
Dissecting the stress granule RNA world: dynamics, strategies, and data
- 1Section of Hematology, Department of Internal Medicine, Yale Comprehensive Cancer Center, Yale University School of Medicine, New Haven, Connecticut 06511, USA
- 2Hematology Unit, Fondazione IRCCS Ca’ Granda Ospedale Maggiore Policlinico, Milan, 20122, Italy
- 3Department of Cellular, Computational and Integrative Biology (CIBIO), University of Trento, Trento, 38123, Italy
- Corresponding author: giulia.biancon{at}yale.edu
-
↵4 These authors contributed equally to this work.
Abstract
Stress granules (SGs) are cytoplasmic ribonucleoprotein granules that commonly nucleate from the interaction of translationally stalled mRNAs and RNA-binding proteins. SGs are involved in the cellular adaptation to stress conditions participating in the regulation of gene expression and cell signaling. While dysregulation of SG dynamics has been increasingly implicated in human disease, a comprehensive understanding of SG composition, particularly of the RNA component, across various conditions remains elusive. Here, we review the physiological and pathological aspects of SGs, discuss current and future experimental strategies to identify SG components, and provide insights into the SG RNA world through the meta-analysis of 26 human SG transcriptome data sets.
Keywords
INTRODUCTION
First discovered in 1983 (Nover et al. 1983), stress granules (SGs) are membraneless organelles that form in the cytoplasm under cellular stress through condensation of RNAs and proteins, mostly RNA-binding proteins (RBPs). SGs orchestrate cellular response to stress by controlling gene expression and cell signaling. Their dysregulation has been linked to neurodegenerative diseases and cancer (Protter and Parker 2016). Given the physiological and pathological roles of SGs, it is therefore critical to disentangle SG composition in different cellular contexts and under different types of stress to better understand SG biology and to identify potential modulators. The tools at our disposal to study SGs are transcriptomics (Khong et al. 2017), proteomics (Youn et al. 2019), and imaging approaches (Van Treeck and Parker 2019).
SGs assemble through protein–protein interactions, such as G3BP(key SG component)–Caprin1–USP10 complexes or G3BP dimerization (Kedersha et al. 2016; Yang et al. 2020), protein–RNA interactions, such as UBAP2L–RNA interactions or G3BP1–RNA interactions (Cirillo et al. 2020; Guillén-Boixet et al. 2020), and RNA–RNA interactions. Recent studies have shown indeed that accumulation and interaction of RNA molecules represent driving forces behind SG formation: (i) RNA condensation can reproduce the SG transcriptome in yeast (Van Treeck et al. 2018); (ii) RNA helicases limit SG formation in cells and RNA condensation in vitro (Tauber et al. 2020); (iii) G3BP1 can promote the formation of new RNA–RNA interactions (Parker et al. 2025). The importance of trans RNA–RNA interactions in SG formation is further reviewed in Roden and Gladfelter (2021) and Zhang et al. (2025). The weight of the RNA component of SGs is also supported by the evidence that, proportionally, more RNAs than proteins are present in SGs compared to the surrounding cytoplasm (Bounedjah et al. 2014; Campos-Melo et al. 2021).
With this review, we aim at providing a useful tool for researchers that are new to the field of SGs, and of ribonucleoprotein (RNP) granule science more broadly. We outline SG dynamics and proposed functions, currently available approaches, as well as possible future strategies to characterize SG components, and we share a list of human SG RNAs with low, medium, or high consensus score (CS) based on their recurrence across 26 published SG transcriptome data sets. While “assembling” this review, the complexity of SG biology came to light, with the involvement of hundreds or even thousands of RNA molecules. This biological complexity is inevitably associated with experimental challenges in the definition of SG composition, dynamics, and cellular functions. However, in the last eight years, the emergence of technologies to accurately purify and characterize SGs significantly propelled our understanding of physiological and pathological SGs forward. Through our meta-analysis, we found 7792 SG-enriched RNAs with low CS; this indicates that as gene expression programs are tissue- and condition-specific, SG composition might also be tissue- and condition-specific, and the fast exchange of RNA molecules between SGs and the cytoplasm is likely to further contribute to SG heterogeneity. On the other hand, we also found 769 SG-enriched RNAs present in 10 or more data sets, suggesting the importance of data integration to identify consistent SG RNAs that could point to a common mechanism in SG ontogeny and offer the rationale for drug development.
Physiological life cycle of stress granules
What triggers SG formation
SG formation in mammals is commonly triggered by the activation of the integrated stress response (ISR) that relies on one or more of the following serine/threonine kinases: (i) heme-regulated initiation factor 2α (HRI) kinase, which senses oxidative stress; (ii) protein kinase double-stranded RNA-dependent (PKR), which senses viral infection; (iii) PKR-like endoplasmic reticulum kinase (PERK), which senses the accumulation of unfolded proteins; and (iv) general control nonderepressible 2 (GCN2) kinase, which senses amino acid starvation. These kinases converge on the phosphorylation of the eukaryotic translation initiation factor 2 alpha subunit (eIF2α) (Kedersha et al. 2013; Pakos-Zebrucka et al. 2016). The detection of stressors is followed by polysome disassembly and translational arrest to prompt cellular adaptation programs. This results in the accumulation of stalled preinitiation complexes (PICs), constituted by small ribosomal subunits, translation initiation factors, and polyadenylated mRNAs (Ivanov et al. 2019; Marcelo et al. 2021). Another pathway of SG formation through inhibition of translation initiation is demonstrated by the chemical targeting of eIF4A, e.g., using hippuristanol (Tauber et al. 2020). It appears that anytime there is a large pool of RNAs not engaged in translation the formation of SGs is favored. Splicing defects that lead to the accumulation of long intron-containing RNAs during mitosis can trigger the formation of a type of noncanonical SGs, even before translation stalling (Zhou et al. 2024). SG assembly can also be promoted by RNA and SG-nucleating proteins that aggregate in response to molecular crowding under osmotic stress (Bounedjah et al. 2012; Jalihal et al. 2020).
RNA/protein features conducive to SG formation
Stress-dependent influx of untranslated RNAs facilitates the recruitment of SG-nucleating RBPs (Panas et al. 2016). Multivalent, weak, dynamic interactions within SGs are favored by specific features at both protein and RNA level (Alberti et al. 2019). In vitro, polyadenylated RNA triggers liquid–liquid phase separation (LLPS) of the SG-nucleator G3BP1 nearly as potently as total RNA; in cells, G3BP1–mRNA interactions increase under stress conditions due to increased availability of mRNA (Yang et al. 2020). The increment in the local concentration of RNA and RBPs, combined with protein–protein, protein–RNA, and RNA–RNA interactions, enhances phase transition and the assembly of SGs (Courchaine et al. 2016; Putnam et al. 2023; Wadsworth et al. 2024).
Protein phase separation relies on the presence of structured domains, such as oligomerization domains and RNA recognition motifs, and on the presence of conformationally flexible intrinsically disordered regions (IDRs), that can contain low-complexity domains (LCDs) characterized by an overrepresentation of only a few amino acids (Alberti and Dormann 2019; Musacchio 2022). In Li et al. (2012), multivalent interactions between SH3–PRM protein modules were sufficient to drive phase separation, and the multivalent RBP PTB was able to form droplets in the presence of an RNA oligonucleotide. In Molliex et al. (2015), the LCD of hnRNPA1 alone is sufficient to mediate LLPS, while the RNA recognition motifs contribute to LLPS in the presence of RNA. Two important classes of IDRs are Prion-like domains (PrLDs), with mostly polar amino acids promoting protein–protein interactions (Wang et al. 2018), and positively charged arginine- and glycine-rich (RGG/RG) regions that can form electrostatic interactions with negatively charged RNAs (Chong et al. 2018). For example, RGG regions in the RNA helicase LAF1 electrostatically interact with RNA promoting Caenorhabditis elegans P granule formation by LLPS (Elbaum-Garfinkle et al. 2015).
In addition to proteins, RNA molecules represent an ideal scaffold molecule for LLPS. RNA phase separation is dependent on length, sequence, and secondary structure (Hofmann et al. 2021; Bevilacqua et al. 2022; Swain et al. 2024). Of note, RNA molecules with repeat expansion sequences can create templates for multivalent base-pairing, leading to RNA gelation without requiring protein components (Jain and Vale 2017).
SG maturation
In the physiological life cycle of SGs, their maturation leads to a hierarchical internal organization (Glauninger et al. 2022) that can be explained by two models: nanoscopic SG seeds coalescence to form mature SGs (Panas et al. 2016), or a more stable “core” is surrounded by a more dynamic “shell” of other proteins and RNAs (Jain et al. 2016). This shell phase, as observed by region-specific probes or single-molecule imaging, may simply be long RNAs that are tethered to the core and extend beyond the protein surface of an SG, promoting the fusion of small SGs (Moon et al. 2019). Finally, SGs tend to grow in microtubule-rich and actin-low regions of the cell (Böddeker et al. 2023).
SG functions
SGs are among the main players in the cellular response to stress. It is suggested that, once formed, SGs sequester translatome components and transiently store mRNAs encoding house-keeping proteins to decrease global translation and conserve anabolic energy (Kedersha and Anderson 2009; Marcelo et al. 2021). An experimental example of the link between SGs and energy modulation is represented by Amen and Kaganovich (2021) showing that, during long-term starvation, SGs downregulate fatty acid β-oxidation by obstructing voltage-dependent anion channels that import fatty acids into mitochondria. At the same time, SGs seem to act as sites of RNA triage: included transcripts are stabilized while excluded transcripts can be targeted for translation or degradation by the processing(P)-bodies to restore cellular homeostasis (Anderson and Kedersha 2008; Luo et al. 2018; Podszywalow-Bartnicka and Neugebauer 2024). SG-dependent subcellular localization might also affect nuclear processes such as transcription, 3′-end processing, splicing, and export (Buchan and Parker 2009). SGs have also antiviral effects inhibiting the translation of viral proteins and controlling innate immune response (McCormick and Khaperskyy 2017; Eiermann et al. 2020; Paget et al. 2023). Finally, SGs influence cell survival by limiting the availability of proteins involved in pro-apoptotic pathways, such as specific RNA helicases or executioner caspases (Samir et al. 2019; Fujikawa et al. 2023).
SG disassembly
Upon stress release, SGs disassemble in a reverse stepwise process. The resumption of translation determines a decreased mRNA influx into SGs, multivalent interactions between proteins and RNAs are disrupted, and SGs are broken into smaller foci by ATP-dependent segregases and molecular chaperones allowing the stored RNA to re-enter the translational pool (Wheeler et al. 2016; Millar et al. 2023; Cui et al. 2024). In fact, single-molecule translation imaging shows that mRNA molecules localized into granules during stress have translational rates similar to their cytosolic counterpart when stress is relieved (Wilbertz et al. 2019). Certain persistent SGs can also be cleared by autophagy (Buchan et al. 2013; Ryan and Rubinsztein 2024).
Protein and RNA modifications
Posttranslational modifications (PTMs) such as poly ADP-ribosylation, phosphorylation, arginine methylation, SUMOylation and ubiquitination further drive both SG assembly and disassembly. PTMs can facilitate or inhibit protein recruitment and protein–RNA multivalent interactions (Yang et al. 2020; Hofmann et al. 2021). The effect of specific PTMs on SG dynamics is thoroughly reviewed in Millar et al. (2023).
Alongside PTMs, RNA modifications can also affect SG dynamics and composition. N6-methyladenosine (m6A), for example, has been reported to increase mRNA partitioning into SGs by providing binding sites for YTHDF proteins; the resulting mRNA-YTHDF complexes are then able to localize into SGs (Ries et al. 2019, 2023). However, the role of m6A in promoting the enrichment of mRNAs in SGs remains debatable. In Khong et al. (2022), m6A modified mRNAs partition similarly into arsenite-induced SGs in both wild-type and m6A-deficient cells. In Sheehan et al. (2023), in situ visualization of m6A sites in target RNAs shows that m6A is not sufficient for transcript localization in arsenite-induced SGs. The interplay between m6A modification and SGs might also depend on other factors, such as SG stability (Li et al. 2024) or RNA homeostasis (Gunage et al. 2024).
Pathological role of stress granules
Persistent or aberrant SGs are associated with various diseases, including neurodegenerative disorders and cancer (Protter and Parker 2016). The study of dynamics and functions of pathological SGs can therefore provide new therapeutic solutions (Wang et al. 2020; Lavalée et al. 2021). For example, in Fang et al. (2019), the application of a high-content screening identified a class of planar small molecules that reduce the accumulation of TDP-43 into persistent cytoplasmic SGs in motor neurons derived from patients with amyotrophic lateral sclerosis (ALS).
With acute stress, heterogeneous nuclear ribonucleoproteins (hnRNPs) such as TDP-43, FUS, TIA1, hnRNPA1, and hnRNPA2B1 translocate to the cytoplasm and condense into SGs. Upon stress resolution, SGs disassemble, and soluble hnRNPs shuttle back to the nucleus. In contrast, chronic stress associated with aging and inflammation leads to the formation of persistent, insoluble, and more fibrillar SGs, which are a hallmark of patients with ALS and frontotemporal dementia (FTD) (Wolozin and Ivanov 2019). These pathological SGs result in impaired stress response and altered RNA transport and translation, disrupting cellular homeostasis and eventually leading to neuron cell death (Reineke and Neilson 2019). Beside chronic stress, genetic mutations and PTMs influence SG formation and clearance and are key drivers in the formation of pathological aggregates (Cui et al. 2024). Genetic mutations frequently affect hnRNPs and are positioned in LCDs, increasing the tendency of proteins to aggregate, or in the nuclear localization signal (NLS), determining the accumulation of proteins in the cytoplasm (Taylor et al. 2016; Mariani et al. 2024). Pathological deposits of TDP-43 in ALS and FTD patients, that can form via SG-dependent or independent mechanisms, are characterized by polyubiquitinated, hyperphosphorylated, truncated TDP-43 (Zbinden et al. 2020) and by dipeptide repeat proteins translated from expanded G4C2 repeats in Chromosome 9 open reading frame 72 (Cook et al. 2020). G4C2 mRNA contributes to pathological aggregates by binding and sequestering nucleocytoplasmic transport regulators (Zhang et al. 2018). Finally, physiological SG clearance in ALS/FTD patients is impaired by genetic mutations in genes involved in the ubiquitin-proteasome system (UPS) and autophagy, such as VCP and optineurin (Wolozin and Ivanov 2019; Nedelsky and Taylor 2022).
Increased SG formation has been proposed to provide a strategy exploited by cancer cells to survive and expand in a hostile microenvironment. Cancer cells are highly proliferative cells with high metabolic demand, which results in extracellular and intracellular stressors such as hypoxia, nutrient scarcity, and endoplasmic reticulum stress causing SG persistence (Anderson et al. 2015). SG formation in cancer cells is also inherently driven by oncogenic-induced stress, including oncogenic mutations or dysregulated signaling cascades, and by pre-existing stress conditions, e.g., a chronic inflammatory status (Redding and Grabocka 2023).
Multiple stressors can therefore lead to SG formation in cancer cells, and SGs can be implicated in multiple processes that are linked to improved cancer cell fitness, cancer progression, immune escape, and chemoresistance (Jia et al. 2024). For example, in pancreatic cancers, the upregulation of SGs is associated with mutant KRAS, which regulates prostaglandin metabolism leading to increased levels of 15-deoxy-Δ12,14-prostaglandin J2 (15-d-PGJ2) and consequent eIF2α-independent SG formation via inactivation of eIF4A (Grabocka and Bar-Sagi 2016). 15-d-PGJ2 can also act through eIF2α phosphorylation as reported in Tauber and Parker 2019. The stress phenotype in pancreatic cancers is drastically heightened in obesity-dependent inflammatory states (Fonteneau et al. 2022). U2AF1 mutations in hematologic malignancies and in lung cancers are responsible for RNA binding and splicing alterations that directly affect SG components and SG-associated processes fostering cell fitness under oxidative stress, resistance to chemotherapy, and oncogenic transformation (Biancon et al. 2022b; Jin et al. 2024; Liang et al. 2024). A last example is represented by the mTORC1 pathway, whose activation in cancer cells contributes to SG assembly and, on the other side, mTORC1 sequestration into SGs prevents its hyper-activation that would lead to cell death (Song and Grabocka 2020). Cancer SG-dependence is therefore a complex process, highlighting an area that requires further investigation.
Experimental methods to isolate SG components
The first step in defining the composition of SGs is their isolation from the rest of the cellular components. Individual RNAs and proteins localizing into SGs can be directly visualized by single-molecule imaging approaches (Moon et al. 2019; Van Treeck and Parker 2019). On a larger scale, transcriptome-wide profiling of SGs, which is the focus of this review, is enabled by the following methods: biochemical purification, proximity labeling, photo-isolation chemistry, RNA editing, and fluorescence-activated sorting (Fig. 1). A total of 32 data sets using these methods are reported in Supplemental Tables 1 and 2.
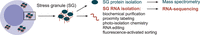
Schematic representation of experimental methods to isolate and characterize SG RNAs.
The biochemical purification approach is based on: (i) successive differential centrifugation to first separate cytoplasmic and nuclear fractions and then to enrich the cytoplasmic fraction for insoluble, stable SG cores; (ii) affinity purification using an antibody against a key SG component; (iii) identification of SG RNAs by RNA-seq (and of SG proteins by mass spectrometry) (Supplemental Tables 1 and 2; Jain et al. 2016; Khong et al. 2017; Wheeler et al. 2017; Khong et al. 2018; Matheny et al. 2019, 2021; Iadevaia et al. 2022; Chen et al. 2023; Curdy et al. 2023; Ries et al. 2023; Sato et al. 2023; Cheung et al. 2024; Kudrin et al. 2024; Mariani et al. 2024). The experiment can be conducted under different stress conditions, but also in unstressed conditions, given the demonstrated presence of a set of RNAs prone to assemble into smaller SGs that can be isolated even in the absence of stress (Jain et al. 2016).
The main advantage of the biochemical purification approach is that cells do not require genetic engineering before proceeding with SG isolation, making this approach ideal for cells that are difficult to engineer, such as suspension cells and primary cells (Namkoong et al. 2018; Ries et al. 2019; Curdy et al. 2023; Cheung et al. 2024). Possible caveats are: preferred isolation of SG cores that occupy ∼20% of the entire SG volume, although they concentrate most of the SG RNAs and proteins (Jain et al. 2016); and loss of weakly associated material during differential centrifugation (Ren et al. 2023). The centrifugation step can be simplified (Namkoong et al. 2018) or even abolished (Weeks et al. 2016; Kershaw et al. 2021), but in this way even free cytoplasmic components are immunoprecipitated. Additionally, weak interactions can be stabilized using a mild cross-linking step before SG isolation, for example by formaldehyde (Jain et al. 2016) or by mild UV-cross-linking, as already adopted with the proximity labeling approach (Benhalevy et al. 2018) to not only stabilize but also map SG RNA–protein interactions as per the enhanced UV-cross-linking and immunoprecipitation (eCLIP)-seq protocol (Biancon et al. 2022a).
The proximity labeling approach is based on: (i) the generation of a cellular system containing a key SG marker fused with an enzyme that can catalyze the labeling of proximal RNAs (and/or proteins); (ii) biotin-based labeling; (iii) affinity purification using streptavidin; (iv) identification of SG RNAs by RNA-seq (and/or of SG proteins by mass spectrometry). Different types of proximity labeling are available, depending on the employed enzyme. APEX-seq uses the engineered ascorbate peroxidase APEX2. In the presence of hydrogen peroxide, APEX2 converts biotin-phenol into biotin-phenoxyl radicals that biotinylate RNAs (and proteins) in close proximity (Supplemental Table 1; Padrón et al. 2019; Somasekharan et al. 2020). BioID is based on the usage of the abortive biotin ligase BirA*. After adding exogenous biotin, BirA* biotinylates proteins in close proximity but is unable to label RNAs (Youn et al. 2018). CAP-seq uses the photocatalyst miniSOG. Under blue light excitation, miniSOG mediates the photo-oxidization of proximal RNA, which can be captured by biotin-conjugated amine probes (Supplemental Table 1; Ren et al. 2023). Proximity labeling approaches allow the study of SG components prior to stress and during stress (Markmiller et al. 2018).
The main advantage of SG isolation by proximity labeling is the capturing of weak and transient interactions, in their native state, that do not withstand biochemical purification (Youn et al. 2019). Possible caveats are: genetic manipulations of target cells to express the fusion construct, limited temporal resolution because some transcripts have an SG-cytoplasm exchange rate that is faster than the proximity labeling time, and contamination from cytoplasmic transcripts given that the majority of the SG bait freely localizes in the cytoplasm instead of inside SGs (Glauninger et al. 2022). Between APEX-seq and CAP-seq, APEX-seq is based on a faster labeling time (∼1 min vs. ∼20 min), but CAP-seq works without cytotoxic hydrogen peroxide (Ren et al. 2023, 2024).
Photo-isolation chemistry is based on: (i) deposition of photo-caged oligodeoxynucleotides and reverse transcriptase onto coverslips with seeded cells; (ii) selection of SG structures by immunostaining; (iii) UV-irradiation of the region of interest to locally uncage oligodeoxynucleotides; (iv) identification of SG RNAs by RNA-seq (Supplemental Table 1; Honda et al. 2021). This in situ reverse transcription-based labeling is less commonly used in comparison to biochemical purification and proximity labeling approaches, but it has several advantages including low input material, possible application on tissue sections, and combination with single-cell RNA-seq technology, while the previously described approaches use standard bulk, short-read, RNA-seq.
The SG transcriptome can also be investigated by the application of an RNA-editing method called hyperTRIBE (Supplemental Table 2; van Leeuwen et al. 2022). Upon oxidative stress, the SG protein FMR1, fused to the catalytic domain of the RNA-editing enzyme ADAR, compartmentalizes into SGs. SG RNAs interacting with the fusion protein are therefore subjected to A-to-I editing that is read as an A-to-G mutation by sequencing. The rationale behind this technology is that the more an RNA is edited, the more likely it was recruited into SGs. Similar to the photo-isolation chemistry approach, given the absence of a previous purification step, hyperTRIBE does not require a large amount of input material and is compatible with single-cell RNA-seq. Possible caveats, besides the need of genetic engineering, are related to: basal activity and sequence preferences of the fusion protein, contamination from cytoplasmic edited RNAs due to the large percentage of SG baits that localize outside of SGs, and certain editing sites might be blocked by the binding with other SG RBPs.
Lastly, a flow cytometry-based technology has been recently developed to purify and characterize a noncanonical type of SGs that form under UV-induced RNA damage (Zhou et al. 2024). This technology, called fluorescence-activated nonmembrane condensates isolation (FANCI), is based on: (i) cell fixation and sonication; (ii) flow cytometry sorting of intact G3BP1-mCherryhigh particles from cell lysates; (iii) identification of SG RNAs by RNA-seq (and of SG proteins by mass spectrometry) (Supplemental Table 1). FANCI stems from the successful isolation of other membraneless organelles via fluorescent-activated sorting in both adherent and suspension cell cultures (Hubstenberger et al. 2017; Kodali et al. 2024), and it helps preventing contamination from ER and nuclear membrane components that can interact and therefore precipitate with SGs. Zhou and colleagues used FANCI technology in epithelial cells under UVA irradiation stress that triggers DHX9 SGs in the presence of thiouracil, allowing RNA–protein cross-linking without DNA damage (Parker et al. 2024). DHX9 SGs are enriched in intronic RNAs, form after mitosis independently from translation shutdown and induce double-stranded RNA (dsRNA)-related immune response. Of note, the authors also showed the presence of UV-induced SGs in a human epidermal model.
Characterization of the stress granule RNA world
SG data sets
SGs are heterogeneous condensates. Their RNA (and protein) content might vary according to the cellular system, the type of stress and the duration of the stress (Cui et al. 2024). Moreover, experimental methods, sequencing strategies, and computational pipelines represent additional sources of variability. While previous reviews have examined the protein composition of SGs (Youn et al. 2019) and a database exists that compiles experimental evidence on SG proteins (http://rnagranuledb.lunenfeld.ca), a comparable analysis or resource for SG RNAs is currently lacking. Among published SG transcriptome studies, some experiments have directly compared the SG transcriptome in the same cellular model under different stressors, using the same SG isolation and computational approach (Padrón et al. 2019; Matheny et al. 2021; Ren et al. 2023; Zhou et al. 2024); this allows to investigate how different stressors affect SG composition. Other experiments focused on a certain biological condition to define SG composition. To integrate currently available data sets of human SG transcriptome, we performed a meta-analysis of 26 data sets (Fig. 2A; Supplemental Table 1) and we here provide a list of RNAs that are more or less consistently enriched in SGs (Supplemental Table 3).
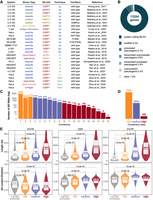
Meta-analysis of the human SG transcriptome. (A) Overview of data sets included in the meta-analysis. Techniques to isolate SG RNAs: bp, biochemical purification = differential centrifugation > immunoprecipitation; bp*, immunoprecipitation; pl, biotin-based proximity labeling > streptavidin pulldown; (pic) photo-isolation chemistry; (fanci) fluorescence-activated nonmembrane condensates isolation. (B) Most represented RNA types among total SG-enriched RNAs. (C) Number of SG RNAs grouped by CS. (D) Number of SG RNAs per consensus category: low, CS < 4; medium, 4 ≤ CS ≤ 9; high, CS > 9. “Not SG enriched” category includes all human-annotated genes except SG-enriched lists. (E) Length in nucleotides and AU content of SG RNAs (canonical transcripts) per consensus category, considering 5′ UTR, CDS, and 3′ UTR. Median values are reported. P-values are based on the Wilcoxon one-tailed test.
Genetic context
Available studies on the SG transcriptome were mostly performed under wild-type conditions (Fig. 2A). Nevertheless, our meta-analysis contains two data sets based on genetically modified cellular systems and one data set characterized by a disease-associated mutation. G3BP1/2 knockout condition in Matheny et al. (2021) was used to compare SG RNA composition after immunoprecipitation with different SG baits: G3BP1, the most frequently used SG bait, and PABPC1. PABPC1 immunoprecipitation was conducted in the absence of the G3BP hetero-multimer to demonstrate that SG RBPs can act in tandem, and the deletion of an individual SG RBP has only limited effect on defining SG RNA composition. NAT10 knockout condition in Kudrin et al. (2024) was pivotal to show the role of the RNA modification N4-acetylcytidine (ac4C) in SG formation. Upon oxidative stress, ac4c-modified mRNAs shuttle into SGs independently from their length and promote the recruitment of RBPs into SGs. Mariani and colleagues investigated changes in SG transcriptome and dynamics in the presence of FUS-P525L mutant protein, accounting for 4% of familial ALS cases (Mariani et al. 2024). FUS-P525L is associated with the recruitment of AU-rich, unstructured, neuronal-specific RNAs into SGs, explaining the causative link between cytoplasmic delocalization of FUS mutant proteins, formation of cytotoxic inclusions and the neurodegenerative phenotype.
Stress type
Arsenite stress is the most widely used stress type in SG studies. In fact, 15 out of 26 data sets in our meta-analysis were conducted under oxidative stress triggered by arsenite treatment (Fig. 2A). Other possible stressors for SG formation are: sorbitol or NaCl, to apply osmotic stress; heat shock, to apply thermal denaturing stress; thapsigargin, to apply endoplasmic reticulum stress; viral infection or antigen stimulation, to induce immune response; hippuristanol, which inhibits translation initiation targeting eIF4A; UV-light inducing RNA damage and the accumulation of long intron-containing RNAs (Supplemental Tables 1 and 2). Of note, SG formation under osmotic stress, hippuristanol treatment and UV stress is independent from phospho-eIF2α; SG formation under osmotic stress and heat shock stress is independent from G3BP (Kedersha et al. 2016; Padrón et al. 2019; Matheny et al. 2021). Different SG subtypes are further reviewed in Hofmann et al. (2021).
SG isolation
SG RNAs are isolated thanks to the visualization and/or pulldown of key SG nucleators, so-called “SG baits.” Especially when using the biochemical purification approach, the SG bait can be fused to a fluorescent protein to validate the effective isolation of SG cores (Wheeler et al. 2017). One exception in our meta-analysis is represented by Matheny et al. (2019). Here, the biochemical purification approach was used first to isolate stress-induced, dense, cytoplasmic fractions without immunoprecipitation for SGs or P-bodies, and then to isolate stress-induced P-bodies by EDC3 immunoprecipitation. The SG transcriptome in Matheny et al. (2019) has therefore been obtained by removing the P-body transcriptome, instead of using a specific SG bait (“ΔEDC3”, Fig. 2A).
Thirteen data sets included in our meta-analysis were generated using the biochemical purification approach followed by RNA-seq (“bp”, Fig. 2A; Khong et al. 2017; Matheny et al. 2019, 2021; Iadevaia et al. 2022; Chen et al. 2023; Curdy et al. 2023; Kudrin et al. 2024; Mariani et al. 2024). Two older data sets were generated using immunoprecipitation without prior centrifugation steps, followed by exon microarrays. However, given the application of immunoprecipitation as in the more recent biochemical purification approach and the absence of chemical labeling, we included this data set in the “bp” category for downstream analyses (“bp*”, Fig. 2A; Weeks et al. 2016). Six data sets were generated using proximity labeling and RNA-seq (“pl”, Fig. 2A), but in three data sets, the labeling was APEX2-mediated (Padrón et al. 2019; Somasekharan et al. 2020), while in the other three data sets, the labeling was miniSOG-mediated (Ren et al. 2023). Furthermore, one data set was generated using photo-isolation chemistry (“pic”), and four data sets were generated using fluorescence-activated nonmembrane condensates isolation (“fanci”, Fig. 2A).
SG RNA identification and classification
Our meta-analysis across 26 data sets identified a total of 13,204 SG-enriched genes. Source files and selection criteria for extracting the list of SG-enriched genes in each data set are specified in Supplemental Table 1. When available, we used the list reported in the reference publication. Otherwise, we started from count data and identified SG-enriched genes by applying a standard computational pipeline implemented in edgeR Bioconductor package (Robinson et al. 2010): TMM normalization and glmQLFTest function for differential analysis with log2 fold change > 0.75 and P-value < 0.05. Considering different expression levels in different data sets, SG-enriched gene lists are based on a direct comparison between “SG” counts and “input” or “cytoplasm” counts, which should scale out overall transcript abundances.
Consistent with SG dynamics and with previous literature on the relative contribution of RNA categories to the SG transcriptome (Khong et al. 2017; Sato et al. 2023), SGs are largely constituted by protein-coding RNAs (89.5%) but also by long noncoding RNAs (5.3%) (Fig. 2B). The number of SG-enriched genes reported in at least one data set is more than half the number of protein-coding genes in the human genome, suggesting little specificity. We therefore grouped the 13,204 SG-enriched genes by consensus score (CS), defined as the number of data sets reporting a specific RNA as SG-enriched. SG RNAs that are captured by more than one experimental method (“bp”, “pl”, “pic”, “fanci”) are rewarded with a + 1. Based on the distribution of the CSs, we classified SG RNAs into three categories: low consensus (CS < 4), medium consensus (4 ≤ CS ≤ 9), and high consensus (CS > 9). High consensus RNAs can be considered as recurrent, solid SG markers, while low consensus RNAs can be considered as condition-specific (Fig. 2C,D). Gene ontology analysis of the 769 high consensus RNAs reports: regulation of transcription, chromatin remodeling, and regulation of gene expression as significantly involved biological processes; intracellular organelles, nucleus, and cytoskeleton as significantly involved cellular components; RNA binding, GTPase regulator activity and protein serine/threonine kinase activity as significantly involved molecular functions. The full list of SG-enriched RNAs, with experimental details and CS for defining the consensus category or for further customized filtering, is available in Supplemental Table 3.
SG RNA features
Finally, we analyzed the length and AU content of SG RNAs by consensus category, separately for 5′ UTR, CDS, and 3′ UTR. These RNA features have been shown to be involved in the RNA compartmentalization into SGs (Khong et al. 2017; Dar et al. 2024). RNA length is suggested to contribute to SG formation because long RNAs can assume more conformations and engage in more multivalent interactions (Sanchez de Groot et al. 2019; Campos-Melo et al. 2021) or can bind more RBPs (Guillén-Boixet et al. 2020; Mann and Donnelly 2021). In our meta-analysis, we confirmed a positive correlation with transcript length, with high consensus RNAs associated with increased length in all three transcript regions (Fig. 2E, top panels). The AU content, instead, shows a less evident trend. GC-rich regions can promote the formation of RNA structures but actually the presence of AU-rich elements, typically located in the 3′ UTRs, favors the interaction with RBPs and therefore compartmentalization into stress-induced RNP granules (Namkoong et al. 2018; Guzikowski et al. 2019). Published studies have shown a strongly positive correlation between AU content and localization into P-bodies, but only a slightly positive correlation with SG localization (Matheny et al. 2019; Curdy et al. 2023). Our meta-analysis confirmed a slight increase (6%–7%) in AU content within CDS and 3′ UTRs of medium and high SG consensus genes. Conversely, 5′ UTRs showed a decrease in AU content with higher CSs, potentially due to other functions of GC-rich regions in the 5′ UTR (Fig. 2E, bottom panels).
SG consensus lists offer valuable tools for studying SG biology. They have the potential to facilitate the analysis of SG RNA expression across diverse cell types, cell differentiation stages, physiological and pathological conditions. For example, they can be used to enable the derivation of SG activity scores, analogous to cell cycle scores, for interpreting single-cell data (Biancon et al. 2022b; Busarello et al. 2024).
Future perspectives
SGs are RNA–protein condensates that are involved in the response to stress conditions and in the preservation of cellular homeostasis. However, SG dynamics, physiologic activity, involvement in human disease and evolutionary history require further investigation. Future research using high-resolution technologies to examine SGs in diverse tissues, and under both normal and disease-associated conditions, will significantly advance our understanding of SG biology and propel the development of effective SG-modulating therapies forward. Below, we summarize key open questions about SGs.
A key challenge lies in characterizing the diversity of RNA within SGs. We need to fully understand how this composition varies across different cell types and tissues, and how it is impacted by diverse stressors. A systematic investigation of SG composition across various cell lines and tissues under a range of stress conditions is the crucial first step toward addressing these questions. The available data, even with our meta-analysis, provide only a glimpse into this complex issue.
A critical related question concerns the heterogeneity of SGs at single-cell and spatial level. How do individual SGs differ from one another? This heterogeneity could exist between different cells within a tissue, or even within a single cell. Is the composition of SGs influenced by subcellular organization? SGs have a distinct spatial organization due to their multistep assembly, they interact with other aggregates such as P-bodies and pathological inclusions, and they rewire signal transduction affecting the homeostasis of the cell itself and potentially of surrounding cells in a tissue (Grabocka and Bar-Sagi 2016). Single-cell and single-granule analyses using spatial approaches hold crucial promise for addressing these fundamental questions. Examples of spatial transcriptomic technologies include (Ren et al. 2024): (i) an antibody-based in situ reverse transcription method, called ARTR-seq (Xiao et al. 2024), which identified RNA targets of G3BP1 with spatiotemporal resolution in stressed and fixed HeLa cells on a slide; (ii) multiplexed error-robust fluorescence in situ hybridization (MERFISH) (Chen et al. 2015), which has been used to visualize untranslated RNAs in G3BP1-positive focal adhesion (Boraas et al. 2025), although the molecular density of SGs may hamper high-plex fluorescence measurement; (iii) spatially indexed next-generation sequencing methods such as the pathology-compatible deterministic barcoding in tissue (Patho-DBiT) (Bai et al. 2024); these methods could reveal expression and localization of SG RNAs in an unsupervised way in pathogenic versus wild-type cells within a tissue.
Unraveling the complexities of SG heterogeneity holds the key to answering crucial questions about RNA: What are the primary RNA factors that determine composition similarities and differences? Length and AU content are some aspects previously considered and confirmed in our meta-analysis. To further dissect RNA properties associated with SG composition, the application of full-length direct RNA-seq would be promising for: (i) RNA decay analysis through the identification of robust RNA length variations under stress (Dar et al. 2024); (ii) RNA splicing analysis, given the link between RNA splicing perturbations and SG formation (Berchtold et al. 2018; Biancon et al. 2022b; Liang et al. 2024); (iii) m6A RNA methylation analysis. The weight of m6A modification on RNA partitioning into SGs is still under investigation (Ries et al. 2019; Khong et al. 2022), but the inhibition of the m6A writer METTL3 is able to successfully reduce FUS inclusions in fibroblasts derived from ALS patients (Di Timoteo et al. 2024).
A compelling question also arises regarding the evolutionary trajectory of the SG RNA world. Given the divergence of RNA processing mechanisms, such as RNA splicing or RNA modifications between organisms like yeast and humans, how are these evolutionary changes in RNA biology reflected in the composition and organization of SGs? A systematic comparative analysis of SGs across diverse species is essential as a first step toward illuminating this evolutionary adaptation.
Addressing these fundamental questions is necessary to unlock our understanding of the intricate relationship between SGs and disease. Studying how disruptions in SG dynamics contribute to the pathogenesis of various diseases will potentially lead to the development of effective therapeutic strategies. In short, a deep comprehension of the basic biology of SGs is a prerequisite for translating this knowledge into clinical applications.
In conclusion, this review summarizes SG pathophysiology and our current understanding of RNA within SGs, including current methodologies to study the SG transcriptome. We provide a robust list of more or less frequently reported RNAs in human SGs and, lastly, we suggest future directions for a more comprehensive understanding of SG biology.
SUPPLEMENTAL MATERIAL
Supplemental material is available for this article.
ACKNOWLEDGMENTS
G.B. was supported by the Italian Association of Cancer Research (AIRC) under Start-Up 2023 - ID. 29035 project, the American Society of Hematology Scholar Award, and the Edward P. Evans Foundation. S.H. was supported by National Institutes of Health, NIH/NIDDK R01DK124788, NIH/NCI R01CA266604, NIH/NCI R01CA222518, and NIH/NCI R01CA253981, the Frederick A. Deluca Foundation, and the Edward P. Evans Foundation. T.T. was supported by AIRC under MFAG 2020 - ID. 24883 project, by the EU (MUR PNRR Project no. CN00000041 CN3 RNA S6 RINGTAIL), by MUR under the “Departments of Excellence 2023–2027” initiative (Law 232/2016), project no. 40613. T.T. and E.B. were supported by Fondazione VRT, AIL Trento, AIL Bolzano.
Footnotes
-
Article is online at http://www.rnajournal.org/cgi/doi/10.1261/rna.080409.125.
-
Freely available online through the RNA Open Access option.
This article, published in RNA, is available under a Creative Commons License (Attribution-NonCommercial 4.0 International), as described at http://creativecommons.org/licenses/by-nc/4.0/.








