
rRFtargetDB: a database of Ago1-mediated targets of ribosomal RNA fragments
- Department of Biology, Center for Computational and Integrative Biology, Rutgers University, Camden, New Jersey 08102, USA
- Corresponding author: andrey.grigoriev{at}rutgers.edu
-
↵1 These authors contributed equally to this work.
-
Handling editor: Eric Westhof
Abstract
rRNA-derived fragments (rRFs) are a class of emerging posttranscriptional regulators of gene expression likely binding to the transcripts of target genes. However, the lack of knowledge about such targets hinders our understanding of rRF functions or binding mechanisms. The paucity of resources supporting the identification of the targets of rRFs creates a bottleneck in the fast-developing field. We have previously analyzed chimeric reads in cross-linked Argonaute1-RNA complexes to help infer the guide-target pairs and binding mechanisms of multiple rRFs based on experimental data in human HEK293 cells. To efficiently disseminate these results to the research community, we designed a web-based database rRFtargetDB that preserves most of the experimental results after the removal of noise and has a user-friendly interface with flexible query options and filters allowing users to obtain comprehensive information on rRFs (or targets) of interest. rRFtargetDB is populated by ∼163,000 experimentally determined unique rRF-mRNA pairs (∼60,000 supported by ≥2 reads). Almost 30,000 rRF isoforms produced >385,000 (>156,000 with ≥2 reads) chimeras with all types of RNA targets (mRNAs and noncoding RNAs). Further analyses suggested hypothetical modes of interactions, supported by secondary structures of potential guide-target hybrids and binding motifs, essential for understanding the targeting mechanisms of rRFs. All these results (ranging from the weakest to the strongest experimental support) are presented in rRFtargetDB, whose goal is to provide a resource for building users’ hypotheses on the potential roles of rRFs for experimental validation. Further, we illustrate the value/application of the database in several examples.
rRFtargetDB is freely accessible at https://grigoriev-lab.camden.rutgers.edu/tardb.
Keywords
INTRODUCTION
The field of regulatory small RNAs has expanded significantly with putative functional fragments derived from previously unexpected sources such as transfer RNAs (tRNAs) and ribosomal RNAs (rRNAs). Although the debate on whether these fragments represent functional molecules or biological by-products continues, tRNA-derived fragments (tRFs) have largely gained acceptance in recent years (see reviews Polacek and Ivanov 2020; Su et al. 2020; Shi et al. 2022). Elucidation of their biogenesis and targeting mechanisms has progressed substantially in both experimental and computational areas (Sharma et al. 2016; Kim et al. 2017; Kuscu et al. 2018; Guan et al. 2021), and several public databases have been developed to store such information (Kumar et al. 2015; Pliatsika et al. 2017; Guan and Grigoriev 2023). In contrast, rRNA-derived fragments (rRFs) have received much less attention, and their function remains largely unknown (Cherlin et al. 2020; Guan and Grigoriev 2021). Sequencing experiments typically remove or ignore rRNAs, but even a limited regulation effect of the most abundant RNA in the cell can have significant consequences. Notably, several publications in 1990s hinted at the regulatory potential of rRNA, with hundreds of mRNAs hybridizing with complementary subsequences of rRNAs in humans and mice (Matveeva and Shabalina 1993; Mauro and Edelman 1997). Yet the ensuing “ribosome filter” hypothesis (Mauro and Edelman 2002) would make such regulation dependent on spatial constraints caused by the differential accessibility of rRNAs inside the ribosome. In contrast, as we have argued earlier (Guan and Grigoriev 2021), the fragment-based regulation model would only be constrained by the abundance of rRFs, just like in other Argonaute-mediated mechanisms.
Argonaute (Ago) proteins are essential components of RNA-induced (RNAi) silencing complex. Like miRNAs and tRFs, numerous rRFs have been found loaded to Ago, supporting hypotheses of their potential regulation of RNA targets in different organisms (Guan and Grigoriev 2020, 2021). The implication of rRFs in posttranscriptional regulatory pathways is supported by their association with RNAi-related proteins, such as QDE proteins in fungus (Lee et al. 2009), and demonstrated by early studies in multiple other species, including Piper nigrum (Asha and Soniya 2017), rice (Chen et al. 2011), Arabidopsis (Wei et al. 2013; Asha and Soniya 2017), flies (Wei et al. 2013; Chak et al. 2015; Guan and Grigoriev 2020), and human (Cherlin et al. 2020; Lai et al. 2023). One review had attempted to generalize and unify such findings (Lambert et al. 2019), whereas another recent one pointed at the value of cross-ligation and sequencing of hybrids (CLASH) Ago1 screens (Helwak et al. 2013), enabling analyses of chimeras of small RNAs and their putative targets on a large scale. Further, several other studies used CLASH or its modifications (Gay et al. 2021), including CLASH of human Ago2 (Hejret et al. 2023). A study of interkingdom involvement of rRFs (Kusch et al. 2023) has opened an interesting view on species interactions.
Lists of rRFs found in small RNA sequencing (RNA-seq) data sets from GEUVADIS and TCGA projects have been organized into a recently developed database (MINRbase) of human rRFs in B cells, tissues, and tumors (Pliatsika et al. 2024). However, despite being a useful resource, MINRbase lacks information on the targets and functions of these rRFs.
Computational prediction of target RNAs by adopting algorithms originally designed for miRNAs remains a frequently used method to study the functional effects of small RNAs, and rRFs are not an exception. However, target prediction methods developed for miRNA do not perform well in such non-miRNA applications. For example, a recent study characterized rRFs as a major class of small RNAs of 15–30 nt in length and ending with 2′,3′-cyclic phosphate (3′-cP) or 3′-OH in Ago2 proteins in mouse and human (Lai et al. 2023). miRanda (Enright et al. 2003) was employed to predict the target genes for such small RNAs (including rRFs), and luciferase assays, qPCR, and immunoblotting experiments were performed in mouse Hepa 1–6 cells to validate the predictions. Of 107 predicted target 3′ UTRs, the authors demonstrated dysregulation in the levels of 11 mRNA and four proteins, a small fraction of the predicted targets. Thus, hypotheses on potential target binding based on already available experimental rRF–target pairs may be very helpful in planning such validations, and a database representing detailed information on these pairs is urgently needed to advance the research on rRFs.
We present here such a database of targets of rRFs, rRFtargetDB. It is based on the earlier transcriptome-scale CLASH Ago1 screens (Helwak et al. 2013), which produced chimeras of small RNAs and their putative targets. We and others have shown that sequencing experimentally hybridized RNA–RNA duplexes in CLASH is a promising approach to investigate tRF targets. Expanding this approach to rRFs, our subsequent rigorous and comprehensive analysis of the CLASH data set has identified Ago1-associated rRF–target pairs in the human HEK293 cell line (Guan and Grigoriev 2021).
We characterized a total of 29,812 rRF isoforms paired with various target RNAs, identified in at least two CLASH chimeric reads. To reduce the redundancy among highly similar rRF isoforms, we clustered the isoforms based on their coordinates on the parental rRNAs. Further, for every rRF isoform, we have uncovered and annotated target RNAs that it pairs within CLASH chimeras and found common target motifs, representing regions likely utilized for binding to these targets by the respective rRFs. Such motifs are spatially compatible with the Ago cross-linking patterns detected in independent PAR-CLIP data sets (Hafner et al. 2010) as we have described previously (Guan and Grigoriev 2021).
rRFtargetDB provides an interactive interface with graphical representations of rRNA secondary structures for intuitive querying. It also has flexible filters that allow users to perform powerful queries on both rRFs and targets to access multiple layers of information. To our knowledge, rRFtargetDB is a pioneering database dedicated to targets of rRFs and we believe it will serve as a useful resource for generating testable hypotheses and conducting systematic functional validation in the rRF field.
RESULTS
rRFtargetDB contains information on 29,812 rRF isoforms (supported by ≥2 reads). Each rRF is matched with a number of target RNAs from pairs found in experimentally derived RNA–RNA chimeras in CLASH (Fig. 1; Helwak et al. 2013). In total, we extracted >385,000 (>156,000 with ≥2 reads) chimeras with all types of RNA targets (mRNAs and noncoding RNAs). These rRFs overlap each other (often differing by only a few bases at the end), and we clustered them based on such overlaps.
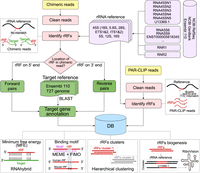
Workflow to identify rRF–target pairs in rRFtargetDB. Reference sequences obtained from NCBI and Ensembl (top right) are used to map chimeric reads and identify rRFs (top left) as described earlier (Guan and Grigoriev 2021). After mapping, targets are identified (center left), forward (rRF on the 5′ end and target on the 3′ end) and reverse (rRF on 3′ end and target on 5′ end) chimeric reads are used for grouping targets of each rRF. PAR-CLIP reads (center right) are used as evidence supporting the potential binding sites detected as common motifs in the targets of that rRF. Bottom dashed boxes illustrate the annotation of rRF–target pairs with MFE and motifs, clustering overlapping rRFs and linking the observed pairs to the rRNA genes and their secondary structure.
We obtained 1051 clusters of rRFs derived from 45S pre-rRNA including three mature rRNAs (18S, 5.8S, and 28S), external and internal transcribed spacers (ETS and ITS). A total of 175 clusters of rRFs were identified from 5S rRNAs, mitochondrial 12S, and 16S rRNAs. For every rRF isoform, we have uncovered and annotated target RNAs that it pairs within CLASH chimeras. If only mRNA targets are considered, the total number of clusters dropped to 799. To further narrow down the set of the most reliable pairs, we analyzed the multiple independent and unique mRNA target sequences pairing with specific rRFs to identify common target motifs. We detected 647 motifs, representing regions likely utilized for binding to these targets by the respective rRFs, and we mapped these motifs back to rRF sequences. We thus derived 160 motif-containing clusters (number of clusters from rRNA regions: 18S—98, 28S—56, 5S—3, ETS—2, 12S—1, and zero from others). All these findings can be accessed via the query interfaces described below.
Graphical query of rRFs
rRNAs are very long, so it is challenging to clearly represent on their large structures the cleavage sites for rRFs, compared to tRFs whose precursor RNAs are small and have clear cloverleaf-shaped secondary structure. To provide users with graphical query options based on rRF location, we designed interactive graphical representations of secondary structures for mature sequences for cytosolic and mitochondrial LSU and SSU rRNAs. Such an intuitive interface allows users to search for rRFs from regions of interest with a single click.
LSU and SSU rRNAs are color-coded (18S—green, 28S—blue, 5.8S—red, 5S—purple, 16S—red, and 12S—green) with the shade of each circle representing the number of rRFs covering the respective nucleotide in all chimeric reads (not only with miRNA targets). To accommodate different analyses, we provided two optional color scales: one based on a linear transformation to emphasize the most prominent rRFs origins (Fig. 2A) and another based on a logarithm transformation to highlight all positions that derived rRFs (Fig. 2B). The graphical display of structures allows users to interact with the database by clicking on specific nucleotides to retrieve all rRFs covering that position. As an example, clicking on a nucleotide A at position 56 of the 28S rRNA will display all mRNA-targeted rRFs containing these positions (Fig. 2C). Here, one can see a default filter of at least five CLASH chimeric reads and five unique hybrids (UHs, which refers a pair of unique rRF sequences and unique target sequences) on the result page.
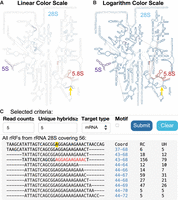
Search rRFs by biogenesis on precursor rRNAs. (A,B) Secondary structures of rRNAs in the large ribosomal subunit. The shade of color in each circle represents the total CLASH chimeric reads supporting the rRFs covering that nucleotide. As the linear scale indicates a few regions of most abundance of rRFs and may seem too faint (A), so users may opt for a differently shaded log scale (B). The colormaps reflect coverage by all chimeric reads, not only those passing the default filters. (C) rRFs presented after clicking the nucleotides pointed by arrows in A,B with default filters for rRFs supported by at least five CLASH chimeric reads and five UHs. Ten first lines with rRFs are shown, red letters indicate a motif for one of the rRFs. As is common, motif significance comes hand in hand with the high read count (RC) and UH support.
CLASH data contained plentiful pairs of rRFs with non-mRNAs and their biological effects are unclear. Hence, the numbers of CLASH chimeric reads and UH that are displayed for each rRF by default include rRF-mRNA pairs only. However, for completeness, the rRF-non mRNA pairs are preserved, and users can see their read count and UH by changing the target type filter to “All” in the text-based search page or in the simplified search menu in rRF isoform pages, described below.
Text-based search for rRFs and targets
In addition to the intuitive graphical query system above, we provided a search interface similar to one we introduced in tatDB (Guan and Grigoriev 2023), so users of both databases can benefit from recognizable interfaces. This option is also available for mitochondrial rRFs. Detailed and flexible options and filters can be specified to query rRFs that bind to specific genes or to reveal all target genes of given rRFs (Fig. 3A). On the top panel, rRFs can be selected from different precursor rRNAs within a specified range of Start to End. Checking “Exact S/E” box will ensure rRFs with exact cleavage positions are returned. Each rRF isoform has a human-readable ID (see Materials and Methods), which can be used to search for corresponding rRFs. Similar options are available for gene-based queries or queries combining rRF and target information.

Search rRFs and targets with flexible filters. (A) The search page with default filters indicated. (B) Output of the search results for rRFs from 28S rRNA that target mRNAs using binding motif (motif filter ON); only the first 20 lines are shown. Matching rRF sequences (Guide Seq) are shown, it is easy to see how they slightly overlap each other in this example. Each line corresponds to a UH (that refers to a pair of unique rRF sequences and unique target sequences) identified in a CLASH chimeric pair involving an rRF isoform and a target sequence. Result table is banded to highlight different pairs of rRFs and target genes. For example, 14 UHs (same rRF sequence, different target sequences) support the interaction between 28S-47-67 and target gene RPL41. Four UHs pass the filters (i.e., read count ≥ 5 and MFE ≤ −15) and shaded in the same color, to distinguish them from other pairs of rRFs and target genes. From-To indicate an rRF coordinate in the host rRNA and serves as a link to the rRF and target information (red box hyperlinks to Fig. 4). Target transcript, region of binding, name, and biotype are indicated, together with the counts of supporting UHs, read counts, and orientation (forward or reverse, see text).
rRFs have been observed to bind to intronic regions (Guan and Grigoriev 2021). Our database offers users the option to explore the interaction of rRFs with target genes on either exonic (including 5′ UTR, CDS, and 3′ UTR) or intronic regions. Users can input partial gene symbols if they unselect the “Exact name” option. Furthermore, users can search both guide and target RNAs by sequence and rRFs or targets containing the entire input sequence without mismatches will be returned by the query. Despite the unclear biological significance of noncoding RNAs paired with rRFs in CLASH chimeras, we kept them in rRFtargetDB for completeness, since these pairs were identified in experimentally derived CLASH reads (Helwak et al. 2013). By default, mRNA targets are displayed on the result page, unless specified otherwise for noncoding targets.
We offer several filters for the users to query rRFs and targets with specific thresholds:
-
Minimum free energy (MFE), calculated by RNAhybrid, assesses the strength of interaction between rRFs and target RNAs (Kruger and Rehmsmeier 2006). Default MFE threshold is set to ≤−15 kcal/mol, which corresponds to 75% of all UHs of rRF and target sequences as we observed in our earlier study (Guan and Grigoriev 2021).
-
In our previous work, we suggested that the rRF and target RNA can be ligated interchangeably due to the removal of 3′-cP end during the CLASH experiment (Guan and Grigoriev 2021). Consequently, an rRF may be found in a forward (rRF on the 5′ end and target on the 3′ end) or in a reverse chimeric read (rRF on 3′ end and target on 5′ end). As such chimeras represent independent biological constructs, this information can be leveraged to search for rRF–target pairs of higher confidence (by setting the “Direction” option to “0” to obtain pairs supported by interactions of both directions).
-
The presence of motifs is another threshold: by default, rRFs with significant motifs are returned on the result page, indicating more reliable data, and this option can be turned off.
-
Additional settings include a minimum CLASH read count supporting each UH and a minimum UH supporting each pair of rRF and target gene (default is 5 to ensure the robustness of presented data but can be changed by a user).
List of rRF–target pairs
Search results are displayed in a text table format, where each line corresponds to a UH identified in CLASH chimeric pair involving an rRF isoform and a target sequence (Fig. 3B). In cases where the chimeric reads involve a particular rRF and several overlapping subsequences of the same target gene, we display them with the same background color. Counts of UHs supporting a pair of rRF and target gene, and count of chimeric reads supporting each UH are shown in separate columns. Column “F/R” indicates whether the UH of an rRF sequence and a target sequence is found in forward or reverse chimeric read, with FR indicating it is found in both directions.
In the “From-To” column, the start and end coordinates uniquely identify an rRF on an rRNA gene. Clicking on this hyperlink directs users to a detailed rRF information (DRI) page (its elements are shown in Fig. 4). The MFE value serves as another hyperlink, directing users to a detail page with the secondary structure of the interaction between given rRF and target RNA, along with additional details such as the counts of raw CLASH chimeric reads and counts of UH.

Detailed information of rRF 28S-47-67. (A) rRF sequence, origin, and group information. (B) Sequence logo of binding motif in the rRF. (C,D) Top 2 target genes for the rRF. With no UH filter at this stage, target genes with at least one UH are displayed. Total number of UH are shown, and only those UH supported by at least five chimeric reads are counted for the number of reads supporting UHs. (E,F) The secondary structures show the potential hybridization pattern between the rRF and target RNAs. Binding motif is highlighted in red, using the whole sequence in B, ignoring its bit scores. Because the motif is established via statistical analysis of all targets of rRF 28S-47-67 with MEME motif finder, this whole sequence does not exactly match a base-pairing region with every single target. Hyperlink “Click to see raw chimeric reads” allows users to see specific CLASH reads utilized in the analysis of these rRFs and targets. A red line in E indicates a hairpin sequence in the target RPL41 (see text for explanations).
rRF isoform page
This page includes the sequence of rRF isoform, cleavage sites on the mature rRNA, and precursor rRNA (if there is a precursor) (Fig. 4). Each rRF is assigned to a cluster including a list of rRF isoforms with similar cleavage sites on the rRNAs. The target RNAs of all rRF isoforms in a cluster were combined to infer a common target motif with MEME (Bailey et al. 2009). Subsequently, FIMO (Grant et al. 2011) was used to detect complementary matches for this target motif in each rRF isoform, which was reported as the potential binding motifs in the corresponding rRF sequences. The hyperlink on the coordinates of the “motif group” directs to a page displaying all rRF isoforms in the cluster (Fig. 4A). If an rRF contains a significant binding motif, the sequence logo of this motif is visualized (Fig. 4B). Throughout all output pages, the motif region is consistently highlighted in the rRF sequence to emphasize its presence.
To indicate potential sites in rRFs where cross-linking to Ago proteins may occur, we provide the T > C conversions identified in human PAR-CLIP Ago1-Ago4 data sets to each rRF. These conversion sites are underlined in the rRF sequence along with the frequency of each conversion shown next to the sequence (Fig. 4A).
It is important to note that due to the variation in library preparation across different experiments, rRFs may vary by a few terminal nucleotides. This variability is also found in other small RNAs (e.g., miRNAs and tRFs) and leads to challenges in directly comparing sequences. Therefore, we aligned T > C conversion sites in PAR-CLIP data sets to rRNA sequences and display them if the rRFs identified in CLASH contain those positions, as such conversion sites often appear in the same locations in different isoforms. Since they are derived computationally in this manner, in many cases, these sites are hypothetical, they do not necessarily imply that the exact rRF isoform is observed in the PAR-CLIP data sets.
Target genes that bind to this rRF are listed below this information, both with summaries (Fig. 4C,D) and with respective binding patterns (Fig. 4E,F). Filters selected in the search page are automatically applied in a simplified menu and can be adjusted on the DRI page to display corresponding sets of rRF–target pairs. If no target meets user-specified filtering criteria, setting more relaxed filters or selecting “All” targets in this menu will show the corresponding hits with weaker support. For each gene, only the target site with the strongest rRF interaction is shown, as determined by MFE (although an rRF can bind to multiple sites in a target gene or the target RNA sequences detected in the chimeric reads may vary in length). Users can access from DRI page-specific CLASH reads used in the analysis of these rRFs and targets. Clicking on the Transcript ID directs users to its Ensembl page where all regions of the target can be explored in detail.
Database interoperability, linking to rRFtargetDB
To facilitate the aggregation of information about known rRFs across databases and studies, we designed a unique URL for each rRF isoform. When the sequence of an rRF is known, users can link from any source to its dedicated rRFtargetDB page to access detailed information about the rRF and its target genes by incorporating the rRF sequence in the URL. For example, the rRF illustrated in Figure 4 may be directly accessed with the URL https://grigoriev-lab.camden.rutgers.edu/tardb/rrf_isoform.php?guide_seq=AGTCAGCGGAGGAGAAGAAAC. Default filters will be selected for such a link if no filter is specified in the URL or in the previous search step. Further, filter settings may be changed after landing on the rRFtargetDB page.
DISCUSSION
To the best of our knowledge, only one database has recently been dedicated to rRFs (Pliatsika et al. 2024). This database, MINRbase, includes 130,238 rRFs identified from the RNA-seq data of human B-cell lines, healthy tissues, and tumor samples. Yet, although it reflects the growing interest in the rRFs, MINRbase currently lacks information on their target genes (and hence, putative functions). This limitation is common across many studies of rRFs, which often focus on characterizing rRFs in RNA-seq and CLIP-seq data sets but fall short in directly identifying pairs of guide rRFs and target RNAs.
Knowing targets of small RNAs is critical for understanding their regulatory function, and computational target prediction remains a common strategy. In the Introduction, we highlighted the low predictive power of tools designed for miRNA target finding. Of note, the basis of such target predictors is the assumption that miRNAs bind to target via a seed sequence located between nucleotide positions 2 and 8, with potential contribution of the 3′ end. However, we have previously suggested that the binding regions (equivalent to “seeds”) of rRFs can be found at both the 5′ and 3′ end of rRFs (Guan and Grigoriev 2021), and rRFtargetDB can provide multiple examples of such cases. Another common notion is the target region being the 3′ UTR. Yet searching rRFtargetDB can help reveal many putative target binding sites in other gene regions. Further experiments can confirm or refute this and rRFtargetDB can be used to formulate specific hypotheses.
rRFtargetDB represents the first database to catalog human rRFs along with their putative target genes, not predicted but detected in experimental Ago1 IP screens. It features a user-friendly interface that enables flexible searching of rRFs, including intuitive graphical search of rRFs based on their location within rRNA secondary structures. We expect the extensive information provided for every rRF in rRFtargetDB, including the novel binding motifs and rRF–target interactions, to help users generate research hypotheses for experimental validation in various fields of biological regulation. For example, we have reported that a 16 nt rRF (TGGAGCGATTTGTCTG) of 18S rRNA sequence was detected inside a minus strand sgmRNA template for the Spike gene in SARS-CoV-2 (Grigoriev et al. 2022). Additionally, in conjunction with our previously released database of targets of tRFs (tatDB), users can easily query both tRFs and rRFs that bind to specific target genes. Investigating the potential synergistic regulatory functions of these fragments on target genes may be intriguing. For example, we have previously suggested that rRFs can bind to RPS28, which is also targeted by the LeuCAG tRF that was shown to enable translation by unwinding the RPS28 mRNA (Kim et al. 2017), and such binding regions overlap for both rRFs and LeuCAG. Figure 4E provides another example of how rRFs may interact with genes encoding ribosomal proteins and enable their translation by affecting their secondary structure: the red line shows a small hairpin in RLP41 mRNA that appears targeted (Guan and Grigoriev 2021).
A recent paper by Lai et al. (2023) has shown the biogenesis and function of small RNAs using TANT-seq to capture small RNAs (≥15 nt in length) with 3′-cP and 3′-OH ends loaded to Ago2. This is perhaps the most advanced experimental rRF study to date as it has validated the target gene regulation of three rRFs. Thus, we were eager to compare it with the CLASH results. However, there were only two validated rRFs ≥16 nt in length (this is a length threshold in rRFtargetDB), and low read counts and poor MFE scores for the respective chimeras did not support these specific isoforms acting via Ago1.
The difference in Ago proteins and in cell lines between the two data sets may explain the nonoverlapping results although it is only two rRFs from a rather small group tested by Lai et al. (2023). This is in contrast to the transcriptome-wide trends of similarity and substantial overlap in rRFs we have detected previously between mouse Ago2-IP (Sarshad et al. 2018) and Ago1 CLASH (Helwak et al. 2013), both in the cytoplasmic and nuclear fractions. Exploring the target groups and mechanisms of common rRFs in Ago1 and Ago2 can help us uncover potential regulatory differences and similarities between the two major Ago proteins.
A limitation of rRFtargetDB is that its target assignment is based on a single type of experiment (CLASH, but with additional data from PAR-CLIP for supporting binding regions), a single cell type, and a single Ago protein. We plan to expand this work by adding data sets of modified CLASH (Gay et al. 2021), those of human Ago2 (Hejret et al. 2023), and other data as they become available to establish a generalized view on possible functionality of various types of RNA fragments in different Ago contexts.
Another possibility to consider is that some rRF-mRNA pairs may represent intriguing cases of mRNA fragments acting as potential regulatory guide molecules, described a decade ago (Zamudio et al. 2014) and mentioned in the original CLASH paper (Helwak et al. 2013). This could affect the interpretation of some rRF-mRNA pairs, making rRNA fragments possible targets.
We also provided a comment option, so that experimentally validated targets can be described manually. Although we had several examples of such validations in tatDB, in rRFtargetDB this feature awaits future use, as the database will hopefully lead to hypotheses that can be tested experimentally to expand our knowledge of potential regulation by rRFs.
MATERIALS AND METHODS
Building rRNA index
In general, we followed the procedures described in our earlier paper (Guan and Grigoriev 2021). Here, we list only the main changes and provide some important clarifications.
We downloaded five 45S precursor rRNA sequences (RNA45SN1 to RNA45SN5) from NCBI RefSeq and a 45S gene sequence (first 13,314 bp of U13369) from GenBank (Fig. 1). U13369 was chosen as a representative isoform to identify rRFs. For other isoforms of 45S, the isoform numbers were appended to the end of each region for naming the rRFs generated from it. For example, 18S, 5.8S, and 28S rRNAs that originated from RNA45SN1 were named as 18S-001, 5_8S-001, and 28S-001, respectively. ETS and ITS were considered as precursors of rRFs; for example, the two ETS regions of U13369 were named as ETS1 and ETS2. In addition to all 5S rRNAs from RefSeq, we incorporated transcripts annotated as 5S rRNAs from the latest Ensembl database (version 110). After collapsing identical sequences, we obtained 25 unique sequences for rRNAs. RNA5S6 was used as a representative 5S sequence, and the other 24 sequences were named as 5S-002 to 5S-025. RNR1 and RNR2 were used as 12S and 16S, respectively (Fig. 1).
Naming convention for rRFs
We gave every rRF isoform informative and extensible names with the format Mature_rRNA-(Isoform_Num)-Start-End. The Isoform_Number field was optional for rRFs that are mapped to the representative rRNA sequences. For example, 28S-47-68 represents an rRF of 22 nt in length, cleaved at positions 47 and 68 of 28S rRNA in U13369 sequence. An alternative ID including the coordinates on precursor rRNA is also assigned, e.g., 45S-7971-7992 is the same as 28S-47-68. We provided one representative ID on the result page and all alternative IDs can be found on the details page.
Analyses of experimental data sets (CLASH and PAR-CLIP)
We followed our pipeline previously utilized with hg38 genome assembly (Guan and Grigoriev 2021), but this time, we utilized the latest haplotype-resolved telomere-to-telomere (T2T, which has additional 4.5 Gbp) reference genome to annotate target RNAs (Fig. 1). The rationale in using the T2T genome is to use the resolved sequence in repeat regions (including rDNA) and to find potential targets among novel T2T-specific genes. We downloaded CLASH data from HEK293 cells from the SRA database (accession numbers SRR959751–SRR959759). After barcode and adapter removal and collapsing identical reads with the FASTX-Toolkit v0.0.13 (http://hannonlab.cshl.edu/fastx_toolkit/), we included an additional filtering step using Bowtie 2 (version 2.4.1) (Langmead and Salzberg 2012), using –very-sensitive setting in end-to-end mode to remove chimeric reads that completely aligned with the T2T genome to reduce artifacts and obtain high-confidence rRF–target interactions. For each hybrid read, we identified the longest perfect match to rRNA reference sequences that began at the 5′ end or ended at the 3′ end of the hybrid read. The remaining portion of each hybrid read was aligned to the complete T2T transcriptome and genome sequences (see https://rapid.ensembl.org/) using BLAST (version 2.6.0) to identify/annotate the rRF targets in each chimeric read. A read was classified as chimeric if the combined length of the rRF and its putative target sequence accounted for at least 75% of the total read length, after barcode and adapter removal. To ensure identification of high-confidence mRNA targets, we excluded targets that exhibited higher than 90% identity in BLAST alignments to noncoding genes. If a target sequence aligned to alternative transcripts of the same gene with identical E values, we prioritized the protein-coding transcript. If an rRF in a read was shorter than 16 nt, we excluded this read from further analysis.
We downloaded the Ago1-4 human PAR-CLIP data set (Hafner et al. 2010) from the SRA database (accession numbers SRR048973–SRR048979). As above, adapter sequences were trimmed using the FASTX-Toolkit (v0.0.13), and reads were aligned to rRNA references using Bowtie (version 1.3.1) in end-to-end mode (Langmead et al. 2009). We allowed for a single T > C mismatch, prioritizing perfect matches. rRFs shorter than 16 nt were excluded from analysis. The abundance of each rRF isoform was normalized to reads per million (RPM) mapped to the human genome in every sample.
Aligning rRFs to rRNA secondary structures
The secondary structures of nuclear rRNAs (18S, 5.8S, 28S, and 5S) were obtained from RiboVision (Bernier et al. 2014). RiboVision uses U13369 as the reference sequence. Therefore, we aligned all other rRNA isoforms used in our study to U13369 with ClustalW (https://www.genome.jp/tools-bin/clustalw). rRFs were then aligned to U13369 with their coordinates on the precursor rRNA isoforms. The secondary structures of mitochondrial rRNAs (12S and 16S) were obtained from RNAcentral with accession numbers URS0000961A7F and URS000080E357 (The RNAcentral Consortium 2016). For every position on the reference rRNAs, we calculated the total number of CLASH reads involving the rRFs covering the position and all their putative targets. A linear or logarithm (user-selected) transformation (matplotlib.colors.Normalize or matplotlib.colors.LogNorm) was used to assign colors based on this total read coverage for each nucleotide on the rRNA secondary structure.
Clustering of rRFs and inference of binding motifs
To classify rRF isoforms with variable ends into clusters, we firstly separated rRFs according to their parental rRNAs. For rRF isoforms on a given rRNA, we used Python scipy fcluster function to perform hierarchical clustering with Ward's linkage. A Euclidean distance matrix based on the start and end coordinates of rRF isoforms on the rRNAs was used to form flat clusters.
For every rRF cluster, we combined all their targets of rRFs supported by ≥2 CLASH chimeric reads. If multiple target RNAs mapped to the same gene, we selected the longest target RNA and discarded the others to avoid redundancy in motif searching. We employed MEME version 4.11.2 (Bailey et al. 2009) to search for significant motifs (e-value < 0.01, P-value < 0.05) in these target RNA sequences with default parameters. We next used FIMO version 4.11.2 (Grant et al. 2011) to look for the significant complementary match (P-value < 0.001) of the motif in the sequences of all rRFs of a cluster.
For every rRF–target pair, we used RNAhybrid 2.1.2 (Kruger and Rehmsmeier 2006) to predict the secondary structure and MFE.
Implementation of rRFtargetDB
A relational database was implemented with MariaDB 10.3.39, Apache v2.4, PHP v7.4.33, and Bootstrap v3.3.7. Interactive plots of rRNA secondary structures were created by appending hyperlinks with positional information and color-coded annotations of total read coverages to the SVG files downloaded from RiboVision (Bernier et al. 2014) and RNAcentral (The RNAcentral Consortium 2016) websites. Sequence motifs were visualized by a jQuery plugin (https://github.com/simonsfoundation/jp_gene_viz/blob/master/jp_gene_viz/sequence_motifs.js).
Footnotes
-
Article is online at http://www.rnajournal.org/cgi/doi/10.1261/rna.080285.124.
-
Freely available online through the RNA Open Access option.
- Received October 10, 2024.
- Accepted December 17, 2024.
This article, published in RNA, is available under a Creative Commons License (Attribution-NonCommercial 4.0 International), as described at http://creativecommons.org/licenses/by-nc/4.0/.
REFERENCES
MEET THE FIRST AUTHOR

Meet the First Author is an editorial feature within RNA, in which the first author(s) of research-based papers in each issue have the opportunity to introduce themselves and their work to readers of RNA and the RNA research community. Sathyanarayanan Vaidhyanathan is co-first author of this paper, “rRFtargetDB: a database of Ago1-mediated targets of ribosomal RNA fragments,” along with Lingyu Guan. Sathyanarayanan is a PhD candidate in the Grigoriev Lab at Rutgers University, Camden. He works on studying small RNA mechanisms, targets, and their interactions.
What are the major results described in your paper and how do they impact this branch of the field?
rRFtargetDB is populated by ∼163,000 experimentally determined unique rRF-mRNA pairs (∼60,000 supported by ≥2 reads). Almost 30,000 rRF isoforms produced >385,000 (>156,000 with ≥2 reads) chimeras with all types of RNA targets (mRNAs and noncoding RNAs). Further analyses suggested hypothetical modes of interactions, supported by secondary structures of potential guide-target hybrids and binding motifs, essential for understanding the targeting mechanisms of rRFs. The database will serve as a key resource to help researchers in this field to characterize rRFs and their targets, to further formulate their hypotheses and to plan experiments exploring various aspects of the rRF function.
What led you to study RNA or this aspect of RNA science?
I worked on sequence data analysis before starting my PhD. After joining the Grigoriev Lab during my PhD, my research shifted to studying RNA, specifically small RNAs.
What are some of the landmark moments that provoked your interest in science or your development as a scientist?
Interning at a children's hospital and enrolling in a PhD program were pivotal experiences that sparked my curiosity and deepened my interest in science. These opportunities taught me the foundational principles of the field and enabled me to give back to the community, helping me grow as a scientist.
Are there specific individuals or groups who have influenced your philosophy or approach to science?
The professors from my undergraduate and PhD programs have significantly impacted my approach to science. My principal investigator, Dr. Grigoriev, has greatly influenced my ideas. He encouraged me to adopt a scientific mindset when exploring research questions and has guided me throughout my PhD journey.
How did you decide to work together as co-first authors?
Lingyu Guan is an alumna of the Grigoriev Lab, where she obtained her PhD. Her research on small RNAs forms the foundation that I further develop in my current work, and this paper builds upon her previous research. She started this project while in the lab, and we have completed it together. She is supportive and helpful, and I have learned much from her.








