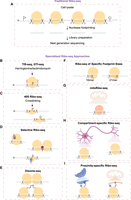
(A) Schematic representation of traditional Ribo-seq, which is optimized to isolate footprints from translating 80S ribosomes. (B–I) Specialized Ribo-seq approaches to study subset of ribosomes: initiating 80S ribosomes (B); 40S subunits resulting from initiation and/or ribosome recycling defect (C); 80S, disomes, or 40S subunits bound to translation factors or carrying posttranslational modifications (D); collided ribosomes or disomes interacting through their nascent chains (E); specific sizes of footprints from ribosomes stalled at truncated mRNAs (16mer) or with open A-site (21mer) (F); mitoribosomes (G); ribosomes at specific cellular compartments (H); and ribosomes that are localized to endoplasmic reticulum and mitochondria (I).
In recent years, Ribo-seq methodology has undergone significant refinements not only to improve library preparation for samples with limited input material, but also to enable the study of specific ribosome subpopulations, thereby enhancing the overall resolution of the technique (see additional complementary reviews on this topic [Brar and Weissman 2015; Ingolia et al. 2019; Wang et al. 2020; Andreev et al. 2021; Wang and Mao 2023; Tomuro and Iwasaki 2025]). These advances have extended Ribo-seq's utility beyond global translational profiling, making it possible to interrogate targeted subgroups of ribosomes or translation events (Fig. 1B–I). In this mini-review, we highlight recent technical innovations in these specialized approaches and discuss the biological insights they have uncovered in eukaryotic cells.
Ribo-seq methods to study a subgroup of ribosomes
Identification of initiating ribosomes
Accurate mapping of translation initiation sites is essential for defining the coding sequences and the regulatory role of translation initiation, in addition to determining which ORFs are translated. However, standard Ribo-seq experiments cannot directly distinguish between initiating 80S ribosomes positioned at start codons and elongating ribosomes distributed across ORFs. This limitation complicates accurate annotation of translation start sites, particularly in transcripts with upstream ORFs (uORFs), near-cognate start sites (non-AUG codons), or in-frame AUG start codons, where signals from elongating footprints can obscure those from initiating ribosomes.
Overcoming this limitation required the development of specialized Ribo-seq approaches—such as translation initiation site-seq (TIS-seq)—that trap ribosomes at translation initiation sites using elongation inhibitors that block the initial rounds of peptide bond formation (Fig. 1B). The pioneering TIS-seq studies were performed in mouse embryonic stem cells pretreated with harringtonine (Ingolia et al. 2011), which is a compound that blocks elongation (Fresno et al. 1977). Harringtonine-facilitated TIS-seq revealed peaks at noncanonical start sites, noncoding RNAs, and uORFs (Ingolia et al. 2011). Intriguingly, uORF translation decreased during the differentiation of mouse embryonic stem cells into embryoid bodies, suggesting a potential role for uORF-mediated translation regulation during development (Ingolia et al. 2011). A related strategy, global translation initiation sequencing (GTI-seq), uses pretreatment of cells with lactimidomycin (Lee et al. 2012), which also inhibits the first round of elongation (Schneider-Poetsch et al. 2010). Similar to the TIS-seq studies in mouse embryonic stem cells, human GTI-seq revealed peaks at non-AUG start codons—particularly within uORFs—and at noncoding RNAs (Lee et al. 2012). Although both studies suggested translation of noncoding RNAs, follow-up work integrating machine learning approaches (Chew et al. 2013) and computational metrics designed to assess protein-coding signatures (Guttman et al. 2013) concluded that the translation events observed in these regions are unlikely to produce proteins. It is likely that Ribo-seq signals in noncoding RNAs occur due to protection from nucleases by RNA-binding proteins. Another intriguing possibility is that ribosome engagement with noncoding RNAs could have a regulatory role other than generating a functional protein, such as modulating RNA stability or structure (Guttman et al. 2013).
Further methodological refinements integrated biochemical and computational strategies. An orthogonal TIS-seq method utilized puromycin, a tRNA analog that releases nascent polypeptides and dissociates elongating ribosomes (Allen and Zamecnik 1962; Nathans 1964), in combination with cycloheximide, which blocks translation elongation (Schneider-Poetsch et al. 2010). When paired with machine learning models trained on annotated start codons, this approach revealed novel uORFs and N-terminal extensions in a human monocytic THP-1 cell line (Fritsch et al. 2012). Additional refinements came from pairing TIS-seq with the ORF-RATER algorithm, which detects ribosome occupancy patterns characteristic of coding sequences (Fields et al. 2015). This integration enabled the discovery of novel start sites in mouse bone marrow–derived dendritic cells stimulated with lipopolysaccharide, as well as in human foreskin fibroblasts during viral infection (Fields et al. 2015). Another variation, quantitative translation initiation sequencing (QTI-seq), involves sequential treatment of cell lysates with lactimidomycin and puromycin (Gao et al. 2015). Unlike earlier TIS-seq approaches that required pretreatment of cells with translation inhibitors, this protocol was performed on lysates, making it suitable for studying tissue homogenates. When applied to liver tissue from nutrient-starved mice, QTI-seq revealed increased initiation at genes involved in proteasome function, as indicated by enriched ribosome occupancy at start codons (Gao et al. 2015). Collectively, these findings underscore the dynamic regulation of translation initiation across diverse cellular and physiological states.
Lactimidomycin-based TIS-seq has also been adapted for yeast (Hollerer et al. 2021), where it uncovered N-terminally extended protein isoforms specifically expressed during meiosis and produced from near-cognate start codons (Eisenberg et al. 2020). These isoforms were enriched under conditions of reduced expression of the translation elongation factor eIF5A—a deficiency that promotes ribosome queuing (Schuller et al. 2017)—which may enhance initiation from suboptimal start sites. Although the exact molecular mechanisms remain unclear, these results suggest a regulatory interplay between elongation dynamics and start codon selection. Another example of how start codon fidelity changes with cellular state came from TIS-seq across the human cell cycle, comparing interphase, cycling mitosis, and mitotic arrest (Ly et al. 2024). This study demonstrated that start codon selection becomes more stringent during mitosis, driven by increased association of ribosomes with the initiation factor eIF1, which relocates from the nucleus to the cytoplasm during mitotic entry. This shift favors initiation at canonical AUG start codons over near-cognate alternatives, linking start codon fidelity to eIF1 localization.
Collectively, these TIS-seq variants have revealed ribosome occupancy across hundreds of previously unannotated start sites, uncovering hidden ORFs and alternative protein isoforms in different species. Yet, TIS-seq captures only the initiating ribosomes, offering little evidence of whether translation continues to produce stable proteins. The use of translation inhibitors may also bias results by promoting initiation at near-cognate or noncanonical sites, complicating interpretation. Determining which of these start sites yield stable proteins, and what biological role these proteins play, remains an exciting avenue for future investigation.
40S Ribo-seq
The assembly of 43S preinitiation complexes (PICs) (40S subunit bound to initiation factors and ternary complex) followed by formation of 48S PIC (43S PIC with cap-binding complex on mRNAs) determines where translation will start. At the end of translation, the 40S ribosomal subunits are recycled to be used for subsequent rounds of translation. Studying both the initiation and recycling processes has been challenging due to the difficulty of capturing these short-lived complexes in the cell. To address this, a formaldehyde cross-linking strategy was introduced (Valasek et al. 2007) to stabilize 40S complexes prior to downstream applications. This cross-linking approach underlies translation complex profile sequencing (TCP-seq), also called 40S Ribo-seq, which uses either formaldehyde alone (Shirokikh et al. 2017) or a combination of formaldehyde and dithio-bis-succinimidyl propionate (Wagner et al. 2022) to stably capture 40S–mRNA complexes (Fig. 1C). First applied in yeast, this technique isolates mRNAs with polysomes and then separates the 40S fraction to sequence footprints associated with the translation initiation complex (Archer et al. 2016). This pioneering 40S Ribo-seq study provided the evidence for scanning ribosomes in 5′ UTRs, identified distinct translation initiation complexes protecting various footprint lengths, and revealed the presence of 40S subunits at stop codons following ribosome recycling (Archer et al. 2016). These observations established 40S Ribo-seq as a powerful tool for directly monitoring both early and late steps of translation. In this section, we highlight how subsequent adaptations of 40S Ribo-seq have expanded its utility, offering new insights into the mechanisms of translation initiation and the processes governing ribosome recycling.
Translation initiation begins with the recruitment of the 43S PIC to the mRNA, although the precise mechanism of mRNA entry into the ribosomal channel has been unclear. Two models have been proposed: a threading mechanism, in which the mRNA is gradually fed into the channel, and a slotting mechanism, in which mRNA is directly loaded. To distinguish between these possibilities, an expanded 40S Ribo-seq protocol, called RCP-seq, was applied to developing zebrafish embryos (Giess et al. 2020). This approach integrated formaldehyde cross-linking followed by fractionation of 40S/80S complexes and a previously established ligation-free, low-input library preparation (Hornstein et al. 2016). Unlike TCP-seq, which only captures transcripts already engaged with 80S ribosomes (Archer et al. 2016; Shirokikh et al. 2017), RCP-seq directly isolates the 40S fraction, enabling detection of scanning complexes across all transcripts (Giess et al. 2020). Notably, RCP-seq revealed a distinct ladder of differently sized 40S footprints aligned to transcription start sites, reflecting progressive recruitment of the mRNA to the 43S PIC complex (Giess et al. 2020). This result is therefore consistent with the threading model of ribosome recruitment during early zebrafish development.
After ribosome recruitment to the mRNA cap, an unresolved question is whether the cap stays attached to the scanning ribosome. Two models have been proposed: In the cap-severed model, the 43S PIC dissociates from the cap after initial recruitment, allowing multiple ribosomes to load and scan the 5′ UTR, whereas the cap-tethered model posits that the cap remains engaged with the scanning ribosome, limiting scanning to a single ribosome at a time. While 40S Ribo-seq in yeast revealed queuing of 40S subunits at 5′ UTRs, supporting a cap-severed model (Archer et al. 2016), similar experiments in mammalian cells showed no evidence of such queuing, consistent with a cap-tethered mechanism in higher eukaryotes (Bohlen et al. 2020a).
As ribosomes scan the 5′ UTR, they can encounter uORFs. After terminating uORF translation, ribosomes may reinitiate translation at the main ORF. To investigate the mechanisms of this poorly understood translation reinitiation process, 40S Ribo-seq was adapted to selectively capture scanning 40S subunits associated with specific translation factors by adding an immunoprecipitation step, termed “Sel-TCP-seq” (more details on immunoprecipitation-coupled selective Ribo-seq are outlined below). The Sel-TCP-seq in mammalian cells, tailored to isolate ribosome populations associated with the initiation factors (eIF2S1, eIF3B, eIF4G1, and eIF4E), showed that eIF3B, eIF4G1, and eIF4E remain bound to 80S ribosomes during uORF translation, a feature proposed to facilitate reinitiation following termination (Bohlen et al. 2020a). A similar Sel-TCP-seq approach in yeast and human cells demonstrated that eIF2 and eIF3 travel with the scanning 40S, whereas eIF3 dissociates during early translation elongation (Wagner et al. 2020). In addition to studying reinitiation mechanisms, Sel-TCP-seq and GTI-seq (pretreatment with lactimidomycin) were used as tools for identifying cognate and near-cognate translation initiation sites (Ichihara et al. 2021). Collectively, these studies demonstrate that 40S Ribo-seq can be used to elucidate translation initiation mechanisms and the role of initiation factors in this process.
Ribosomal subunits must be recycled after each translation cycle to be reused in further rounds of translation. The first stage of recycling is the splitting of the 80S ribosome into 60S and 40S subunits, followed by the removal of the 40S subunit from the mRNA. When recycling fails, 40S subunits remain bound to the mRNA at stop codons, leaving a diagnostic footprint that can be captured by 40S Ribo-seq (Archer et al. 2016). This signal provides a metric for ribosome recycling efficiency and can be used to study the role of translation factors responsible for recycling 40S subunits. Using this metric, yeast studies provided direct evidence that the translation factors Tma20 and Tma22 (the homologs of human MCTS1 and DENR, respectively) serve as key players in 40S recycling in yeast, while Tma64 (EIF2D in human) plays a relatively minor role (Young et al. 2021). Importantly, this work also linked ribosome recycling to human health: Introducing an autism-associated mutation into yeast Tma22 caused 40S recycling defects, suggesting a potential connection between impaired ribosome recycling and autism (Young et al. 2021). Recycling of 40S ribosomal subunits is also important for translation reinitiation. In line with this, 40S Ribo-seq in mammalian cells revealed that DENR promotes reinitiation after uORFs (Bohlen et al. 2020b). This activity is crucial for the expression of stress-responsive genes containing uORFs such as ATF4, which encodes a transcription factor central to the integrated stress response pathway (Bohlen et al. 2020b). More recently, human studies identified recycling defects associated with MCTS1 deficiency. These defects impair reinitiation in the transcript encoding JAK2, a key component of interferon-γ signaling, and underlie Mendelian susceptibility to mycobacterial disease (Bohlen et al. 2023). Taken together, these studies illustrate how 40S Ribo-seq has emerged as a powerful method to uncover mechanistic roles of ribosome recycling factors and to connect recycling defects with disease phenotypes.
Selective Ribo-seq approaches
Translation is coordinated by a dynamic network of proteins interacting with the translation machinery, including canonical translation factors, enzymes, chaperones, and other regulatory components. To identify their mechanistic roles, Ribo-seq can be combined with an immunoprecipitation step (Oh et al. 2011) targeting a specific ribosome-associated protein, as introduced earlier in the context of 40S Ribo-seq. This general strategy, broadly defined as “selective Ribo-seq,” has been applied to characterize a wide-range of translation-associated proteins (Fig. 1D; Galmozzi et al. 2019; Wagner et al. 2022).
One notable application of selective Ribo-seq involves probing the CCR4–NOT complex, which mediates degradation of mRNAs enriched in nonoptimal codons (codons with limited cognate tRNAs), in both yeast and mammalian cells. In yeast, selective Ribo-seq targeting Not4, a CCR4–NOT complex subunit, demonstrated that low optimality codons are enriched in the A-site of Not4-bound ribosomes (Buschauer et al. 2020). Structural analysis of CCR4–NOT-bound ribosomes further revealed that the presence of a nonoptimal codon in the A-site leads to a ribosome with empty E- and A-sites lacking bound tRNAs. This conformation allows for Not5, another CCR4–NOT subunit, to bind to the ribosomal E-site (Buschauer et al. 2020). A complementary study in human cells showed that ribosomes associated with CNOT3 (the human Not5 homolog) display increased representation of P-site arginine codons, particularly when paired with nonoptimal A-site codons, underscoring the conserved relationship between codon optimality and mRNA stability (Zhu et al. 2024). In addition to protein factors that interact with ribosomes, posttranslational modifications of the ribosomal proteins may modulate translation. For example, Ribo-seq of human and mouse ribosomes containing phosphorylated RPS6, a downstream target of mTORC1, showed that these modified ribosomes are preferentially enriched near the 5′ region of ORFs and on transcripts with short coding sequences (Bohlen et al. 2021). These phosphorylated ribosomes displayed enhanced translational efficiency, suggesting that RPS6 modification modulates translation initiation to favor the expression of select mRNAs. Collectively, these findings highlight the utility of selective Ribo-seq in uncovering translation mechanisms shaped by regulatory factors and posttranslational modifications.
Another layer of translational regulation emerges from the proteins that interact with the nascent peptides. A selective Ribo-seq known as SeRP (Oh et al. 2011; Becker et al. 2013; Galmozzi et al. 2019) has been used to study cotranslational nascent peptide folding dynamics by immunoprecipitating the chaperone Ssb in Saccharomyces cerevisiae (Doring et al. 2017). SeRP revealed that Ssb binds to the ribosome surface when the emerging nascent peptide contains both positively charged and hydrophobic amino acids. Ssb-bound ribosomes translate faster, presumably due to the lack of mRNA and nascent peptide features that typically slow down the ribosome. SeRP has also been employed to investigate cotranslational assembly of protein complexes, indicating that many multisubunit proteins begin assembling during translation via direct interactions between nascent peptides (Shiber et al. 2018; Seidel et al. 2022). In contrast, complexes that do not assemble cotranslationally appear to rely on specialized chaperones, possibly to prevent nascent peptide misfolding or premature interactions with partner proteins (Shiber et al. 2018).
Disruption of translation-associated interactions can have profound physiological consequences. Spinal muscular atrophy (SMA) neuromuscular disease occurs due to loss of SMN protein. Ribo-seq coupled with SMN immunoprecipitation in the mouse brain showed that SMN-bound ribosomes are enriched near the start of the coding sequences and at 5′ UTR regulatory elements of mRNAs connected to SMA pathogenesis (Lauria et al. 2020). These findings suggest that SMN could contribute to proper translation initiation on specific mRNAs, providing a potential mechanistic link between defective translational regulation and SMA.
While these studies highlight the power of selective Ribo-seq to uncover new layers of translational regulation, the specificity of the captured footprints remains an important consideration, since immunoprecipitation-based approaches may recover secondary or indirect interactions. Inclusion of appropriate controls such as isotype-matched immunoglobulin G or lysates from corresponding knockout cells will be critical for distinguishing genuine factor–ribosome associations from nonspecific interactions. As selective Ribo-seq approaches continue to evolve, expanding their application across different cellular contexts, developmental stages, and disease models will be essential for uncovering the mechanisms of translation-associated proteins.
Disome-seq of ribosome pairs
When a translating ribosome stalls due to obstacles on the mRNA, such as slowly decoded codons, inhibitory nascent peptide sequences, chemically damaged nucleotides, or limited translational resources, trailing ribosomes collide with the stalled ribosome, forming tightly packed ribosome collision complexes (Juszkiewicz et al. 2018; Ikeuchi et al. 2019) consisting of two or three ribosomes (disomes or trisomes, respectively). The disome interface is recognized by cellular surveillance pathways that can trigger stress response signaling, including integrated stress response via GCN1 and GCN2 (Meydan and Guydosh 2020; Wu et al. 2020; Yan and Zaher 2021) and ribotoxic stress response through ZAK (Wu et al. 2020; Sinha et al. 2024). Disomes also engage the ribosome quality control (RQC) pathway (Brandman et al. 2012; Simms et al. 2017), initiated by ZNF598 (Hel2 in yeast), which ubiquitinates the stalled ribosome and recruits the RQC trigger complex to rescue the stalled ribosome (Joazeiro 2019; Meydan and Guydosh 2021; Inada and Beckmann 2024).
Since disomes act as hubs for cellular surveillance signaling, defining the principles governing their formation and recognition in the cell requires isolating them for Ribo-seq. Collided ribosomes resist nuclease digestion, making them suitable for footprinting (Wolin and Walter 1988), though they are typically excluded from conventional Ribo-seq. To address this, modified Ribo-seq protocols have been developed to isolate disomes or trisomes using sucrose gradients or gel-based size selection to enrich for longer footprints (Fig. 1E; Guydosh and Green 2014; Meydan and Guydosh 2020; Mito et al. 2020; Ayres-Galhardo et al. 2025). This approach, known as disome profiling or Disome-seq, revealed ribosome collisions across various systems, including bacteria (Fujita et al. 2022), yeast (Meydan and Guydosh 2020; Zhao et al. 2021), mouse liver (Arpat et al. 2020), mouse embryonic stem cells (Tuck et al. 2020), zebrafish, and human cells (Han et al. 2020).
Under basal conditions, a substantial fraction of translating ribosomes is found in “endogenous” disome complexes, estimated at ∼10% for highly translated genes in mouse liver cells using spike-in-based Ribo-seq/Disome-seq quantification (Arpat et al. 2020). It is likely that some of these disomes may engage in productive roles such as cotranslational nascent peptide folding or subcellular targeting (Stein and Frydman 2019; Collart and Weiss 2020). Supporting this idea, Disome-seq in S. cerevisiae showed that the endogenous disomes may promote cotranslational protein folding since they are enriched in chaperones (Zhao et al. 2021). Disomes were also detected in the vicinity of frameshifting sites in viral RNAs in human cells (Kibe et al. 2025) or in vitro translation systems (Bhatt et al. 2021), indicating a potential role in programmed frameshifting. It is unclear, however, whether a “productive” disome would still be targeted by surveillance pathways. Early studies used reporters containing polybasic amino acid tracts to induce RQC-targeted collisions (Dimitrova et al. 2009). Cryo-EM analysis of the endogenous disomes (Zhao et al. 2021) revealed a conformation distinct from the RQC-targeted disomes formed on a collision-inducing reporter (Ikeuchi et al. 2019). These findings suggest that some endogenous disomes may have regulatory roles and can perhaps evade recognition by surveillance mechanisms.
In contrast, several studies suggest that endogenous disomes are frequently targeted by surveillance pathways. For example, Ribo-seq of Hel2-bound ribosomes showed enrichment of transcripts encoding secretory proteins, particularly when the signal recognition particle expression was suppressed (Matsuo and Inada 2021), suggesting that Hel2 targets ribosomes lacking signal recognition particle interaction. Disome-seq in S. cerevisiae revealed Hel2-mediated stabilization of disomes via ubiquitination, supporting downstream RQC recruitment (Meydan and Guydosh 2020). Similarly, in Caenorhabditis elegans, ribosome and disome footprints were reduced at polyproline motifs in GCN1-deficient cells, consistent with a model in which GCN1 stabilizes disomes and increases their availability to recruit chaperones for protein folding or membrane protein assembly (Muller et al. 2023). Selective Disome-seq of GCN1 in human cells also demonstrated enrichment at nonoptimal codons and on mRNAs encoding transmembrane domains and collagen proteins. When protein folding fails, GCN1 recruitment leads to CCR4–NOT-mediated mRNA decay (Muller et al. 2023). A related mechanism links disomes to mRNA decay through SKIV2L–AVEN pathway, as demonstrated by Disome-seq in mouse embryonic stem cells (Tuck et al. 2020). Finally, selective Disome-seq of ZAK revealed enrichment at stop codons and proline-rich motifs (Wu et al. 2020). Collectively, these data suggest that cellular surveillance pathways can engage with endogenous disomes even in the absence of environmental stress.
Environmental perturbations to cellular homeostasis, such as oxidative stress, can enhance disome formation and trigger disome-mediated stress signaling. For example, a high-fat, high-sugar diet induced oxidative stress and elevated disome footprints at highly translated transcripts in mouse livers (Snieckute et al. 2023). Similarly, hydrogen peroxide treatment in S. cerevisiae triggered stalling at isoleucine–proline motifs in a manner dependent on Rad6, a ubiquitin conjugase implicated in stress signaling (Meydan et al. 2023). Ribosome composition can also affect disome formation: Disome-seq in mouse myoblasts lacking RPL3L, a paralog of RPL3 expressed in heart and muscle, showed increased disome footprints relative to wild-type cells (Shiraishi et al. 2023). Furthermore, selective Ribo-seq of human ribosomes harboring a decoding-deficient 18S rRNA (due to A1824C mutation) detected unique 43S–80S collision complexes at start codons (Coria et al. 2025), echoing yeast observations of monosome stalling caused by nonfunctional rRNAs (Li et al. 2022b). Notably, the 43S–80S collisions have also been identified in yeast (Ferguson et al. 2023), supporting the idea that ribosome queuing at start sites may represent a conserved and regulated feature of translation initiation stress. Together, these studies illustrate that both environmental stresses and intrinsic ribosome features shape disome formation, highlighting collisions as sensitive readouts of cellular and ribosomal state.
Unlike the disomes that form when ribosomes physically collide at a stall site, another class of disome-like complexes arises when adjacent ribosomes remain spatially close but are functionally connected through their nascent peptides. In these cases, the growing peptides emerging from neighboring ribosomes directly interact, enabling cotranslational assembly of protein complexes on the same mRNA. This coupling does not reflect ribosome collisions but instead represents a productive strategy to coordinate folding and assembly. Such events were uncovered using disome selective ribosome profiling (DiSP), which captures monosome footprints linked by nascent peptide interactions (Fig. 1E; Bertolini et al. 2021). This method revealed widespread cotranslational homomer formation in human cells, in which nascent peptides from adjacent ribosomes assemble into protein complexes on the same mRNA, highlighting how ribosome pairs can cooperate through their peptide products to enhance efficiency and specificity of protein complex assembly (Bertolini et al. 2021).
These disome-focused approaches have reshaped our view of translation by showing that ribosome collisions encompass both endogenous, potentially regulatory, events and stress-induced signals. These studies highlight that disomes can influence nascent peptide folding, quality control, and mRNA stability. Yet major questions remain about the molecular determinants that distinguish productive, regulatory collisions from those that trigger surveillance pathways. In addition, future research should clarify how cellular conditions, such as nutrient availability, stress, developmental stage, and ribosome composition, shape disome dynamics, and how disome formation impacts translation efficiency, proteostasis, and cellular health.
Ribo-seq of specific footprint sizes
The functional state of the ribosome is closely coupled to its conformation, which determines the length of the protected mRNA fragment recovered in Ribo-seq (Fig. 1F). Although typical Ribo-seq results in 28–30 nt footprints, technical modifications yield other footprint sizes, indicative of alternative ribosomal states. For example, ribosomes stalled on truncated mRNAs, such as those at the 3′ ends of decay intermediates, produce characteristically short 16 nt footprints (16-mers) (Guydosh and Green 2014, 2017; Guydosh et al. 2017; D'Orazio et al. 2019). These shorter footprints arise because the mRNA exit channel is unoccupied, allowing nuclease cleavage closer to the P-site. In the absence of rescue factors such as the yeast proteins Dom34 and Hbs1, ribosomes stalled at the 3′ ends of truncated mRNAs can be detected by 16-mer footprints. Endogenous decay intermediates producing these footprints include endonucleolytically cleaved HAC1 mRNA, 3′ UTRs of some protein-coding genes (Guydosh and Green 2014), and targets of Ire1 endonuclease (Guydosh et al. 2017).
Distinct footprint sizes also mark other ribosome conformations. Ribosomes with an open A-site generate 21-mers (Lareau et al. 2014; Wu et al. 2019), which can be captured using Ribo-seq in the presence of the translation inhibitors cycloheximide and tigecycline (Wu et al. 2019). Similarly, anisomycin treatment in S. cerevisiae yields 51-mers (Meydan and Guydosh 2020) corresponding to disome footprints, thought to reflect a trailing 80S ribosome (30-mer) collided with an anisomycin-stalled ribosome that has an open A-site (21-mer) (Lareau et al. 2014). Combining 16-mer and 21-mer Ribo-seq methods has further dissected ribosome collision mechanisms. In cells expressing a disome-inducing polyarginine reporter, the leading ribosome in the disome complex stalls in an unrotated conformation, resulting in 21-mers, while Cue2 endonuclease cleaves between collided ribosomes generating truncated mRNAs that produce 16-mers (D'Orazio et al. 2019).
Other Ribo-seq variations have revealed unique ribosome-protected fragments. For example, Ribo-seq of ribosomes bound to Ski3, a component of the mRNA decay-associated Ski complex, identified a subpopulation of 35–40-mer footprints, likely due to extended protection from Ski complex binding (Schmidt et al. 2016). Together, these nuanced footprint signatures and the methods that enrich for them have uncovered previously hidden translation intermediates, distinct ribosome conformations, and regulatory events. Future studies building on these approaches will be well positioned to define fundamental principles of translational control across cellular states.
MitoRibo-seq to study mitochondrial translation
Mitochondrial ribosomes (mitoribosomes) are distinct from cytoplasmic ribosomes and translate a limited set of mitochondrial mRNAs encoding oxidative phosphorylation proteins. While mitoribosome footprints can occasionally be detected in standard Ribo-seq data sets (Iwasaki et al. 2016; Muller et al. 2019; Li et al. 2022a), their low abundance relative to cytoplasmic ribosomes necessitated the development of specialized profiling methods (referred to as MitoRibo-seq, Fig. 1G). These methods include sucrose-gradient fractionation of mitoribosomes (Rooijers et al. 2013; Li et al. 2022a), affinity purification of tagged mitoribosomes (Couvillion et al. 2016), and isolation of intact mitochondria prior to footprinting (Wakigawa et al. 2025). All these approaches have provided insights into the operation of mitochondrial translation, its disruption in disease, as well as its coordination with cytoplasmic translation ensuring the assembly of dual-origin oxidative phosphorylation complexes (Couvillion et al. 2016; Soto et al. 2022).
In mammalian cells, mitochondrially encoded mRNAs are processed from polycistronic precursors and polyadenylated. Most are leaderless transcripts (lacking long 5′ UTRs) and therefore are not translated by scanning mechanisms used in the cytoplasm or bacterial Shine–Dalgarno sequences, raising the question of how mitoribosomes initiate translation. Cryo-EM structures addressed this conundrum by showing that LRPPRC, a protein required for mitochondrial translation and mutated in Leigh syndrome French-Canadian type (LSFC), forms a complex with mitoribosomes and SLIRP to deliver transcripts to mitoribosomes (Singh et al. 2024). Consistently, MitoRibo-seq in LRPPRC-deficient cells revealed altered translation efficiencies and shorter mitoribosome footprints, supporting a model where LRPPRC stabilizes mRNA–mitoribosome interactions (Singh et al. 2024).
After recruitment to the mRNAs, mitoribosomes rely on mtIF2 and mtIF3 for initiation, although the precise role of mtIF3 has been unclear. In mice, conditional knockout of mtIF3 caused cardiomyopathy and led to mitoribosome accumulation at the 5′ ends of transcripts, suggesting a role for mtIF3 in initiation fidelity (Rudler et al. 2019). In human cells lacking mtIF3, translation of the ATP6 gene, which is encoded in the bicistronic ATP8/ATP6 transcript, was severely reduced, while translation of leaderless transcripts was largely unaffected (Remes et al. 2023). These findings indicate that mtIF3 is critical for initiation on bicistronic or structured transcripts, but dispensable for leaderless initiation. Beyond studying the role of mitochondrial translation factors, MitoRibo-seq mapping of initiation sites in mitochondrial mRNAs has led to reannotation of mitochondrial ORFs and identification of novel initiation sites in mitochondrially encoded transcripts (Soto et al. 2022; Marks et al. 2024; Wakigawa et al. 2025), although the functional significance of these initiation sites and their products remains unresolved.
Beyond initiation, MitoRibo-seq has provided insights into elongation dynamics. A run-off assay using bacterial inhibitors retapamulin to trap ribosomes at initiation (Meydan et al. 2019), followed by chloramphenicol to arrest elongation (Marks et al. 2016), revealed that mitochondrial elongation rates vary across cell types (Wakigawa et al. 2025). As observed in bacterial and cytosolic ribosomes, mitoribosomes stall at mRNA modification sites or problematic codons. For example, combined with N1-methyladenosine (m1A) profiling, m1A was shown to induce mitoribosome stalling (Li et al. 2017). Similarly, MitoRibo-seq revealed the role of a translation elongation factor TACO1, which relieves stalling at polyproline stretches (Brischigliaro et al. 2024), equivalent to its homologs EF-P in prokaryotes and EIF5A in eukaryotes. Interestingly, mitoribosomes did not exhibit the pausing associated with wobble base-pairing (Gao et al. 2017), highlighting their distinction from cytoplasmic ribosomes.
During translation termination, human mitoribosomes follow a modified genetic code in which UGA is assigned as tryptophan, while UAA, UAG, as well as AGA and AGG codons serve as stop codons. MitoRibo-seq combined with cryo-EM structure demonstrated that mtRF1, a mitochondrial release factor, decodes these noncanonical stop codons, as loss of mtRF1 leads to ribosome accumulation at AGA/AGG sites (Saurer et al. 2023).
Environmental and genetic perturbations can disrupt mitochondrial translation. Due to their bacterial ancestry, mitoribosomes remain sensitive to antibiotics that inhibit bacterial translation. Several studies have leveraged MitoRibo-seq to dissect similarities and differences in drug action, providing knowledge that could be important for designing less harmful therapeutics (Marks et al. 2024; Bibel et al. 2025; Wakigawa et al. 2025). Patient-derived cybrid studies further linked specific mutations to translational defects, such as a pathogenic tRNA(Trp)5556G>A mutation that causes stalling at tryptophan codons (Rooijers et al. 2013). MitoRibo-seq has also revealed how tRNA processing and modification affect mitochondrial translation fidelity. For instance, PDE12, a poly(A)-specific exoribonuclease, prevents translation arrest by regulating tRNA polyadenylation (Pearce et al. 2017). Additional studies of tRNA-modifying enzymes (SHMT2, QTRT1, QTRT2, METTL8, NSUN3, GTPBP3, OSGEPL1, and MTU1) showed that their loss causes mitoribosome pausing at cognate codons (Morscher et al. 2018; Suzuki et al. 2020; Scholler et al. 2021; Wakigawa et al. 2025). Looking ahead, applying MitoRibo-seq to investigate mitochondrial diseases and cell type–specific mitochondrial function presents an exciting frontier. As methodologies continue to evolve, MitoRibo-seq will remain an indispensable tool for exploring the unique biology of the mitochondrial translation system.
Localized translation of cytoplasmic ribosomes
In highly polarized cells such as neurons, translation is spatially compartmentalized. Specific mRNAs localize to dendrites and axons (neuropil), far from the cell body (somata), enabling localized protein synthesis tailored to the unique functional demands of each compartment (Fig. 1H; Hafner et al. 2019; Holt et al. 2019). Supporting this, Ribo-seq of physically separated neuropil and somata from rat brain slices revealed thousands of mRNAs that are differentially translated between compartments, underscoring the precision of spatial translational control (Glock et al. 2021). Interestingly, Ribo-seq comparing monosome- and polysome-associated mRNAs showed that neuropil-enriched transcripts were more frequently associated with monosomes, while somata-enriched transcripts mostly engaged polysomes (Biever et al. 2020). This suggests a distinct translational mode in neuropil, potentially adapted to a limited pool of local ribosomes at synapses, though the functional implications of monosome-associated translation remain unclear.
Complementary studies integrating Ribo-seq, RNA-seq, and proteomics in mouse neuronal cultures confirmed the significance of neurite-localized translation: Neurite-enriched mRNAs accounted for the majority of proteins detected in neurites, highlighting the role of local translation in shaping compartment identity (Zappulo et al. 2017). More recently, a proximity-labeling strategy fused TurboID to the postsynaptic protein PSD95, enabling Ribo-seq of dendritic ribosomes in resting and depolarized cortical neurons (Hacisuleyman et al. 2024). This revealed that neuronal activity rapidly reprograms local translation through EIF4G2 binding to uORF-containing 5′ UTRs, providing a mechanism for activity-dependent remodeling of the synaptic proteome (Hacisuleyman et al. 2024).
Neurons also employ a unique strategy of transporting stalled ribosome–mRNA complexes. Translation often initiates in the soma, after which ribosomes stall and are packaged into RNA granules for transport to synapses (Graber et al. 2013; Sun et al. 2025). Ribo-seq of RNA granules isolated from rat brains showed that stalled ribosome footprints are enriched for sequences bound by the RNA-binding protein FMRP (Anadolu et al. 2023). Loss of FMRP causes fragile X syndrome, suggesting that ribosome stalling in granules may play an important role in neuronal development.
In nonneuronal cells, proximity-specific Ribo-seq (Fig. 1I) has enabled dissection of subcellular translation at organelle surfaces. These approaches, which use biotinylated ribosomes and proximity-based biotin ligases tethered to organelle membranes, showed that a large fraction of mRNAs is cotranslationally targeted to the endoplasmic reticulum (Jan et al. 2014) and mitochondria (Williams et al. 2014; Vardi-Oknin and Arava 2019). An advanced light-activated approach tethered to mitochondrial outer-membrane proteins allowed controlled labeling of ribosomes near mitochondria (Luo et al. 2025). This method showed distinct localization routes of proteins into the mitochondria, including cotranslational targeting driven by a bipartite signal with mitochondrial targeting sequence and a downstream region (Luo et al. 2025). The bipartite signaling mechanism was also observed with selective Ribo-seq of mitochondrial TOM complex (Zhu et al. 2025), which is responsible for importing cytoplasmic proteins into the mitochondria. Together, these Ribo-seq studies reveal diverse strategies by which cells can spatially organize translation. Local translational control may also be modulated by spatial variation in translational surveillance mechanisms (Meydan and Guydosh 2023). How these pathways interact with translation machinery within specific cellular microenvironments remains a compelling direction for future research.
Monitoring translation in low-abundance samples
Understanding translational control in biological systems often requires studying samples that are small, heterogeneous, or experimentally limited, such as early developmental tissues, sorted cell populations, or subcellular compartments. Conventional Ribo-seq protocols require mRNA footprints from 1 to 10 million cells, limiting application to large tissue samples or bulk cultures. In this final section, we highlight the biology that we learned from technical innovations that have adapted Ribo-seq to low-input conditions.
One major refinement is ligation-free library preparation, which omits adapter ligation and gel purification steps, reducing RNA input requirements to 1–40 ng and shortening overall protocol time (Hornstein et al. 2016; Xiong et al. 2022; Ferguson et al. 2023). Ligation-free Ribo-seq enabled cell type–specific profiling in the mouse brain and revealed that translation regulation predominantly affects neuronal genes, correlating with their functional roles (Hornstein et al. 2016). Another approach, Ribo-lite, allowed analysis of gene expression at the single-oocyte level during the mouse oocyte-to-embryo transition, a developmental process governed almost entirely by posttranscriptional regulation (Xiong et al. 2022). Ribo-lite showed that cytoplasmic polyadenylation elements (CPEs) near the poly(A) tail determine translational fate: CPE-containing transcripts are repressed in oocytes but activated after meiotic resumption, whereas CPE-less transcripts remain silenced (Xiong et al. 2022).
RiboLace, an orthogonal approach, selectively captured actively translating ribosomes using biotinylated puromycin and can be performed with as few as 200,000 cells (Clamer et al. 2018, 2021). More recently, a protocol combining ligation-free library preparation with P1 nuclease digestion instead of RNase I further enhanced sensitivity and reduced rRNA contamination (Ferguson et al. 2023). Other low-input strategies include the use of RNase A + T1 digestion (Liu et al. 2019) and a modified QIA-seq miRNA library kit capable of generating small RNA libraries as little as 1 ng input RNA (Froberg et al. 2023). This latter method, termed nanoRibo-seq, revealed differences in translational efficiency between related cortical projection neuron subtypes, highlighting the role of translation in shaping neuronal identity.
Pushing the Ribo-seq sensitivity even further, recent advancements have enabled its application at the single-cell level. One approach performs traditional Ribo-seq library preparation in a one-pot reaction of individual cells, allowing single-cell Ribo-seq without extensive sample handling steps (VanInsberghe et al. 2021). An alternative strategy, based on microfluidic isotachophoresis (Ribo-ITP), substantially enhanced the sensitivity of ribosome occupancy measurements at both the single-cell and single-embryo levels (Ozadam et al. 2023). These innovations bring Ribo-seq into the realm of single-cell translatomics, capturing cellular translation regulation with unprecedented resolution.
SUMMARY AND OUTLOOK
Here we summarized how Ribo-seq approaches (Fig. 1) have been tailored to study diverse aspects of translational mechanisms. Advancements in the Ribo-seq protocols have markedly improved its sensitivity, enabling its application to limited biological samples, including low-abundance brain tissues, spatially distinct subcellular compartments, and even individual cells. These developments have transformed Ribo-seq from a bulk tool to a technique capable of resolving translational events at the level of individual cell types and subcellular compartments.
Nevertheless, our understanding of the complexity that underlies tissue- and cell type–specific translation remains incomplete. Moving forward, we anticipate that applying highly sensitive Ribo-seq methods to multicellular organisms holds the promise of defining translation status at the single-cell level, complementing the existing single-cell transcriptomic and proteomic approaches. Such integration will not only deepen our understanding of how gene expression is regulated posttranscriptionally but may also uncover translational features that cannot be detected by RNA or protein-level measurements alone. As techniques continue to mature, we envision that specialized Ribo-seq strategies, such as sequencing of 40S and disome-protected mRNA fragments, can be adapted for single-cell applications. Resolving these layers of translational control in tissues or dynamic developmental systems would represent a major leap in understanding gene expression mechanisms.
Translation is highly sensitive to cellular context and physiological state. Factors such as stress, nutrient availability, developmental stage, metabolic status, and cell cycle progression all have the potential to reshape the translational landscape. Future applications of specialized Ribo-seq approaches may shed light on how these conditions impact distinct translational steps, from ribosome recruitment and scanning to elongation, stalling, disome formation, and interactions with translation factors. As Ribo-seq technologies become increasingly refined and integrative, we anticipate that they will play a pivotal role in decoding the layered complexity of translational regulation across different cell types and physiological conditions.
ACKNOWLEDGMENTS
We thank Ruby Pitts, Manny Ascano, and Nicholas Guydosh for the critical reading of the manuscript. This work is supported by National Institutes of Health (NIH) 4R00GM144688-02 to S.M. Figures were prepared in BioRender.
Footnotes
-
Article is online at http://www.rnajournal.org/cgi/doi/10.1261/rna.080654.125.
-
Freely available online through the RNA Open Access option.
This article, published in RNA, is available under a Creative Commons License (Attribution-NonCommercial 4.0 International), as described at http://creativecommons.org/licenses/by-nc/4.0/.








