
Role of the sarcin-ricin loop of 23S rRNA in biogenesis of the 50S ribosomal subunit
- Sepideh Fakhretaha Aval1,2,
- Amal Seffouh3,4,
- Kyung-Mee Moon5,
- Leonard J. Foster5,
- Joaquin Ortega3,4 and
- Kurt Fredrick1,2,6
- 1Ohio State Biochemistry Program, The Ohio State University, Columbus, Ohio 43210, USA
- 2Center for RNA Biology, The Ohio State University, Columbus, Ohio 43210, USA
- 3Department of Anatomy and Cell Biology, McGill University, Montreal, Quebec H3A 0C7, Canada
- 4Centre for Structural Biology, McGill University, Montreal, Quebec H3G 0B1, Canada
- 5Department of Biochemistry and Molecular Biology, Michael Smith Laboratories, University of British Columbia, Vancouver, British Columbia V6T1Z4, Canada
- 6Department of Microbiology, The Ohio State University, Columbus, Ohio 43210, USA
- Corresponding author: fredrick.5{at}osu.edu
-
Handling editor: John Woolford
Abstract
The sarcin-ricin loop (SRL) is one of the most conserved segments of ribosomal RNA (rRNA). Translational GTPases (trGTPases), such as EF-G, EF-Tu, and IF2, form contacts with the SRL that are critical for GTP hydrolysis and factor function. Previous studies showed that expression of 23S rRNA lacking the SRL confers a dominant lethal phenotype in Escherichia coli. Isolated ΔSRL particles were found to be not only inactive in protein synthesis but also incompletely assembled. In particular, block 4 of the subunit, which includes the peptidyl transferase center, remained unfolded. Here, we explore the basis of this assembly defect. We find that 23S rRNA extracted from ΔSRL subunits can be efficiently reconstituted into 50S subunits, and these reconstituted ΔSRL particles exhibit full peptidyl transferase activity. We also further characterize ΔSRL particles purified from cells, using cryo-EM and proteomic methods. These particles lack density for rRNA and r-proteins of block 4, consistent with earlier chemical probing data. Incubation of these particles with excess total r-protein of the large subunit (TP50) fails to restore substantial peptidyl transferase activity. Interestingly, proteomic analysis of control and mutant particles shows an overrepresentation of multiple assembly factors in the ΔSRL case. We propose that one or more GTPases normally act to release assembly factors, and this activity is blocked in the absence of the SRL.
Keywords
INTRODUCTION
Protein synthesis involves GTPases of the classical translation factor family (translational GTPases or trGTPases) in all organisms (Leipe et al. 2002). In Escherichia coli, there are eight trGTPases: IF2, EF-Tu, EF-G, RF3, SelB, LepA, BipA, and CysN (Margus et al. 2007). Aside from CysN, which is a component of the enzyme sulfate adenylyltransferase, these trGTPases are affiliated with ribosomes. Five play well-known roles in translation. IF2 promotes fMet-tRNA selection and subunit joining during initiation. EF-Tu delivers aminoacyl-tRNA (aa-tRNA) to the A site during decoding. SelB functions much like EF-Tu but specifically facilitates incorporation of selenocysteine in response to programmed UGA codons. EF-G catalyzes translocation, the movement of tRNAs (and paired codons) to their adjacent sites within the ribosome. RF3 catalyzes dissociation of the primary release factor (RF1/RF2) from the A site during termination. The other two trGTPases, LepA and BipA, are nonessential proteins that resemble EF-G. These proteins were initially proposed to regulate translation elongation in some way, but more recent studies indicate that they function primarily in ribosome biogenesis (Gibbs and Fredrick 2018). LepA aids in assembly of the 30S subunit (Gibbs et al. 2017), while BipA aids in assembly of the 50S subunit (Krishnan and Flower 2008; Choudhury and Flower 2015; Gibbs et al. 2020). Common to all trGTPases is an invariant histidine of the switch II motif of the GTPase (G) domain and an architecture in which the G domain is followed by a β barrel domain (domain II) (Leipe et al. 2002; Margus et al. 2007). TrGTPases bind the 70S ribosome in a similar (and mutually exclusive) way, interacting with the intersubunit cleft on the A-site side of the ribosome (Voorhees et al. 2010; Zhou et al. 2012; Pulk and Cate 2013; Gagnon et al. 2014; Kumar et al. 2015; Fischer et al. 2016; Kaledhonkar et al. 2019). The G domain contacts the sarcin-ricin loop (SRL) and neighboring components of the 50S subunit, while domain II interacts with helix h5 and neighboring components of the small subunit. These interactions promote the activation of GTP hydrolysis, a key aspect of factor function in all cases.
The SRL is a universally conserved sequence located on helix H95 of 23S rRNA (nt 2654–2665; E. coli numbering) (Cannone et al. 2002), named after the two toxins that act on it. α-Sarcin cleaves the phosphodiester bond between G2661 and A2662, and ricin depurinates A2660 (Endo et al. 1983; Endo and Tsurugi 1987). Treatment of ribosomes with either toxin strongly inhibits trGTPase function and shuts down protein synthesis. An X-ray crystal structure of an elongation complex containing EF-Tu and the nonhydrolyzable GTP analog β-γ-methyleneguanosine 5′-triphosphate (GDPCP) shed light on how activation of GTP hydrolysis occurs on the ribosome (Voorhees et al. 2010; Liljas et al. 2011). When EF-Tu·GDPCP·aa-tRNA binds the ribosome, a key contact between the phosphate group of A2662 of the SRL and His84 of EF-Tu orders the active site. Amino acids of the switch II (Gly83, His84) and switch I (Thr61) motifs form H-bonds that orient a water molecule for attack of the γ-phosphate of GTP. Structures of elongation complexes containing either EF-G or SelB bound to nonhydrolyzable GTP analogs have also been determined (Pulk and Cate 2013; Tourigny et al. 2013; Zhou et al. 2013; Fischer et al. 2016), and remarkably similar active-site conformations are seen, with equivalent switch II and switch I residues ordered via A2662 of the SRL. Nucleotides of the SRL also coordinate Mg2+ ions (two in the EF-G case; one in the SelB case), which also contribute to the ordering of the active site residues. Collectively, these data suggest a common mechanism of trGTPase activation on the ribosome, involving conserved features of the factors and the ribosome.
In 2008, Lancaster et al. showed that the SRL contributes not only to translation but also to the assembly of the 50S subunit (Lancaster et al. 2008). An aptamer-tagged variant of 23S rRNA lacking the SRL was expressed in E. coli, and the corresponding 50S particles were purified via affinity chromatography. These mutant subunits were defective in protein synthesis and factor-dependent functions, as expected. In addition, these particles were not fully formed. They lacked uL16, and nucleotides of helices H37, H39, H42, H72, H80, H89-H92, and H97 showed increased accessibility to chemical probes (Lancaster et al. 2008). These nucleotides correspond well to block 4, one of five structural modules of the subunit, which cooperatively folds during 50S assembly in the cell (Davis et al. 2016). Block 4 includes uL16 and components of the peptidyl transferase center (PTC).
Ribosome assembly is a complex process, which in E. coli entails the folding of three large rRNA molecules (4566 nt total) and the binding of 54 ribosomal proteins (r-proteins). Early studies showed that each ribosomal subunit can self-assemble in vitro from purified components (Mizushima and Nomura 1970; Nomura and Erdmann 1970; Nierhaus and Dohme 1974), indicating that the process is intrinsically wired in the rRNA and r-proteins themselves. However, subunit reconstitution is slow, inefficient, and requires nonphysiological conditions such as high temperature and high Mg2+ concentration. In the cell, subunit assembly begins cotranscriptionally and involves several nonribosomal proteins, termed assembly factors (AFs) (Davis and Williamson 2017; Naganathan and Culver 2022). These include ribonucleoprotein-binding proteins, rRNA and protein modification enzymes, rRNA and protein chaperones, RNA helicases, and GTPases. These AFs collectively facilitate subunit assembly and prevent particles from entering the translationally active pool of ribosomes before their assembly is completed. The GTPases involved in ribosome assembly include Der, YihA, ObgE, Era, RsgA, LepA, BipA, and IF2. The latter three are trGTPases that hydrolyze GTP in the context of the 70S ribosome, suggesting that they act late during the biogenesis process (Gibbs and Fredrick 2018).
Lancaster et al. (2008) discussed ways in which the SRL could contribute to 50S subunit assembly. They hypothesized that, by contacting adjacent rRNA elements, the SRL may facilitate organization of the PTC. They also raised the alternative possibility that the SRL interacts with a GTPase whose activity is needed for efficient 50S assembly in the cell (Lancaster et al. 2008). In this work, we show that 23S rRNA lacking the SRL can be efficiently reconstituted into 50S subunits, which exhibit full peptidyl transferase (PT) activity. These data suggest that the defect conferred by ΔSRL is specific to the in vivo process of assembly. Using cryo-EM and proteomic methods, we further characterize affinity-purified ΔSRL particles isolated from cells. These particles lack PT activity and density for block 4, and incubation of these particles with excess total protein of the 50S subunit (TP50) only marginally improves their activity. Interestingly, multiple proteins, including AFs, copurify specifically with ΔSRL particles. Collectively, these data suggest that the SRL is needed to promote the dissociation of several AFs during 50S subunit biogenesis in the cell.
RESULTS
Efficient 50S assembly occurs without the SRL in vitro
To investigate whether the SRL helps fold the PTC via tertiary rRNA–rRNA interactions, we performed subunit reconstitution experiments. A mutation that removes the SRL, ΔSRL, was introduced into p278MS2 (Youngman and Green 2005), a plasmid that encodes an aptamer-tagged 23S rRNA in the context of the rrnB operon. Plasmid-encoded rRNA (control or ΔSRL) was expressed transiently, and corresponding 50S subunits were purified via affinity chromatography (see Materials and Methods). Based on poisoned primer extension and RNase H assays, the purity of aptamer-tagged particles exceeded 90% (Supplemental Fig. S1), in line with previous studies (McGarry et al. 2005; Youngman and Green 2005; Shi et al. 2012). Ribosomal RNA was extracted from each preparation and used in reconstitution experiments.
50S particles were reconstituted using the two-step method of Nierhaus (Nierhaus 1990). A fourfold excess of TP50, obtained from E. coli strain AT9 (Reiner 1969), was incubated with control or ΔSRL rRNA for 25 min at 44°C in the presence of 4 mM Mg2+ (step 1). Then, the Mg2+ concentration was increased to 20 mM, and reactions were incubated at 50°C for 90 min (step 2). Reaction products were subjected to sucrose gradient sedimentation analysis (Fig. 1A). Virtually identical single peaks, migrating at 50S, were seen in both cases, suggesting highly efficient reconstitutions. Note that natural modifications of nucleotides in the PTC region of 23S rRNA are known to be critical for the efficient reconstitution of E. coli 50S subunits (Green and Noller 1996; Bao et al. 2024), implying that both control and ΔSRL rRNA carried these key modifications.

Loss of SRL inhibits assembly of 50S subunits in vivo but not in vitro. (A) Sucrose gradient traces of reconstituted control subunits (blue), reconstituted ΔSRL subunits (orange), and control 50S subunits isolated from cells (black). The term “control” is used here to specify the aptamer-tagged wild-type case. (B) PT activity of isolated and reconstituted 50S particles, containing or lacking the SRL (as indicated). Purified 30S subunits were used as a negative control (gray, as indicated). (C) Gain of PT activity during step 2 of the reconstitution protocol, for control and ΔSRL cases (as indicated). Shown are data from four independent experiments, and corresponding best-fit exponential curves. Observed rates (control, 0.022 ± 0.006 min−1; ΔSRL, 0.012 ± 0.006 min−1) are statistically indistinguishable. (D) PT activity of control and ΔSRL particles isolated from cells before and after incubation at 50°C in the presence or absence of excess TP50 (as indicated). Bars represent the mean ± SEM of three independent experiments, and individual data points (circles) are superimposed. Asterisks indicate P < 0.05, based on an unpaired two-tailed t-test. (ns) Not significant.
To assess the functional integrity of the reconstituted particles, we compared their PT activity (Fig. 1B). We found that reconstituted ΔSRL subunits are as active as the reconstituted control subunits and isolated control subunits. In contrast, ΔSRL particles isolated from cells exhibit much lower activity, ∼10% that of isolated control particles, in line with previous work (Lancaster et al. 2008). The fact that the rRNA of the reconstituted ΔSRL particles comes from the isolated ΔSRL particles strongly suggests the assembly defect conferred by ΔSRL is specific to the in vivo process.
Previous studies of control reconstitutions (Nikolay et al. 2018; Qin et al. 2023) have shown that step 1 results mainly in 48S particles, which lack PT activity but contain virtually all r-proteins of the large subunit. Step 2 entails rRNA rearrangements and complete folding of the PTC. To further evaluate our reconstitution reactions, we measured the rate with which PT activity is gained during the second incubation step (Fig. 1C). Aliquots were removed at various time points and flash-frozen in liquid nitrogen to stop assembly. Then, all samples were assayed in parallel for PT activity. The observed rates of assembly for the control (0.022 ± 0.006 min−1) and ΔSRL (0.012 ± 0.006 min−1) reactions were very similar, in fact statistically indistinguishable.
Ribosomal protein composition of isolated and reconstituted particles
To compare the protein composition of the isolated (affinity-purified) and reconstituted particles, we used SILAC (stable isotope labeling of amino acids in culture) and LC-MS/MS (see Materials and Methods). Isolated control and ΔSRL particles showed similar near-stoichiometric levels of most proteins (Fig. 2; Supplemental Table S1). One clear exception is uL16, which was underrepresented in ΔSRL particles compared to control particles. This result is generally consistent with Lancaster et al. (2008), although the degree of underrepresentation is smaller in our case. Levels of bL31 were substoichiometric (∼0.7) for both control and ΔSRL particles, probably due to the dissociation of this protein, which usually spans the subunit interface (Liu and Fredrick 2016), during the purification process. The reconstituted particles also showed similar levels of most proteins. In both control and ΔSRL cases, bL31, bL33, and uL16 were present at substoichiometric levels. Such low levels of L31 came as no surprise, based on previous work (Nikolay et al. 2018; Qin et al. 2023), but the reduced levels of uL16 and bL33 were unexpected. Both uL16 and bL33 bind late in the assembly process (Nikolay et al. 2018; Qin et al. 2023), so we suspect that our reaction conditions did not allow for the complete assembly of all particles. Importantly, no difference in the levels of these proteins was seen between the control and ΔSRL cases, consistent with equivalent PT activities of these reconstituted particles (Fig. 1B). In fact, despite substoichiometric uL16, the PT activities of the reconstituted particles were as high as the control isolated particles and much higher than the isolated ΔSRL particles. One protein, uL6, was specifically underrepresented in the ΔSRL reconstituted particles (Fig. 2). Protein uL6 binds near H95, and the C-terminal domain of the protein normally makes multiple contacts to the SRL.
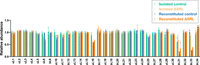
Ribosomal protein composition of control and ΔSRL particles isolated from cells or reconstituted in vitro. Shown are normalized isotope ratios, indicating the relative abundance of each 50S protein with respect to the spike-in standard. Bars represent the mean ± SEM of three independent experiments, and individual data points (circles) are superimposed. Green, isolated control; gold, isolated ΔSRL; blue, reconstituted control; orange, reconstituted ΔSRL.
Effect of heat and TP50 on isolated ΔSRL particles
Efficient reconstitution requires nonphysiological conditions, including high temperature, which can destabilize nonnative rRNA conformations and resolve kinetic traps. To determine whether ΔSRL particles isolated from cells could be rescued with respect to block 4 folding, we incubated these and control particles at 50°C for 90 min, with or without TP50 (in fourfold excess), under conditions akin to standard reconstitutions. The PT activity of these particles was then compared (Fig. 1D). ΔSRL particles isolated from cells gained a small amount of activity due to incubation at 50°C and gained slightly more activity when incubated at 50°C in the presence of TP50. However, the maximal activity observed was still low, ∼20% that of the control subunits. High-temperature incubation slightly enhanced the PT activity of the control particles, an effect deemed significant only in the absence of TP50 (Fig. 1D). These data suggest that the isolated ΔSRL particles correspond to kinetically trapped intermediates in which block 4 folding is inhibited in some way.
Cryo-EM analysis of isolated ΔSRL particles
We performed cryo-EM on ΔSRL particles isolated from cells to understand the basis of the functional deficiencies observed. Using image classification approaches (Supplemental Fig. S2), we found five distinct classes of particles that we named from class 1 to class 5. Their cryo-EM maps were refined to resolutions ranging from 2.6 to 2.9 Å (Supplemental Figs. S3 and S4). Density for the SRL at the tip of H95 was missing in the five maps (Supplemental Fig. S5A). Overall, this analysis showed that 50S assembly is strongly inhibited without the SRL, particularly with respect to the formation of the A site. The five classes observed can be arranged by assembly stage, from least mature to most mature (Fig. 3A). The structural motifs adopting their mature conformation in each transition from class 1 to class 5 are indicated in Figure 3A. Most of the particles belonged to classes 1 (26%) and 2 (42%), which lack multiple features. No density was seen for rRNA helices forming the A, P, and E functional sites or the uL1 and bL12 stalks in these particles, indicating that these elements remained dynamic. Class 1 cryo-EM map also lacked most of the densities for the rRNA helices and r-proteins forming the central protuberance, the base of the E site, and the interface region of the body below the E site. Structural elements forming the E (H68) and P (H93 and H71) sites were ordered in class 3 and class 4, which represented 18% and 10% of the total particles, respectively. Density for components of the A site (H38, H89, and H91-92) remained absent in these two classes. In fact, the A site was only visible in class 5, representing 4% of the total population (Fig. 3A).
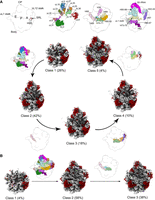
Cryo-EM structures of isolated pre-50S particles lacking or containing the SRL. (A) Cryo-EM maps representing the ΔSRL particles isolated from cells. The five classes of particles (classes 1–5) are arranged on a pathway of increasing complexity (bottom panel). In parentheses are the percentages of each class within the total population. The rRNA is shown in gray, and the r-proteins are shown in dark red. The diagrams of the 50S subunit shown between classes in the pathway indicate the structural motifs adopting their mature conformation in each transition. These structural motifs are color-coded according to the diagrams in the top panel. The left-most diagram in the top panel indicates the landmarks of the mature 50S subunit. (B) Cryo-EM maps representing the three classes of control particles isolated from cells, annotated as in A.
In contrast, control 50S particles adopted three different states that we named class 1 to class 3 (Supplemental Fig. S6). The cryo-EM maps for these three classes were refined to resolutions ranging from 2.6 Å to 4 Å (Supplemental Figs. S7 and S8). These maps confirmed that 50S assembly is more efficient when the SRL is present. However, immature particles still outnumbered mature particles, which might be a consequence of the artificially induced overexpression of aptamer-tagged rRNA. A substantial proportion of particles in the data set (38%) were fully mature (class 3) (Fig. 3B). Most of the remaining particles (58%) represented a late assembly intermediate (class 2) with the A site intact and only H68 of the E site and H69 and H71 disordered. Density for the SRL was visible in the cryo-EM map for classes 2 and 3 (Supplemental Fig. S5B). Only 4% of the total particles were less mature, equivalent in structure to class 1 particles in the ΔSRL data set (Fig. 3B). Importantly, we did not observe populations of particles equivalent to classes 2–4 of the ΔSRL data set, suggesting that the maturation of the A site is fast when the SRL motif is present. Overall, these results suggest that the assembly of the 50S subunit, and the A site in particular, depends on the SRL motif.
To further evaluate the cryo-EM data, we calculated difference maps between the cryo-EM map of the mature 50S subunit (control sample, class 3) and each of the maps obtained for the five classes of ΔSRL ribosomal particles isolated from cells (Supplemental Fig. S9). The resulting difference maps were consistent with progressive maturation from class 1 to class 5 as detailed above. No unexpected difference density, reflecting for example a unique conformational state or ancillary component, was seen in any of the classes.
Isolated ΔSRL particles are bound by multiple AFs
The A, P, and E sites of most particles isolated from ΔSRL cells were largely unformed (Fig. 3A), raising the question of whether one or more bound nonribosomal proteins prevent these particles from completing their maturation. To investigate this possibility, we used label-free quantification (LFQ) mass spectrometry to compare the samples of affinity-purified control and ΔSRL particles (Fig. 4; Supplemental Table S1). Many nonribosomal proteins were identified in both cases, although the overall level of associated protein was substantially higher in the ΔSRL case (Fig. 4A, inset). The main panel of Figure 4A shows all nonribosomal proteins present at stoichiometries >0.01 in either case. Most of these proteins are overrepresented in ΔSRL particles compared to the control (Fig. 4B), and many have been previously implicated in ribosome biogenesis and/or degradation (El Hage et al. 2001; Charollais et al. 2003, 2004; Jiang et al. 2006; Barkan et al. 2007; Jagessar and Jain 2010; Sulthana and Deutscher 2013; Ni et al. 2016; Andrade et al. 2018; Dos Santos et al. 2020; Ero et al. 2024). These include the DeaD-box helicases DeaD and SrmB, the pseudouridine synthases RluC and RluB, the 3′-to-5′ exoribonucleases RNase R and PNPase, the rRNA methylases RlmN and RlmE, the RNA-binding protein YhbY, the RNA chaperone Hfq, the protein chaperone GroEL, and the protease Lon. The cold-shock proteins CspC and CspE, which are known to bind RNA and to be expressed at 37°C (Benhalevy et al. 2015), were also overrepresented in the ΔSRL samples. RNA polymerase subunits (RpoC, RpoA, RpoB) were also overrepresented, as were other proteins with no obvious affiliation with ribosomes, such as components of the pyruvate dehydrogenase complex (AceF, AceE, and LpdA). Relatively few nonribosomal proteins were underrepresented in ΔSRL samples, including trigger factor (TF) and IF3, proteins known to interact with mature ribosomes. Ribosome silencing factor (RsfS), which may regulate ribosome activity during growth-state transitions and contribute to late-stage ribosome biogenesis (Hauser et al. 2012; Nikolay et al. 2021), was also underrepresented in ΔSRL samples. Collectively, these data suggest that isolated ΔSRL particles represent a heterogeneous mix of trapped assembly intermediates, some on route to degradation. The incomplete folding of the A, P, and E sites in these particles is likely due to one or more bound AFs, which fail to dissociate in the absence of the SRL.
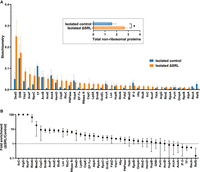
Nonribosomal proteins associated with affinity-purified 50S particles. (A) Shown are the predicted stoichiometries of each nonribosomal protein detected at a level of >0.01 for either control (blue) or mutant (orange) particles. Bars represent the mean ± SEM of three biological replicates. Inset shows the collective abundance of all nonribosomal proteins bound. Units represent ribosome stoichiometry, where 1.0 corresponds to the median r-protein value. Thus, isolated control subunits contain ∼1.5 nonribosomal proteins on average, while isolated ΔSRL particles contain ∼2.7. Based on a two-tailed paired t-test, P < 0.05. (B) For the same 41 nonribosomal proteins, fold enrichment (ΔSRL/control) values are shown. Data represent the quotient of two means ± standard error. For IlvC, ArgG, and NuoF, no protein was detected in any of the control sample replicates, so an enrichment value of 100 was assigned arbitrarily.
Effect of ΔSRL on EF-Tu-dependent decoding
In 2012, Joseph and coworkers isolated and characterized ribosomes with G2655C, A2660U, or ΔU2653ΔC2667 in the SRL (Shi et al. 2012). These mutations had surprisingly small effects on EF-Tu-dependent decoding. Even the latter mutation, which removes a base pair near the center of H95 and is predicted to rotate and reorient the SRL, conferred only moderate effects. While ΔU2653ΔC2667 decreased the affinity of the ternary complex (EF-Tu.GTP.aa-tRNA) by 16-fold, this mutation reduced the maximal rate of GTP hydrolysis (kGTP) by only approximately fourfold and reduced the overall rate of decoding (kpep) by only approximately twofold. The authors hypothesized that either (i) the interaction between the phosphate group of A2662 and His84 of EF-Tu is dispensable for GTPase activation or (ii) another phosphate group can substitute for that of A2662 in the context of truncated SRL. However, they could not readily distinguish these possibilities using affinity-purified ribosomes, because larger deletions of the SRL inhibit 50S assembly in the cell (Shi et al. 2012).
To help clarify the role of the SRL in EF-Tu function, we compared reconstituted control and ΔSRL ribosomes in single-turnover decoding assays (Fig. 5). Reconstituted or isolated 50S particles were incubated with 30S subunits, f-[35S]-Met-tRNA, and message m293 (Shoji et al. 2006) (codon 2 = UAC) at 37°C to nonenzymatically form 70S initiation complexes (70S ICs). Initiation factors were intentionally omitted, as the inclusion of IF2 (which also interacts with the SRL) would complicate interpretation of the results. To assess these 70S ICs, puromycin was added, and the extent of fMet-puromycin (fMet-puro) formation was compared (Fig. 5A). Levels of fMet-puro were similar for the reconstituted control and ΔSRL cases (47% and 48%, respectively), somewhat lower for the isolated control case (32%), and considerably lower for the isolated ΔSRL case (4%). These data are reminiscent of Figure 1B, which reports PT activities of 50S particles themselves, suggesting that mRNA, tRNA, and 30S subunits do little to enhance the function of ΔSRL isolated particles. Next, we measured decoding in the four cases. EF-Tu·GTP·Tyr-tRNA was added at time t = 0, aliquots were removed and quenched at various time points, and reaction products were resolved and quantified (Fig. 5B,C). In the isolated and reconstituted control cases, formation of fMet-Tyr was rapid, too fast to measure manually. The reaction amplitudes were 29% and 47%, respectively (Fig. 5C), consistent with the extent of fMet-puro formation (32%, 47%) independently observed (Fig. 5A), implying efficient delivery of Tyr-tRNA to the A site by EF-Tu in both cases. In contrast, very little fMet-Tyr formation was seen in the ΔSRL cases (Fig. 5B,C), with reaction amplitudes of 2.9% (isolated) and 8.6% (reconstituted). A slow increase in dipeptide was apparent in the reconstituted ΔSRL case, which we reasoned could be due to factor-independent decoding. To evaluate this, all reactions were repeated in the absence of EF-Tu under otherwise identical conditions (Fig. 5D). For the control cases, removal of EF-Tu dramatically reduced the reaction rate and amplitude, as expected. For the ΔSRL cases, the data looked virtually identical in the presence or absence of EF-Tu (compare orange curves of Fig. 5C vs. 5D). Collectively, these data indicate that removal of the SRL abolishes EF-Tu-dependent decoding.
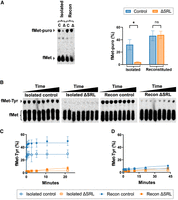
EF-Tu function depends on the SRL. (A) Evaluation of 70S ICs formed with isolated or reconstituted (recon) 50S particles (as indicated), as determined by puromycin reactivity. (C) control; Δ, ΔSRL. A representative experiment is shown on the left, and results of three independent experiments (mean ± SEM) are shown on the right. (*) P < 0.05, (ns) not significant. (B,C) Measures of EF-Tu-dependent decoding in control and mutant 70S ICs, as indicated. (B) Representative raw data showing fMet-Tyr formation as a function of time. (C) Plots of fMet-Tyr formation versus time, for various 70S ICs, as indicated in the key. Data points represent mean ± SEM values of three independent experiments. In some cases, error bars are smaller than (and hidden by) the symbols. Curves represent single-exponential fits of the data. (D) Data from analogous reactions that lack EF-Tu.
DISCUSSION
In this work, we investigate the mechanism by which the SRL contributes to the assembly of the 50S subunit. We find that 23S rRNA lacking the SRL supports efficient reconstitution in vitro, yet the same ΔSRL rRNA forms incompletely assembled particles in vivo. The ΔSRL particles isolated from cells lack PT activity and fail to gain activity when exposed to high temperature, high Mg2+ concentration, and TP50 (i.e., optimal conditions of reconstitution). These data strongly suggest that the problem caused by ΔSRL is specific to the in vivo pathway of assembly. LFQ analysis of particles isolated from cells shows that nonribosomal proteins are substantially overrepresented in the ΔSRL case. These proteins include DeaD, SrmB, RluB, RluC, RlmN, Hfq, and YhbY—AFs known to participate in 50S biogenesis. We propose that one or more SRL-dependent GTPases normally catalyze the release of AFs during 50S assembly in the cell. Thus, when these GTPases cannot act, AFs get stuck on the pre-50S particles, resulting in the collection of trapped intermediates that we observe.
Five GTPases have been implicated in 50S biogenesis—Der, YihA, ObgE, BipA, and IF2. Structural studies have shown that Der binds the subunit well away from the SRL. Der occupies the 50S E and P sites, occluding tRNA in both sites (Zhang et al. 2014). YihA's binding site is still unknown; however, structures of mitochondrial pre-50S intermediates show the homologous protein, mt-EngB, interacting “below” mt-EngA (Der), also well away from the SRL (Jaskolowski et al. 2020). The latter three GTPases—ObgE, BipA, and IF2—all interact with the SRL. The trGTPases BipA and IF2 bind the ribosome similarly (Kumar et al. 2015; Sprink et al. 2016; Kaledhonkar et al. 2019; Basu et al. 2022), with contacts between the switch motifs of the G domain and the SRL. The ObgE binding site spans from the 50S A site to the base of the bL12 stalk (Feng et al. 2014; Nikolay et al. 2021). The G domain of ObgE contacts the SRL in a unique way, using residues distal from the switch motifs. Based on these observations, we hypothesize that ObgE, BipA, and/or IF2 normally promote the release of various AFs during 50S assembly, and these steps are blocked by the removal of the SRL.
The idea that SRL-dependent GTPases catalyze the release of AFs is consistent with previous genetic data from the Flower laboratory (Krishnan and Flower 2008; Choudhury and Flower 2015). Deletion of bipA (ΔbipA) in E. coli inhibits 50S biogenesis and cell growth at suboptimal temperatures (Choudhury and Flower 2015; Gibbs et al. 2020). These phenotypes are suppressed by the loss of RluC or its enzymatic activity (Krishnan and Flower 2008). RluC targets three nucleotides of 23S rRNA—U955, U2504, and U2580—converting each to pseudouridine. Triple mutation of all three RluC targets also suppresses the phenotypes of ΔbipA, whereas single mutations at each position fail to do so (Krishnan and Flower 2008). Thus, the phenotypes conferred by the loss of BipA clearly involve RluC. We envision that BipA normally promotes RluC release, so RluC gets stuck on pre-50S particles in the absence of BipA, causing the ΔbipA phenotypes. Secondary loss of RluC resolves the issue, as does eliminating RluC's activity or targets. Notably, RluC is one of the nonribosomal proteins associated with ΔSRL particles (Fig. 4), which may reflect BipA's inability to act. However, ΔbipA phenotypes are seen only at low temperatures (e.g., 20°C), and our experiments were performed at 42°C, suggesting that other SRL-dependent GTPases also contribute to AF release.
A growing body of evidence suggests that IF2 contributes to ribosome biogenesis. Brown and coworkers showed that the antibiotic lamotrigine (Ltg) strongly inhibits the growth of E. coli at low temperature and causes accumulation of pre-30S and pre-50S particles (Stokes et al. 2014). Ltg targets IF2 (residues 189–203) and binds the protein in a guanosine-nucleotide-dependent manner. Ltg appears to specifically inhibit ribosome biogenesis since the compound fails to impact translation in vitro or in vivo (Stokes et al. 2014). Independent work showed that mutations ΔN and E571K of IF2 (which truncate the factor or ablate its GTPase activity) inhibit ribosome biogenesis and growth, particularly at low temperature (Brandi et al. 2019). Another study (Campbell and Brown 2008) showed that overexpression of IF2 can partially compensate for the loss of RsgA, a small GTPase involved in 30S biogenesis. RsgA catalyzes the release of another factor, RbfA, during late-stage ribosome biogenesis (Goto et al. 2011; Guo et al. 2011; Lopez-Alonso et al. 2017). Interestingly, Ltg can also suppress phenotypes of the ΔrsgA strain, effects that depend on rbfA (Singh et al. 2024). It was proposed that excess IF2 or Ltg-stabilized IF2 can displace RbfA and hence functionally substitute for RsgA (Singh et al. 2024). Collectively, these findings suggest that IF2 contributes to ribosome biogenesis and may do so by helping to promote AF release.
ObgE is an essential protein strongly implicated in 50S biogenesis. Structures of ObgE bound to pre-50S particles and mature 50S subunits have been determined (Feng et al. 2014; Nikolay et al. 2021). In all these structures, ObgE spans from the PTC to the SRL, occupying a site that overlaps that of BipA. The N-terminal domain of ObgE reaches into the 50S A site, while the G domain lies between the bL12 stalk base and the SRL. ObgE directly contacts the SRL, but in a manner distinct from the trGTPases. Residues 198–202 of the G domain, distal from the switch motifs, interact with the opposite side of the SRL. These contacts are probably important for factor function, even if they are unnecessary for GTPase activation. Strains with mutations that compromise ObgE function accumulate pre-50S intermediates, which lack uL16 (Jiang et al. 2006). Affinity purification and cryo-EM analysis of ObgE-bound native complexes revealed similar pre-50S intermediates lacking L16 and rRNA elements of PTC and CP regions. These ObgE-bound complexes also contained RsfS and, in more mature intermediates, RluD and YjgA. Based on these and additional findings, it was proposed that ObgE facilitates late-stage rRNA folding, in concert with the other AFs. Interestingly, pre-50S particles isolated from mutant cells defective in ObgE function showed overrepresentation of DeaD, RluB, and YhbY and underrepresentation of TF (Jiang et al. 2006). Our current work shows that ΔSRL particles isolated under similar (1 mM Mg2+) conditions also contain elevated levels of DeaD, RluB, and YhbY, and reduced levels of TF. These observations suggest the assembly defect conferred by ΔSRL may be due, at least in part, to the inability of ObgE to function normally.
Our cryo-EM analysis reveals the consequences of SRL loss at high resolution. Four classes of pre-50S particles accumulate, none of which exhibit an ordered A site. The functional A, P, and E sites are unformed in classes 1–3 (88% of the total population), and rRNA nucleotides invisible in these maps correspond remarkably well to nucleotides found to be hyperreactive to chemical probes in analogous ΔSRL particles (Supplemental Fig. S10A–D; Lancaster et al. 2008). The disordered rRNA regions also correspond well to assembly blocks 4 and 5 (Supplemental Fig. S10E; Davis et al. 2016), modules of the subunit believed to fold late in the assembly process. These observations suggest that most ΔSRL particles are blocked at a late stage of 50S biogenesis.
Notably, none of the cryo-EM maps obtained from the isolated ΔSRL particles showed any additional density that could be assigned to one of the nonribosomal proteins. This can be explained by the low stoichiometry (below 0.1 in most cases) of these proteins (Fig. 4). Structural determination of macromolecular assemblies in cryo-EM relies on averaging thousands of particle images representing the same structure. In this case, the existence of a maximum of 10% of the particles in the data set with a specific nonribosomal protein bound caused these densities to be averaged out in the process of generating the 3D structure. The spreading of the particles containing the nonribosomal protein bound among the five different classes in the population further compounds this issue. Observing the density of these nonribosomal proteins bound to the ribosome assembly intermediates would require tagging of one of these factors at its native locus with FLAG- or StrepII tags in the ΔSRL cells and subsequent affinity purification of these particles (Nikolay et al. 2021). The much higher occupancy of these particles with the tagged nonribosomal protein compared to our samples in this study would likely reveal a density for this factor when observed by cryo-EM.
Finally, using reconstituted ΔSRL subunits, which contain a fully functional PTC, we revisited the question of SRL's role in decoding. We found that removal of the SRL abolishes EF-Tu-dependent decoding, indicating that EF-Tu-SRL contacts are crucial for factor function. Much smaller effects were conferred by mutations G2655C, A2660U, or ΔU2653ΔC2667. Presumably, those mutations do not fully disrupt EF-Tu-SRL interaction. In the case of ΔU2653ΔC2667, another phosphate group may indeed substitute for that of A2662, contact His84, and enable GTPase activation, as hypothesized previously (Shi et al. 2012).
MATERIALS AND METHODS
Purification of aptamer-tagged 50S particles
A mutation that removes the SRL was introduced into plasmid p278MS2, which contains rrnB encoding an aptamer-tagged 23S rRNA (Youngman and Green 2005), using Phusion Site Directed Mutagenesis (Thermo Scientific). The ΔSRL mutation replaces nt 2653–2667 of 23S rRNA with four pyrimidines (CUUU), analogous to the deletion mutation described previously (Lancaster et al. 2008), which replaces the same nucleotides with GAAA. The CUUU substitution was made inadvertently, a rookie mistake discovered months after the fact. However, subsequent work showed that expression of either mutant rRNA (Δ2653–2667::CUUU or Δ2653–2667::GAAA) confers the same dominant negative growth phenotype, and reconstituted particles formed from either mutant rRNA show full PT activity (Supplemental Fig. S11).
Aptamer-tagged control and mutant (ΔSRL) 50S particles were purified as described (McGarry et al. 2005; Youngman and Green 2005; Liu and Fredrick 2013). In brief, POP2136 recA (F- glnV44 hsdR17 endA1 thi-1 aroB mal− cI857 lambda PR recA56 srlD3000::Tn10) cells harboring p278MS2 (control) or its derivative p278dSRL (ΔSRL) were grown at 30°C in LB medium containing 100 μg/mL ampicillin (Amp) until mid-logarithmic phase and then diluted fivefold with pre-warmed fresh media, and grown for two more hours at 42°C. Cells were harvested and lysed by French press. The clarified lysate was layered onto a 10 mL sucrose cushion (38%) in buffer A (20 mM Tris-HCl pH 7.1, 10.5 mM MgCl2, 500 mM NH4Cl, 0.5 mM EDTA, 6 mM 2-mercaptoethanol), and total ribosomes were pelleted by centrifugation (100,000g for 21 h) at 4°C. Crude ribosomes were dissolved in buffer B (50 mM Tris-HCl pH 7.1, 1 mM MgCl2, 100 mM NH4Cl, 6 mM 2-mercaptoethanol), and dialyzed against 0.3 L of the same buffer for 1 h, three times. The 50S particles were then affinity purified in buffer B, using a GSTrap FF column, preloaded with GST-MS2 fusion protein. Glutathione (10 mM) was used to elute the column, and Amicon ultra (100,000 MWCO, Millipore) filtration units were used to concentrate the fractions containing ribosomal subunits and to exchange the buffer. Depending on the subsequent step, the buffer was either changed to buffer C (50 mM Tris-HCl pH 7.1, 10 mM MgCl2, 100 mM NH4Cl, 6 mM 2-mercaptoethanol) or buffer D (50 mM Tris-HCl pH 7.5, 400 mM KOAc, 20 mM MgCl2).
Wild-type ribosomal subunits were purified from strain AT9 (Reiner 1969) via conventional sedimentation methods as described (Qin et al. 2007). Homogeneous aptamer-tagged ribosomes were similarly purified from the Δ7 prrn strain KLF2066 (Shoji et al. 2009), which harbors p278MS2 as the only source of rRNA.
Evaluating the purity of aptamer-tagged particles
We used poisoned primer extension, essentially as described (Abdi and Fredrick 2005), to measure the purity of tagged subunits. Briefly, a labeled oligonucleotide (5′-[Cy5]-TCAACGTTCCTTCAGGACCCT-3′; 0.05 µM) was annealed to 23S rRNA (0.2 µM rRNA extracted from 50S particles) in 10 µL of 50 mM K-HEPES (pH 7.6),100 mM KCl. Then, 10 µL of AMV reverse transcriptase (three units, New England Biolabs), dATP (300 µM), dGTP (300 µM), dTTP (300 µM), and ddCTP (300 µM) in 2X AMV RT buffer (New England Biolabs) was added. Reactions were incubated at 42°C for 10 min, and products were resolved by 8% denaturing PAGE. Gels were imaged using a Typhoon 5 (Cytivia), and data were quantified using ImageQuant software (Cytivia).
We also used an RNase H assay to assess the purity of tagged subunits. In a 10 µL reaction, oligonucleotide #2685 (5′-ATGACAACCCGAACACC-3′) was annealed to rRNA (1 µg) in 10 mM Tris-HCl (pH 8), 100 mM KCl, 1 mM EDTA. The primer-annealed RNA was then treated with RNase H (two units, New England Biolabs), as recommended by the manufacturer. Reaction products were resolved by 8% denaturing PAGE, stained with SYBR Gold (Invitrogen), and quantified as described above.
50S subunit reconstitutions
Total protein of the 50S subunit (TP50) was obtained from conventionally purified 50S subunits of strain AT9 as described (Nierhaus 1990). Wild-type and ΔSRL rRNA were extracted from affinity-purified control and mutant 50S particles, respectively, as described (Nierhaus 1990). Reconstitutions were performed essentially as described (Nierhaus 1990). Briefly, rRNA (5S and 23S; 2 A260 units) was incubated with fourfold excess TP50 (8 equivalent units) in 100 μL buffer E (20 mM Tris-HCl [pH 7.5], 4 mM Mg(OAc)2, 400 mM M NH4Cl, 0.2 mM EDTA, 5 mM 2-mercaptoethanol) for 25 min at 44°C (step 1). Next, the Mg(OAc)2 concentration was increased to 20 mM, and the reaction was incubated at 50°C for 90 min (step 2). Reaction products were resolved by sucrose gradient sedimentation, and 50S particles were collected. Amicon ultra filtration units (100,000 MWCO, Millipore) were used to concentrate the particles and exchange the sucrose-containing buffer for buffer C or D.
Isolated particle rescue experiments
Isolated control and ΔSRL particles were subjected to conditions of step 2 of the reconstitution protocol. Briefly, subunits (0.6 µM) were incubated in buffer F (20 mM Tris-HCl pH 7.5, 20 mM Mg(OAc)2, 400 mM M NH4Cl, 0.2 mM EDTA, 5 mM 2-mercaptoethanol) for 90 min at 50°C in the absence or presence of fourfold excess TP50. Amicon filtration units (100,000 MWCO, Millipore) were then used to exchange buffer F for buffer D.
Puromycin reactivity assay
To measure PT activity of 50S particles, the puromycin-based “fragment reaction” (albeit with full-length fMet-tRNA) was used (Monro 1967). 50S subunits (0.6 µM) were combined with f-[35S]-Met-tRNA (0.6 µM) and puromycin (125 µM) in 25 µL buffer D at 4°C. Then, methanol (33%) was added to start the reaction, and aliquots were removed at various time points and quenched with KOH (250 mM). The product f-[35S]-Met-puromycin was extracted with ethyl acetate and quantified using a scintillation counter. Initial experiments showed that, under these conditions, the reaction reaches completion within ∼30 sec. Throughout this paper, we report the extent of product formation at 1 min, which should reflect the amplitude of the PT reaction.
SILAC analysis
For SILAC analysis, isotopically labeled polysomes were used as the spike-in standard (Gibbs et al. 2020). Strain MRG01 (Gibbs et al. 2017), a Lys− Arg− auxotroph, was grown in 50 mL enriched M9 minimal media with glucose and labeled arginine (Arg 13C6) and lysine (Lys 2H4) to mid-log phase, the culture was rapidly chilled by pouring over crushed ice, and cells were pelleted and lysed via a lysozyme freeze-thaw method. The lysate was clarified and subjected to sucrose gradient sedimentation to isolate polysomes (Qin and Fredrick 2013). Polysome fractions were pooled, and Amicon ultra filtration units (100,000 MWCO, Millipore) were used to concentrate the particles and exchange the sucrose-containing buffer for buffer C. Equivalent amounts of these labeled polysomes were added to each of the isolated control, isolated ΔSRL, reconstituted control, and reconstituted ΔSRL particles. Proteins were precipitated using trichloroacetic acid (TCA) at a final concentration of 13% (w/v). The protein pellets were boiled in 4% SDS in 100 mM Tris-HCl (pH 8.0), reduced and alkylated at 95°C for 5 min in 10 mM Tris(2-Carboxyethyl)Phosphine hydrochloride (TCEP) and 40 mM 2-chloroacetamide, and run on a short 10% SDS-PAGE gel. The proteins were digested out of the gel using two additions of trypsin with a total 20 h incubation at 37°C. The resulting peptides were cleaned with C-18 STop And Go Extraction (STAGE) tips (Ishihama et al. 2002) using 40% (v/v) acetonitrile in 0.1% (v/v) formic acid as the elution buffer. The peptides were resuspended in 0.5% acetonitrile, 0.1% formic acid in MS grade water, and peptide concentration was estimated by UV absorbance at 205 nm using a NanoDropOne (Thermo Scientific, A205nm scopes). Approximately 100 ng of peptides were loaded onto Thermo Orbitrap Exploris 480 coupled to EASY nLC 1200 (Thermo Scientific) with IonOpticks’ Aurora Series 25 cm × 75 μm C18 1.6 μm column heated to 40°C, using a column oven controlled by Sonation COControl version 3.4.8. (sonation.com). Solvent A consisted of 2% acetonitrile and 0.1% formic acid; solvent B consisted of 80% acetonitrile and 0.1% formic acid. Forty-five minutes of separation was set from 3% to 25% B and an additional 15 min to reach 35% B before the necessary column wash. LC flow rate was set at 250 nL/min. Instrument polarity was set to positive mode; the spray voltage was set to 1900 V, the ion transfer tube temperature was set to 290°C, and expected peak width was set to 15 sec. RF lens was set to 50% and FAIMS was not enabled with data acquisition. Data dependent mode was set for 20 scans and the Orbitrap resolution was set to 120,000 for full MS and 15,000 for fragment MS, with AGC target set to 100% with 20 msec injection time (IT) for full MS and 50% with auto IT for fragment MS. Profile data type was acquired at full MS level, and centroid at fragment MS level. Intensity threshold for fragmentation was set at 8000, isolation window for fragmentation at 2 m/z, with normalized collision energy at 28%. Dynamic exclusion was enabled to exclude after one time for 45 sec.
The data was searched with MaxQuant version 2.4.3.0 (Tyanova et al. 2016), using UniProt's E. coli proteomes (UP000002032 and/or UP000000625) with common contaminant sequences. The search setting was set with variable modifications of oxidation of methionines and acetylation of protein N-termini, and fixed modification of carbamidomethylation of cysteines. SILAC labels of arginine (13C6) and lysine (2H4) were set for quantitation enabling iBAQ, requantify, match-between-runs, and label-free quantitation options. The protein and peptide false discovery rates were set to 1%.
Isotope ratios (H/L, corresponding to standard/sample) of all large subunit ribosomal proteins were obtained from the MaxQuant data file. To convert these to sample/standard ratios, the reciprocal of each value was calculated. Then, these sample/standard ratios were normalized with respect to the median ratio for each sample.
LFQ analysis
Affinity-purified particles were TCA precipitated, and peptides were prepared from the pellets as detailed in the SILAC section above. Approximately 75 ng of peptides were loaded onto timsTOF Pro 2 (Bruker Daltonics) coupled to NanoElute UHPLC (Bruker Daltonics) using Aurora Series Gen 25 cm analytical column (IonOpticks). The CaptiveSpray ionization source was operated at 1800 V capillary voltage, 3 L/min drying gas, and 180°C drying temperature. The analytical column was heated to 50°C using a column toaster M (Bruker Daltonics). The NanoElute thermostat temperature was set at 7°C. Solvent A consisted of 0.5% acetonitrile and 0.1% formic acid in MS grade water; solvent B consisted of 0.1% aqueous formic acid and 0.5% water in acetonitrile. Before each run, the analytical column was conditioned with four column volumes of solvent A and the analysis was performed at 0.30 μL/min flow rate. A 30 min gradient was run from 2% B to 12% B over 15 min, then to 33% B from 15 to 30 min, then to 95% B over 0.5 min, held at 95% B for 7.72 min.
During analysis, the timsTOF Pro 2 was operated with Parallel Accumulation-Serial Fragmentation (PASEF) scan mode for DDA acquisition. The MS and MS/MS spectra were collected in positive mode, from m/z 100 Th to m/z 1700 Th, and from ion mobility range (1/ K0) 0.7 V*sec/cm2 to 1.35 V*sec/cm2. A polygon filter was applied to the mass-to-charge and the ion mobility plane to include the most likely peptide precursors and to reduce singly charged background ions.
TIMS-MS scan was set at equal ramp time and accumulation time of 100 msec, at a rate of 9.42 Hz (100% duty cycle). Active exclusion was enabled with a 0.4 min release and reconsidered if their intensity increased more than 4 times. For each TIMS cycle, five PASEF MS/MS scans were recorded (total cycle time 0.64 sec). Target intensity for parent ions was set to 10,000 cts/sec with a threshold of 1000 cts/sec. Isolation widths were set at 2.07 m/z starting m/z 400 Th and 3.46 m/z ending m/z 1000 Th. The collision energy was ramped linearly as a function of mobility value from 27 eV at 1/k0 = 0.7 V·sec/cm2 to 55 eV at 1/k0 = 1.35 V·sec/cm2.
The data were searched with MaxQuant version 2.4.3.0 (Tyanova et al. 2016), using UniProt's E. coli proteomes (UP000002032 and UP000000625) with common contaminant sequences. The search setting was set with variable modifications of oxidation of methionines and acetylation of protein N-termini, and fixed modification of carbamidomethylation of cysteines. Label-free quantitation, match-between-runs, and iBAQ modes were enabled and the protein and peptide false discovery rates were set to 1%.
LFQ intensity values were obtained from the unlabeled (L) peptides of SILAC samples (replicate 1 and 2) or from samples without any isotopic labeling (replicate 3). For each sample, LFQ intensity values were normalized with respect to the median of the 50S r-proteins.
Dipeptide formation assays
Dipeptide formation was manually measured in single-turnover decoding assays, essentially as described (Liu and Fredrick 2015). 70S ICs were formed nonenzymatically. First, 30S subunits were activated by incubation at 42°C for 20 min in the presence of 20 mM Mg2+. Then, activated 30S subunits (0.2 μM) were incubated with f-[35S]-Met-tRNA (0.15 μM) and m293 mRNA (0.5 μM) at 37°C for 20 min in buffer G (50 mM Tris-HCl pH 7.5, 30 mM KCl, 70 mM NH4Cl, 15 mM MgCl2, 1 mM DTT, 1 mM GTP), 50S subunits (0.4 μM) were added, and the reaction was further incubated for 10 min. The Mg2+ concentration was then adjusted to 7.5 mM. To form the ternary complex, EF-Tu (5 μM) was incubated at 37°C with Tyr-tRNATyr (1 μM), phosphoenolpyruvate (2 mM), and pyruvate kinase (50 μg/mL; Sigma) in buffer H (50 mM Tris-HCl pH 7.5, 30 mM KCl, 70 mM NH4Cl, 7 mM MgCl2, 1 mM DTT, 1 mM GTP) for 5 min. To measure decoding, equal volumes of 70S IC and ternary complex (preequilibrated at 25°C) were mixed at time t = 0, and aliquots were removed at various times and quenched with KOH (250 mM). Reaction products were resolved using electrophoretic TLC and quantified using a phosphor imager (McClory et al. 2014).
In parallel, the functional integrity of each 70S IC was measured. In brief, before [Mg2+] adjustment, a portion of the reaction (3 μL) was combined with puromycin (125 μM) and further incubated at room temperature for 25 min. One volume of 500 mM KOH was added, and f-[35S]-Met-puromycin was extracted with ethyl acetate and quantified using a scintillation counter.
Cryo-electron microscopy
To prepare the cryo-EM grids, purified ribosomal particles isolated from ΔSRL and wild-type cells were diluted to a concentration of 250 nM in buffer C (pH 7.5). Then, 3.6 μL of these dilutions was applied directly to the grid.
Samples were deposited in holey grids (c-flat CF-2/2–2C-T) where we previously had a continuous layer of thin (2–5 nm) carbon. Before applying the samples, grids were washed overnight in chloroform and treated with glow discharged in air at 5 mA for 20 sec. The grids were vitrified in liquid ethane using a Vitrobot Mark IV (Thermo Fisher Scientific). Each grid was blotted once for 2 sec and a blot force of +1. The temperature in the Vitrobot chamber was set at 25°C, and relative humidity was equilibrated at 100% humidity for 30 min before plunging started. All data sets were collected at FEMR-McGill using a Titan Krios microscope at 300 kV equipped with a Gatan BioQuantum LS K3 direct electron detector. The software used for data collection was SerialEM (Schorb et al. 2019). Images were collected in counting mode according to the parameters described in Supplemental Tables S2 and S3.
Image processing
All image processing steps were done using CryoSPARC v4 (Punjani et al. 2017). All frames in the cryo-EM movies were corrected using beam-induced motion to produce a merged micrograph. In the Patch Motion Correction job, we used default settings that included using information up to 5 Å resolution when aligning frames, a B-factor of 500, and a 0.5 calibrated smoothing constant applied to the trajectories. CTF parameter estimation was done using Patch CTF estimation using default parameters. The minimum and maximum resolution considered to estimate the CTF parameters were 25 Å and 4 Å, and the minimum and maximum defocus values were set up at 1000 and 50,000 Å. Images with an estimated resolution of 6 Å or better were kept for further processing. The curated set of micrographs was split into exposure groups using Exposure group utilities.
In all data sets, the particle picking on the selected micrographs was done in two steps: 500 randomly selected micrographs were first picked using a Blob Picker, and the selected particles were used to generate templates for subsequent template picking in all micrographs. Blob Picker was done using a circular blob and a minimum and maximum particle diameter of 256 Å and 290 Å. Maximum resolution considered in the micrographs was 20 Å. The angular sampling used was 5°, and the distance between particles in units of particle diameter was 0.5. Picked particles were extracted using a box size of 420 pixels. Particle curation after Blob Picker and template picking was done using two rounds of 2D classification. In the 2D classification step, 50 classes were requested, the circular mask diameter was 382 Å, and we used the default parameters for the rest of the settings. Before the final curated particles were used for 3D reconstructions, we performed Local Motion Correction using default settings that included maximum alignment resolution of 5 Å, B-factor during alignment of 500, smoothing lambda of 20, and spatial smoothing standard deviation of 400 Å. Next, ab initio reconstruction was used to generate an initial 3D model directly from the local motion-corrected particles without requiring an input model using the job default settings. The obtained volume was used as the initial reference for a Non-Uniform Refinement using default settings, C1 symmetry and with optimized per-particle defocus, optimized per-group CTF parameters, fit Spherical Aberration, fit Tetrafoil, and Fit Anisotropic Mag turned on. This 3D reconstruction was used to obtain a consensus cryo-EM map before going into 3D particle classification. Particle heterogeneity was explored in all data sets through 3D classification analysis using particle images in the original size (0.855 Å/pixel, 420-pixel box size). The 3D classification analysis routines were always run, requesting three classes, target resolution of 5 Å, and number of O-EM epochs 5. All other settings were set at default values. The number of iterative hierarchical 3D classifications requested varied between experiments and ranged from three to five. The resulting maps from the exhaustive 3D classification were visually inspected in Chimera. High-resolution refinements were then performed for each class using the particle numbers indicated in Supplemental Tables S2 and S3. The particles from each class were first subjected to ab initio reconstruction with the same parameters as described above. The resulting maps were used as the initial model for Non-Uniform Refinements run in the same manner as explained above for the consensus refinement. Average resolution estimation and local resolution analysis were done with cryoSPARC using the gold-standard approach (Henderson et al. 2012). Cryo-EM map visualization was performed in UCSF Chimera and Chimera X (Pettersen et al. 2004, 2021).
Difference map analysis was conducted using the Relion image handler command from RELION-5 (Zivanov et al. 2022). First, both maps were low-pass filtered to 20 Å and adjusted to a similar power spectrum. Subsequently, the two maps were subtracted to generate the difference map. Visualization was carried out in Chimera X (Pettersen et al. 2021).
DATA DEPOSITION
The cryo-EM maps obtained in this study have been deposited in the Electron Microscopy Data Bank (EMDB), and the accession codes are detailed in Supplemental Tables S2 and S3. The mass spectrometry proteomics data have been deposited to the ProteomeXchange Consortium via the PRIDE (Perez-Riverol et al. 2022) partner repository with the data set identifier PXD056260 (www.ebi.ac.uk/pride/archive).
SUPPLEMENTAL MATERIAL
Supplemental material is available for this article.
ACKNOWLEDGMENTS
We thank S. Joseph for suggesting the decoding experiments; J. Jackman for helpful discussion; and H. Noller, L. Lancaster, and B. Warner for comments on the manuscript. This work was supported by grants from the National Institutes of Health (GM072528 to K.F.) and the Canadian Institutes of Health Research (PJT-180305 to J.O.). Cryo-EM data were collected at the Facility for Electron Microscopy Research (FEMR) at McGill. FEMR is supported by the Canadian Foundation for Innovation, Quebec Government and McGill University. The Centre de Recherche en Biologie Structurale at McGill University is funded by the Fonds de Recherche du Québec—Santé (FRQS).
Footnotes
-
Article is online at http://www.rnajournal.org/cgi/doi/10.1261/rna.080335.124.
-
Freely available online through the RNA Open Access option.
- Received November 22, 2024.
- Accepted January 11, 2025.
This article, published in RNA, is available under a Creative Commons License (Attribution-NonCommercial 4.0 International), as described at http://creativecommons.org/licenses/by-nc/4.0/.








