
eIF4F-mediated dysregulation of mRNA translation in cancer
- 1Department of Biochemistry, McGill University, Montreal, QC H3G 1Y6, Canada
- 2Rosalind and Morris Goodman Cancer Institute, McGill University, Montreal, QC H3A 1A3, Canada
- 3Department of Pharmacology and Regenerative Medicine, University of Illinois College of Medicine, Chicago, Illinois 60612, USA
- Corresponding author: nahum.sonenberg{at}mcgill.ca
-
↵4 These authors contributed equally to this work.
Abstract
Messenger RNA (mRNA) translational control plays a pivotal role in regulating cellular proteostasis under physiological and pathological conditions. Dysregulated mRNA translation is pervasive in cancer, in which protein synthesis is elevated to support accelerated cell growth and proliferation. Consequently, targeting the mRNA translation machinery has emerged as a therapeutic strategy to treat cancer. In this Perspective, we summarize the current knowledge of translation dysregulation in cancer, with emphasis on the eukaryotic translation initiation factor 4F complex. We outline recent endeavors to apply this knowledge to develop novel treatment strategies to combat cancer.
Keywords
INTRODUCTION
mRNA translation is a tightly regulated, energy-intensive process divided into four phases: initiation, elongation, termination, and ribosome recycling (Pelletier and Sonenberg 2019), with initiation serving as the rate-limiting step (Hershey et al. 2019). To initiate mRNA translation, a ternary complex (TC) consisting of the initiator methionyl tRNA (Met-tRNAi), guanosine-5′-triphosphate (GTP), and eukaryotic translation initiation factor 2 (eIF2) is assembled on the small 40S ribosomal subunit, which, together with several other initiation factors (eIF1, eIF1A, eIF3, eIF5), forms the 43S preinitiation complex (PIC). The PIC is recruited to the mRNA 5′ cap structure (cap; m7GpppN, where N is any nucleotide and m is a methyl group) (Pelletier et al. 2021) by the eIF4F complex, and scans the mRNA 5′ untranslated region (UTR) in the 5′ to 3′ direction to recognize an initiation codon (Pelletier and Sonenberg 2019).
eIF4F is composed of three subunits: eIF4E (cap-binding subunit), eIF4G (scaffolding subunit), and eIF4A (an RNA helicase) (Pelletier and Sonenberg 2019). eIF4E, the major cap-binding protein, was discovered nearly 50 years ago by its chemical crosslinking to the reovirus mRNA cap (Sonenberg et al. 1978). eIF4E acts as the critical limiting factor in translation (Sonenberg 1996; Marcotrigiano et al. 1997). Because of its pivotal role as a rate-limiting factor, multiple mechanisms evolved to regulate the activity of eIF4E, either through posttranslational modifications by kinases or by direct protein–protein interactions. eIF4E is controlled primarily by two key signaling pathways: (1) the phosphoinositide 3-kinase (PI3K)/mechanistic target of rapamycin (mTOR), and (2) the mitogen-activated protein kinase (MAPK)/extracellular signal-regulated kinase (ERK) (Sonenberg and Hinnebusch 2009). In response to intra- and extracellular cues, the mTOR complex 1 (mTORC1, see below) is activated and phosphorylates the translational repressor eIF4E-binding proteins (4E-BPs) at multiple sites, leading to the dissociation of 4E-BPs from eIF4E, thus promoting eIF4F complex formation and translation initiation (Pause et al. 1994; Fadden et al. 1997; Hay and Sonenberg 2004). In addition, the activity of eIF4E is regulated by the phosphorylation at Ser209 by MAP kinase-interacting kinases (MNK1/2), which act downstream from ERK and p38 in response to growth signals and stress, respectively (Fukunaga and Hunter 1997; Waskiewicz et al. 1997; Ueda et al. 2004; Buxade et al. 2008). By manipulating the two pathways, tumor cells enhance the selective translation of mRNAs encoding proteins which are required for cancer growth and metastasis, such as oncogenic proteins, metabolic regulators, and ribosomal proteins (Holland et al. 2004). The involvement of protein synthesis in cancer was first adumbrated as early as 1896 when hypertrophic nucleoli—nuclear sites of ribosome production—were marked as a characteristic feature of cancer cells (Pianese 1896). In 1990, eIF4E was reported as an oncogene product (Lazaris-Karatzas et al. 1990). Aberrant mRNA translation is now recognized as a key characteristic of cancer (Biffo et al. 2024). As the role of mRNA translational control in cancer has been extensively reviewed (Bhat et al. 2015; Robichaud et al. 2019; Xu and Ruggero 2020; Fabbri et al. 2021; Bartish et al. 2023), this article summarizes current advancements in the field and strategies to target eIF4F-mediated dysregulated translation in cancer (Fig. 1).
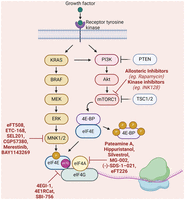
A schematic diagram of the signaling pathways regulating the eIF4F complex and potential therapeutic compounds. The assembly and activity of the eIF4F complex are regulated by the mTOR and MNK1/2 kinases, which phosphorylate 4E-BPs and eIF4E, respectively. Many inhibitors have been developed that target RAS/MEK/ERK and PI3K/Akt/mTOR pathways in cancer (Montagut and Settleman 2009; Hua et al. 2019; He et al. 2021). In addition to the compounds mentioned here, other inhibitors of translation initiation have been discussed in a recent review (Li et al. 2023). Created in BioRender (https://BioRender.com/p92z707).
TRANSLATION CONTROL BY THE MNK/p-eIF4E AXIS IN CANCER
In response to growth signals or stress, MNK1/2 kinases are activated and phosphorylate eIF4E at Ser209, which leads to increased translation of a subset of mRNAs encoding critical proteins involved in cell survival and metastasis, such as c-Myc, matrix metalloproteinases 3 and 9 (MMP3, MMP9), SNAIL, myeloid cell leukemia-1 (MCL1), and Cyclin D1 (Furic et al. 2010).
BRAFV600E is a frequent mutation of BRAF in melanoma, which activates the MNK/p-eIF4E pathway via the MAPK pathway (Boussemart et al. 2014). Similarly, NRAS mutations, most notably NRASQ61L, lead to hyperactivation of the MAPK–MNK1/2–eIF4E pathway along with the PI3K/Akt pathway (Prabhu et al. 2020). Because of the hyperactivation of these signaling cascades, the translation of pro-metastatic and pro-survival mRNAs is upregulated, leading to cancer resistance and progression (Prabhu et al. 2020). Loss-of-function mutations of Neurofibromin 1 (NF1), a tumor suppressor that encodes a Ras GTPase-activating protein (GAP), result in sustained RAS activation, which drives hyperactivation of both the MAPK–MNK1/2–eIF4E and PI3K/Akt signaling pathways (Prabhu et al. 2020). Recent findings reveal that eIF4F-dependent translation of dual specificity phosphatase 6 (DUSP6, also known as MKP3) is crucial for maintaining the optimal activity of the MAPK signaling pathway in BRAF- and NRAS-mutant melanoma cells (Valcikova et al. 2024). Inhibitors targeting the eIF4F complex, such as Rocaglamide A, disrupt this balance, leading to ERK hyperactivation and subsequent cell death (Valcikova et al. 2024).
Elevated levels of p-eIF4E are reported in several types of cancer, including prostate cancer (Furic et al. 2010), non-small cell lung cancer (NSCLC) (Lu et al. 2020), nasopharyngeal carcinoma (Zheng et al. 2014), astrocytoma (Fan et al. 2017), and melanoma (Carter et al. 2016). From a clinical and therapeutic standpoint, it is noteworthy that the p-eIF4E is dispensable for the viability of the organism, as mice harboring a whole-body homozygous Ser209Ala mutation in eIF4E (eIF4ES209A) or lacking both MNKs (MNK1/2 double knockout [Mnk-DKO]) are viable (Furic et al. 2010; Ueda et al. 2010). However, eIF4ES209A and Mnk-DKO mice are resistant to cancer development and metastasis (Furic et al. 2010; Ueda et al. 2010). This is consistent with the finding that p-eIF4E does not affect general translation (Amorim et al. 2018). These findings have prompted numerous preclinical and clinical trials of p-eIF4E inhibitors to treat cancer (see below).
Cancer metabolism
Metabolic reprogramming is a hallmark of cancer, enabling cancer cells to adapt to nutrient limitations and hypoxia in order to thrive in hostile environments (Hanahan 2022). One of the strategies that cancer cells use to promote metabolic reprogramming is the translational upregulation of mRNAs encoding key metabolic proteins (Topisirovic and Sonenberg 2011; Leibovitch and Topisirovic 2018). In response to fasting or ketogenic diets, p-eIF4E is induced via a fatty acid-AMPK-–MNK–eIF4E pathway (Yang et al. 2024). p-eIF4E selectively promotes the translation of 3-hydroxy-3-methylglutaryl-CoA synthase 2 (Hmgcs2) and peroxisome proliferator-activated receptor alpha (Ppara) mRNAs (Yang et al. 2024). HMGCS2 catalyzes the rate-limiting step of ketogenesis and PPARA is a transcription factor involved in lipid metabolism. Pancreatic cancer cells use the AMPK–MNK–eIF4E pathway to promote the utilization of ketone bodies as an alternative energy source (Yang et al. 2024). Targeting p-eIF4E with MNK inhibitors such as eFT508 (tomivosertib) (Reich et al. 2018), disrupts metabolic adaptation and reduces tumor growth in a mouse model of pancreatic cancer (Yang et al. 2024). When combined with a ketogenic diet, eFT508 limits the availability of ketone bodies, which are vital for sustaining tumor growth (Yang et al. 2024). These findings suggest that combining dietary interventions with MNK1/2 inhibitors may offer a therapeutic strategy to target cancer metabolism.
MNK1-null cells undergo a marked metabolic change by shifting from glycolysis toward oxidative phosphorylation (OxPhos) (Preston et al. 2024). The metabolic rewiring impairs the ability of MNK1-null pancreatic and breast cancer cells to metastasize to the liver, an organ that favors glycolytic metabolism (Preston et al. 2024). These data indicate that targeting MNK1 could be an effective strategy for preventing metastasis to the liver, where they have a tissue-specific metabolic dependency.
Translational control of oncogenes
Increased translation of mRNAs encoding cancer-promoting factors (such as c-Myc, MMP-3, SNAIL, MCL-1, and Cyclins) plays a key role in several malignancies (Robichaud et al. 2019). We have previously shown that eliminating the eIF4E phosphorylation site (eIF4ES209A) in mice confers resistance to the development of cancer (Furic et al. 2010). Lack of p-eIF4E decreases the translation of mRNAs implicated in cancer growth, including C–C motif chemokine ligand 2 (Ccl2), Ccl7, baculoviral IAP repeat-containing protein 2 (Birc2), vascular endothelial growth factor C (Vegfc), Mmp3, and Mmp9 (Furic et al. 2010). In glioblastoma, the MNK1 inhibitor BAY1143269 reduces VEGF, Cyclin D1 and A1, and cyclin-dependent kinase (CDK)-2 protein levels, thereby suppressing angiogenesis and tumor growth in vivo (Wan et al. 2022). In melanoma, p-eIF4E promotes the translation of mRNA encoding nerve growth factor receptor (NGFR), which renders the cells more invasive (Huang et al. 2021). Inhibiting p-eIF4E reverses the invasive phenotype and enhances immunotherapy response in multiple mouse models of melanoma (Huang et al. 2021). In soft tissue sarcomas (STS), MNK1/2 play a pivotal role in supporting tumor growth through oncogenic translation (Ke et al. 2021). Elevated levels of MNK1/2 drive the expression of critical transcriptional regulators like E2F transcription factor 1 (E2F1), forkhead box protein M1 (FOXM1), and WEE1 G2 Checkpoint Kinase (WEE1), thereby sustaining the proliferation of STS cells (Ke et al. 2021). Inhibition of MNK1/2 with the novel compound ETC-168 shows substantial antiproliferative effects in STS models, especially when combined with MCL1 inhibitors, which enhance apoptosis (Ke et al. 2021). Thus, dual inhibition strategies targeting MNK1/2 and other key oncogenic pathways could provide a potent treatment for aggressive cancers, especially for those resistant to standard treatments (Ke et al. 2021). We documented PPM1G as a phosphatase that dephosphorylates eIF4E at Ser209, leading to the suppression of Mcl-1 mRNA translation (Wang et al. 2024). Enhancing PPM1G activity or mimicking its effect offers a novel therapeutic strategy for cancers manifesting high p-eIF4E.
Cancer stem cells
Cancer stem cells (CSCs) constitute a very small population of cancer cells (generally between 0.01% and 1%) that possess the ability to self-renew, differentiate, and contribute to cancer growth, metastasis, relapse, and therapeutic resistance (Yang et al. 2020a). CSCs provide phenotypic plasticity to the cancer cells and are a major driver of cancer heterogeneity (Marjanovic et al. 2013). A key posttranscriptional mechanism that facilitates the phenotypic plasticity of CSCs is the reprogramming of mRNA translation (Fabbri et al. 2021). In breast cancer, hypoxia triggers the translation of selective mRNAs that encode stem cell factors such as NANOG, SNAIL, and NODAL (Jewer et al. 2020). This process bolsters the plasticity of breast cancer cells, promoting metastasis and reducing their sensitivity to treatment (Jewer et al. 2020). Breast cancer stem cells (BCSCs) exhibit a high level of MNK1 activity (Preston et al. 2024). MNK1 drives the expression of factors such as aldolase A (ALDOA), lactate dehydrogenase A (LDHA), and Enolase 1 (ENO1), which are essential for maintaining stem cell-like properties and promoting breast tumor growth (Preston et al. 2024). MNK1 deletion in breast cancer cells results in markedly reduced stem cell-like properties, as evidenced by decreased expression of stem cell markers and diminished tumorsphere formation (Preston et al. 2024). MNK1 inhibition by the small molecule eFT508 mimics the effects of genetic knockout, reducing metastasis (Preston et al. 2024).
Tumor microenvironment
Translational control by the MNK/p-eIF4E pathway plays a critical role in the maintenance of the tumor microenvironment (TME; reviewed by Bartish and colleagues (Bartish et al. 2023). p-eIF4E impacts tumor-associated macrophages (TAMs) by selectively enhancing the translation of mRNAs encoding proteins that support an anti-inflammatory, tumor-promoting TAM phenotype (Bartish et al. 2020). Inhibiting MNK2 shifts TAMs toward a proinflammatory state, augmenting their ability to activate CD8+ T cells and promote antitumor immune responses (Bartish et al. 2020). Translational reprogramming of TAMs represents a novel strategy for enhancing the efficacy of immunotherapies by targeting the MNK2–eIF4E axis. Inhibiting the MNK/p–eIF4E pathway reduces prometastatic neutrophils in the TME by decreasing the translation of mRNAs encoding anti-apoptotic proteins such as MCL-1 and BCL-2 (Robichaud et al. 2018). In a mouse model of breast cancer, treatment with merestinib, a multikinase inhibitor that suppresses MNK activity, reduces the formation of lung metastasis by suppressing neutrophil accumulation (Robichaud et al. 2018).
Cancer invasion and metastasis
Translational adaptation is a key characteristic of metastatic cells (Chen et al. 2024). Ample evidence exists that translational dysregulation plays an important role in different stages of cancer invasion and metastasis, as previously summarized (Micalizzi et al. 2021). Blocking p-eIF4E (eIF4ES209A) in a transgenic mouse model of breast cancer (eIF4ES209A-PyMT mice) prevents the development of spontaneous lung metastasis by suppressing the translation of Mmp3 and Snail (Robichaud et al. 2015), which are required in epithelial-mesenchymal transition (EMT). Targeting MNK1 in glioblastoma cells reduces the expression of key EMT markers, including zinc finger E-box-binding homeobox 1 (ZEB1), SNAIL, and SLUG (Wan et al. 2022). MNK1 is also implicated in driving tumor invasion and metastasis by upregulating the translation of mRNAs encoding pro-invasive proteins such as angiopoietin-like 4 (ANGPTL4) and MMPs in melanoma (Yang et al. 2020b). Constitutive activation of MNK1 promotes the expression of the genes involved in extracellular matrix remodeling, cell adhesion, and cancer metastasis. Inhibiting MNK1/2 with SEL201 reduced melanoma metastasis in mice, highlighting the therapeutic potential of MNK1/2 inhibition in aggressive cancers (Yang et al. 2020b).
Cancer immune evasion
Immune evasion is a key hallmark of cancer (Ghorani et al. 2023). A subset of mRNAs encoding proteins important for immune evasion are translationally regulated (Cerezo et al. 2021). In breast cancer, the inhibition of the MNK1/2–eIF4E axis reduces the expression of fibroblast-derived interleukin 33 (IL-33), which is an alarmin molecule that promotes immune suppression and invasion of cancer cells (Guo et al. 2021). Inhibiting the MNK1/2–eIF4E pathway relieves immune suppression, enhancing the infiltration of CD8+ T cells into the lung, which reduces breast cancer metastasis (Guo et al. 2021). In prostate cancer, the MNK1/2–eIF4E axis plays a pivotal role in immune evasion by promoting the translation of mRNAs encoding immunosuppressive factors (Brina et al. 2023). Activation of the MNK1/2–eIF4E pathway in PTEN-null prostate cancer cells upregulates hepatocyte growth factor (HGF), osteopontin (SPP1), and biglycan (BGN), thereby suppressing antitumor immune responses via the recruitment of myeloid-derived suppressor cells (MDSCs) (Brina et al. 2023). Blocking the MNK/eIF4E axis using eFT508 decreased MDSC infiltration and enhanced CD8+ T-cell activity in prostate cancer mouse models (Brina et al. 2023). Thus, targeting the MNK1/2–eIF4E axis not only disrupts tumor growth, but also restores immune surveillance, rendering it a potential therapeutic approach in prostate cancer. Furthermore, combining eFT508 with immunotherapies that target MDSCs or T cells may produce synergistic effects, improving patient outcomes by counteracting immune evasion.
Cancer resistance
MNK1/2 activation plays an important role in cancer drug resistance. For example, bromodomain and extraterminal (BET) inhibitors (BETi), which target transcription factors like MYC, activate MNK1/2, leading to increased p-eIF4E (Pham et al. 2019). p-eIF4E provides a feedback mechanism that confers resistance to BETi-induced proliferation inhibition (Pham et al. 2019). MNK1/2 inhibition in combination with BETi impairs cancer cell survival, particularly in 3D culture models (Pham et al. 2019). The MNK1/2–eIF4E axis also contributes to resistance against the CDK4/6 inhibitor, palbociclib, in melanoma and breast cancer (Prabhu et al. 2023). p-eIF4E promotes the translation of survival proteins in response to CDK4/6 inhibition, enabling cancer cells to evade cell-cycle arrest (Prabhu et al. 2023). MNK1/2 inhibition, in combination with palbociclib, enhances antitumor activity, reduces cancer cell survival, and increases cellular senescence (Prabhu et al. 2023). The dual targeting resensitizes resistant cells to CDK4/6 inhibitors, offering a promising therapeutic combination for treating patients with resistant melanoma and breast cancer.
In summary, p-eIF4E is a key player in cancer progression, metabolism, immune evasion, and metastasis in multiple cancer types. Targeting the MNK1/2–eIF4E axis offers an ambidextrous approach to cancer therapy in combination with existing treatments such as metabolic interventions, CDK4/6 inhibitors, and immunotherapy (Table 1). The use of functional genomic screens should provide new combinatory treatments with MNK inhibitors for cancer therapy. For example, in a genome-wide CRISPR interference (CRISPRi) screen, the interaction of eIF4E with various cellular processes is critical for cancer cell survival (Kuzuoglu-Ozturk et al. 2021). Notably, the anti-apoptotic protein B-cell lymphoma-extra large (Bcl-xL) is a synthetic lethal partner of eIF4E (Kuzuoglu-Ozturk et al. 2021). Cotargeting p-eIF4E and Bcl-xL induces apoptosis and reduces tumor growth (Kuzuoglu-Ozturk et al. 2021). It is anticipated that cotargeting p-eIF4E in combination with its synthetic lethal partners will uncover vulnerabilities in cancer cells.
Targeting eIF4F-dependent mRNA translation for cancer treatment
TRANSLATION CONTROL BY THE mTORC1-eIF4F AXIS IN CANCER
mTOR complexes (mTORC1 and mTORC2) are cardinal signaling hubs regulating cell growth, proliferation, survival, and metabolism (Kim et al. 2017), and are implicated in 80% of cancers (Liu and Sabatini 2020). mTORC1 promotes eIF4E-mediated translation of mRNAs encoding c-Myc, cyclin D1, Snail, and Mcl-1 through the phosphorylation of 4E-BPs (Sun 2021). Although direct mutations of the mTOR kinase are rare, mutations of its upstream regulators lead to a hyperactive mTORC1 state in many cancers (Wood and Gutkind 2022). More than 30% of tumors have genetic alterations (copy number changes, mutations, and epigenetic changes) in the key components of the PI3K–Akt–mTOR pathway (Wood and Gutkind 2022). These mutations include the gain-of-function mutations of G protein-coupled receptors, receptor tyrosine kinases, Ras-MAPK, and PI3K–Akt pathways, or loss-of-function mutations of negative regulators such as phosphatase and tensin homolog (PTEN) and Tuberous sclerosis proteins 1 and 2 (TSC1 and TSC2) (Mak and Yeung 2004).
mTOR hyperactivation is a hallmark of KRAS-mutant lung cancer (Liang et al. 2019). KRAS mutations are prominent in NSCLC, with KRASG12C being a common variant (Kim et al. 2020). Combining inhibitors of KRASG12C (RM-018) and mTORC1 (RMC-6272) shows synergistic anticancer effects in NSCLC by inhibiting cap-dependent translation of mRNAs encoding cyclin D1 and MCL-1, leading to cell cycle arrest and tumor regression (Kitai et al. 2024). The dual inhibition strategy negates compensatory mechanisms by cosuppressing the ERK and PI3K/mTOR pathways, highlighting the therapeutic potential of combining KRASG12C and mTORC1 inhibitors to achieve durable tumor suppression (Kitai et al. 2024). Considerable efforts were made to inhibit the upregulated mTOR signaling in cancer. A comprehensive list of mutations in the various regulators of mTOR signaling and therapeutic targeting of the axis is reviewed elsewhere (Rodrik-Outmezguine et al. 2016; Álvarez-Garcia et al. 2019; Popova and Jücker 2021; Wood and Gutkind 2022). Here, we summarize recent reports examining the mTOR-eIF4F axis in cancer.
Regulation of ferroptosis in cancer
Ferroptosis is an iron-dependent form of cell death that promotes lipid peroxidation and plays a critical role in cancer suppression (Zhang et al. 2022). Ovarian cancer develops resistance to ferroptosis by maintaining high levels of the cystine/glutamate antiporter SLC7A11 protein (Hong et al. 2021; von Krusenstiern et al. 2023; Fantone et al. 2024). The mTORC1 increases the synthesis of SLC7A11 via 4E-BP1, which promotes resistance to MEK inhibitor-induced ferroptosis (Yin et al. 2024). Inhibition of mTOR/4E-BPs signaling restored sensitivity to ferroptosis by reducing the synthesis of SLC7A11 protein (Yin et al. 2024). Dual inhibition of the mTOR and MEK pathways or combining eIF4E inhibitors with ferroptosis inducers could be effective strategies for treating cancers.
Cancer metabolism
Dietary modification has emerged as a key therapeutic strategy for cancers (Kanarek et al. 2020). The interplay between mRNA translation and cancer metabolism in relation to mTOR signaling has been extensively reviewed (Topisirovic and Sonenberg 2011; Biffo et al. 2018). A recent study demonstrated the importance of 4E-BPs in supporting cancer cell survival under glucose deprivation by regulating fatty acid metabolism (Levy et al. 2024). Inhibition of mTORC1 in response to glucose deprivation leads to activation of 4E-BP1/2, which suppresses the translation of mRNA encoding Acetyl-CoA Carboxylase1 (ACC1), a key enzyme in fatty acid synthesis (Levy et al. 2024). ACC1 inhibition conserves the levels of the universal electron donor nicotinamide adenine dinucleotide phosphate (NADPH), reducing oxidative stress and promoting cell viability (Levy et al. 2024). Tumor cells, particularly gliomas and glioblastoma, exploit this pathway by upregulating 4E-BP1 expression to adapt to glucose deprivation (Levy et al. 2024).
Cancer immunity
The link between mTOR/eIF4F pathway and immune evasion is well-established (Cerezo et al. 2021). Tumors resist mTORC1 kinase inhibitors (mTORkis) by maintaining PD-L1 expression via an internal ribosome entry site (IRES)-mediated translation mechanism (Cao et al. 2023). This bypass mechanism from translational inhibition allows tumors to evade immune detection by sustaining PD-L1 expression while cap-dependent translation is suppressed. Combining mTORkis with anti-PD-L1 immunotherapy effectively restored immune activity and improved tumor suppression in mouse models (Cao et al. 2023).
TARGETING THE eIF4F COMPONENTS, eIF4A and eIF4G, IN CANCER
eIF4A and eIF4G, which are core members of the eIF4F complex, play significant roles in cancer development.
eIF4A
eIF4A is a DEAD-box RNA helicase that unwinds secondary structures in the mRNA 5′ UTR to facilitate ribosome recruitment and scanning (Pause and Sonenberg 1992; Chu and Pelletier 2015). The eIF4A family consists of three members: eIF4A1 (DDX2A), eIF4A2 (DDX2B), and eIF4A3 (DDX48, which is a nuclear protein involved in NMD) (Nielsen and Trachsel 1988; Weinstein et al. 1997; Bartkowska et al. 2018). Given eIF4A's cardinal role in the translation of mRNAs encoding oncoproteins such as c-Myc, MYB, NOTCH, CDK6, and BCL2 (Wolfe et al. 2014), it has been the subject of extensive research as a therapeutic target in cancer (Chu and Pelletier 2015; Modelska et al. 2015). The landmark discoveries of Pateamine A (Low et al. 2005), Hippuristanol (Bordeleau et al. 2006), and silvestrol (Bordeleau et al. 2008) paved the way for targeting eIF4A for the treatment of cancers (Kuznetsov et al. 2009; Tsumuraya et al. 2011; Cencic and Pelletier 2016). Silvestrol converts to an inactive silvestric acid when given intravenously (Saradhi et al. 2011), which prompted the exploration of a new generation of eIF4A inhibitors. The second generation of eIF4A inhibitors, MG-002 (Cencic et al. 2024), and eFT226 (zotatifin) (Pelli et al. 1988), suppress the growth of several cancers in mice, including triple-negative breast cancer (TNBC) (Cencic et al. 2024), non-small cell lung cancer (NSCLC) (Nardi et al. 2023), and osteosarcoma (OS) (Lizardo et al. 2024).
MG-002 contains a rocaglate scaffold, which clamps eIF4A onto selective mRNAs, particularly those enriched in purine stretches, causing inhibition of cap-dependent mRNA translation initiation (Cencic et al. 2024). MG-002 induces apoptosis and reduces TNBC proliferation by inhibiting the translation of mRNAs encoding critical oncogenic proteins such as c-Myc and cyclin D1 (Cencic et al. 2024). MG-002 impairs breast tumor growth and metastasis in mouse models, particularly the colonization of lung tissue, the preferred site of TNBC metastasis (Cencic et al. 2024). Furthermore, its oral bioavailability is of potential for use in combination with standard chemotherapy, such as doxorubicin, as it exhibits synergistic effects in reducing primary tumor growth and metastatic burden (Cencic et al. 2024).
eFT226 synergizes with the KRASG12C inhibitors MRTX849 in NSCLC by promoting apoptosis through suppression of BCL-2 family proteins (Nardi et al. 2023). A recent study illustrated the potential of combining eIF4A inhibitors with the DNA repair-targeting agent carboplatin (Zhao et al. 2023). The eIF4A inhibitor, (-)-SDS-1–021, enhances the radiosensitivity of tumor cells by impairing DNA damage repair, offering a novel approach to improving radiotherapy efficacy (Lehman et al. 2022).
A recent study uncovered an intricate oncogenic signaling where mTORC1 promotes the phosphorylation of the inhibitor of Bruton's tyrosine kinase (IBTK) via ribosomal protein S6 kinase B1 (S6K1) (Jiao et al. 2024). Phosphorylated IBTK forms a complex with the Cullin 3-RING ubiquitin ligase (CRL3) (CRL3IBTK), which monoubiquitinates eIF4A1 at multiple lysines (Jiao et al. 2024). The ubiquitination does not mark eIF4A1 for degradation but instead enhances its activity. The ubiquitinated eIF4A1 promotes cap-dependent translation of oncogenic mRNAs involved in the STAT1-IRF1-PD-L1 pathway, thereby supporting cancer cell growth, proliferation, and immune evasion (Cerezo et al. 2018; Jiao et al. 2024). Given the centrality of IBTK in controlling eIF4A1, its targeting offers a novel therapeutic approach to impair cap-dependent translation and enhance antitumor immunity.
eIF4G
The eIF4G family of scaffolding proteins comprises three members: eIF4G1 (first named eIF4GI), eIF4G2 (commonly referred to as Death Associated Protein 5, or DAP5, and originally called p97) (Imataka et al. 1997), and eIF4G3 (first named eIF4GII) (Gradi et al. 1998). The first member of the eIF4G family, eIF4G1, was discovered in 1983 through its interaction with eIF4E (Grifo et al. 1983). eIF4G1 is the most abundant member of the eIF4G family (Kim et al. 2023), and its amplification has been identified in multiple cancers, including prostate, ovarian, head and neck, and cervical cancer (Jaiswal et al. 2019). 4EGI-1 and 4E1RCat are compounds that interfere with the interaction of eIF4G1 with eIF4E (Moerke et al. 2007). Application of both compounds as mono- or dual-therapy shows anticancer effects in a wide range of malignancies (multiple myeloma, prostate cancer, breast cancer, NSCLC, hepatocellular carcinoma, glioma) (Descamps et al. 2012; Wang et al. 2015; Wu et al. 2016; Jaiswal et al. 2018; De et al. 2019; Fang et al. 2021). 4EGI-1 not only blocks eIF4E-dependent translation, but also eIF4E-independent translation, potentially by activating stress response mechanisms (Willimott et al. 2013). Disrupting the eIF4F complex with SBI-756, which blocks eIF4G–eIF4E interaction, sensitizes B-cell lymphomas to venetoclax (a BCL-2 inhibitor) (Herzog et al. 2021). SBI-756 induces cancer cell apoptosis more effectively when combined with venetoclax (Herzog et al. 2021). In pancreatic ductal adenocarcinoma (PDAC), eIF4G1 drives tumor progression and immune evasion by augmenting the translation of mRNAs encoding oncogenic and immune-modulatory proteins (He et al. 2024). Targeting eIF4G1 with SBI-756 disrupts the desmoplastic (highly fibrotic and dense) and immunosuppressive TME by reducing collagen production and depleting TAMs, MDSCs, and regulatory T cells (Tregs) (He et al. 2024). This leads to increased infiltration of CD8+ T cells, thus restoring antitumor immunity. Combining eIF4G1 with anti-PD-1/PD-L1 immune checkpoint inhibitors and chemotherapy demonstrated synergistic effects, suppressing tumor growth, and improving survival in preclinical settings (He et al. 2024).
DAP5 cannot bind eIF4E, precluding it from initiating eIF4E-dependent translation (Pelletier and Sonenberg 2019). Instead, DAP5 controls IRES-dependent translation of mRNAs encoding proteins that function in cell invasion, apoptosis, and cancer progression, such as p53, BCL-2, and Apaf-1 (Hundsdoerfer et al. 2005; Weingarten-Gabbay et al. 2014; Haizel et al. 2020). DAP5 is also implicated in eIF4E-independent cap-dependent translation through its interaction with eIF3d (Lee et al. 2016; de la Parra et al. 2018). The DAP5/eIF3d complex facilitates the synthesis of EMT-related proteins and survival factors in TNBC under stress conditions (Alard et al. 2023). Therefore, targeting the DAP5–eIF3d axis offers an attractive approach to halt cancer progression.
CONCLUSIONS AND FUTURE PERSPECTIVES
The eIF4F complex is a major convergence point for key signaling pathways, and is central to many critical processes in cancer biology, including tumor growth, immune evasion, and drug resistance. Targeting eIF4F with mTOR, MNKs, and eIF4F inhibitors holds potential for treating many types of cancers. The development of combination therapies (consisting of mTOR inhibitors with immunotherapies or MEK inhibitors) is a promising strategy to overcome drug resistance and enhance treatment efficacy.
The novel inhibitors of eIF4A, MG-002, and eFT226, demonstrate therapeutic efficacy on TNBC (Cencic et al. 2024) and NSCLC (Nardi et al. 2023) in preclinical models, respectively. While the eIF4G inhibitor, SBI-756, effectively sensitizes aggressive lymphomas to existing therapies like venetoclax (Herzog et al. 2021). These findings highlight the versatility and therapeutic spectrum of eIF4F inhibitors, particularly when combined with other treatment modalities. Future studies should pursue the application of these inhibitors across various cancer types and resistance mechanisms, and optimize combination strategies to improve clinical outcomes.
To better understand the mechanisms of action of eIF4F inhibitors and to optimize their therapeutic use, ribosome profiling and other omics approaches must be employed. These technologies should unveil the specific mRNA targets and pathways disrupted by eIF4F inhibitors. The findings presented in this Perspective document the seminal role of mRNA translational control in the pathophysiology of cancer. Substantial advancements have been made in identifying and developing small molecule inhibitors that remedy dysregulated translation in different cancers (Herzog et al. 2021; Nardi et al. 2023; Cencic et al. 2024). Future research should focus on maximizing their potential through functional genomics screens. CRISPR knockouts (CRISPRko), activation (CRISPRa), and interference (CRISPRi) could uncover new combinatory strategies to enhance the efficacy of anticancer treatment.
ACKNOWLEDGMENTS
The cancer research in the laboratory of N.S. is funded by a grant from the Canadian Cancer Research Society (CRS; #1051778). Research in the laboratory of S.T. is funded by National Institutes of Health (NIH) R01 HL163806. N.M. is supported by a postdoctoral fellowship from the Charlotte and Leo Karassik Foundation. M.A was supported by a doctoral award from Fonds de recherche du Québec - Santé (FRQS). We thank Michael A. Bellucci for his review of the manuscript.
Footnotes
-
Article is online at http://www.rnajournal.org/cgi/doi/10.1261/rna.080340.124.
-
Freely available online through the RNA Open Access option.
This article, published in RNA, is available under a Creative Commons License (Attribution-NonCommercial 4.0 International), as described at http://creativecommons.org/licenses/by-nc/4.0/.








