
Exploring the therapeutic potential of modulating nonsense-mediated mRNA decay
- 1ReviR Therapeutics, Brisbane, California 94005, USA
- 2Department of Biochemistry and Biophysics, School of Medicine and Dentistry, University of Rochester, Rochester, New York 14642, USA
- 3Center for RNA Biology, University of Rochester, Rochester, New York 14642, USA
- Corresponding authors: mary.mcmahon{at}revirtx.com, lynne_maquat{at}urmc.rochester.edu
Abstract
Discovered more than four decades ago, nonsense-mediated mRNA decay (NMD) plays a fundamental role in the regulation of gene expression and is a major contributor to numerous diseases. With advanced technologies, several novel approaches aim to directly circumvent the effects of disease-causing frameshift and nonsense mutations. Additional therapeutics aim to globally dampen the NMD pathway in diseases associated with pathway hyperactivation, one example being Fragile X syndrome. In other cases, therapeutics have been designed to hijack or inhibit the cellular NMD machinery to either activate or obviate transcript-specific NMD by modulating pre-mRNA splicing. Here, we discuss promising approaches employed to regulate NMD for therapeutic purposes and highlight potential challenges in future clinical development. We are optimistic that the future of developing target-specific and global modulators of NMD (inhibitors as well as activators) is bright and will revolutionize the treatment of many genetic disorders, especially those with high unmet medical need.
Keywords
- NMD
- pre-mRNA splicing
- small-molecule modulators of splicing
- site-directed RNA editing
- small-molecule modulators of translation termination
- gene therapy
NMD OVERVIEW: A SPLICING- AND TRANSLATION-DEPENDENT PATHWAY THAT OPENS SEVERAL NEW AVENUES FOR THERAPEUTIC INTERVENTION IN DISEASE
Acting at the interface of pre-mRNA processing in the nucleus and mRNA translation in the cytoplasm, nonsense-mediated mRNA decay (NMD) is a conserved eukaryotic RNA surveillance pathway best characterized for its disease-associated role in selectively degrading transcripts arising from a nonsense or frameshift mutation (Lykke-Andersen and Jensen 2015; Kurosaki et al. 2019). Key NMD regulators, including the Up-frameshift proteins (UPFs) and suppressor with morphogenetic effect on genitalia proteins (SMGs), recognize and eliminate aberrant cytoplasmic mRNAs containing a premature termination codon (PTC) arising from mutations and also from errors in gene transcription or pre-mRNA splicing, thereby protecting cells against the production of potentially deleterious or otherwise toxic truncated proteins (Hug et al. 2016). In humans, as in all mammals examined, PTCs are generally distinguished from physiological stop codons by the cellular NMD machinery based upon their location relative to a downstream exon–exon junction, “marked” during the process of splicing by an exon-junction complex (EJC). A PTC located more than ∼50–55 nt upstream of an exon–exon junction that is marked by an EJC triggers NMD of the transcript (Nagy and Maquat 1998), with cis-elements, such as an upstream open reading frame (uORF) or an intron within the 3′ untranslated region (UTR), also harnessing the potential to trigger NMD (Jagannathan and Bradley 2016). A long and/or structured 3′ UTR downstream from either a PTC or a native stop codon can also trigger NMD by a poorly defined mechanism (Mendell et al. 2004; Bühler et al. 2006). With advances in understanding the basic mechanisms of the NMD pathway, tremendous progress has been made in the development of therapeutics to treat NMD-associated diseases or “NMD-opathies.” Continuous efforts to improve the understanding of the molecular and cellular basis of NMD, particularly in disease contexts, are critical to propel novel innovations that will lead to the development of disease-modifying drugs.
As a major source of transcriptome plasticity and proteome diversity, alternative splicing and NMD are closely coupled (Weischenfeldt et al. 2012), particularly in response to cellular stress (Karam et al. 2015; Neumann et al. 2020). This expansion of coding potential, however, is accompanied with many mistakes: it is estimated that almost one-third of alternatively spliced mRNA isoforms contain a PTC that triggers isoform degradation by NMD (Lewis et al. 2003; Pan et al. 2006). NMD regulates splicing factor expression and additional proteins involved in RNA processing, suggesting the existence of tight autoregulatory loops. For example, splicing factors such as SR proteins downregulate their own production by generating an NMD-sensitive mRNA isoform from the pre-mRNA that encodes them, thereby regulating splicing more globally (Ni et al. 2007; Saltzman et al. 2008). In a number of genetic diseases, the close coupling of alternative splicing and NMD has prompted a wave of new therapeutic developments to regulate splicing as a means of modulating NMD. Splicing of pre-mRNA from a disease-causing gene, for example, may be modulated to eliminate a deleterious nonsense mutation as in the case of Duchenne muscular dystrophy (DMD). In haploinsufficiency disorders, alternative splicing of pre-mRNA from the normal allele that results in a naturally occurring PTC-containing isoform a fraction of the time may be corrected to augment full-length protein expression from this allele (McNally and Wyatt 2016; Neil et al. 2022). Small-molecule splicing modulators have been used to promote the inclusion of a PTC-containing cassette exon, termed a “poison exon,” into the mature transcript to induce selective degradation of that transcript by NMD, such as in Huntington's disease (HD) (Bhattacharyya et al. 2021; Keller et al. 2022). Methods and therapeutics that alter pre-mRNA splicing hold tremendous potential for the treatment of diseases caused by nonsense or frameshift mutations as well as other diseases wherein varying transcript levels by modulating NMD may be beneficial.
As NMD is dependent on translation, a major determinant of whether a transcript is degraded by NMD is translation termination. Various factors, including the dynamics of translation termination, the translation termination site, and the protein composition of the mRNA-containing ribonucleoprotein complex can be used to predict the sensitivity of an mRNA to NMD (Karousis and Mühlemann 2019). The RNA helicase UPF1 is crucial for NMD activation through its interaction with the eukaryotic release factor (eRF)1 and eRF3 at the terminating ribosome and by its phosphorylation by SMG1, AKT1, or both (Maquat 2004; Kashima et al. 2006; Sato and Singer 2021; Cho et al. 2022). Despite several questions remaining regarding the mechanism and criteria for the selection of an mRNA for NMD, it is clear that effective suppression of translation termination at a PTC (also referred to as readthrough) impedes NMD (Howard et al. 1996; Spelier et al. 2023). While early efforts mainly focused on the development of small molecules to induce translational readthrough at PTCs, a new wave of therapeutics focuses on nonsense suppression by other means. These include exciting RNA-based therapeutics that convert a functional PTC to a coding codon, thereby evading NMD and generating full-length protein, ideally in a transcript-specific manner. Here, we delve into and share our opinion on recent developments in the field of NMD therapeutics (Holbrook et al. 2004; Kuzmiak and Maquat 2006) with an emphasis on the latest advances in splicing modulators and nonsense suppression approaches.
THERAPEUTIC APPROACHES FOR DISEASE-CAUSING NONSENSE MUTATIONS
The NMD pathway plays an important role in diverse cellular processes, including hematopoiesis (Weischenfeldt et al. 2008), neurodevelopment (Petrić Howe and Patani 2023), and tumorigenesis (Tan et al. 2022). Since NMD is always “on” in cells (Lou et al. 2016), it is not surprising that NMD underlies and is associated with numerous disorders, including Duchene muscular dystrophy (DMD), cystic fibrosis, hemophilia, and other more common diseases, like cancers and neurological disorders. In fact, it is estimated that more than 10% of inherited genetic diseases are caused by nonsense mutations in coding regions that produce an in-frame PTC, rendering the transcripts NMD targets that fail to produce functional protein (Mort et al. 2008). NMD-sensitive transcripts arise from insertions or deletions, some of which are due to splice-site mutations, that lead to shifts in the translational reading frame and generate a downstream stop codon. In this section, we discuss approaches to dampen or inhibit NMD of disease-causing nonsense or frameshift mutations (Fig. 1). We are optimistic that novel therapeutic modalities will continue to be developed that will lead to life-saving treatments for patients. We find great value in the continuous discovery of more selective therapeutic interventions for existing modalities, testing in a broader range of genetic diseases, including ultrarare diseases, with a focus on those with no current treatment options, and learning from previous challenges in drug development to ensure future clinical success.

Emerging therapeutic strategies for diseases caused by frameshift and/or nonsense mutations. The main types (top panel) and features (bottom panel) of promising therapeutic modalities either already developed or in clinical and preclinical development for a range of diseases associated with PTC-causing mutations are shown. These include methods for gene replacement, RNA replacement or editing, including splicing modulators, and nonsense-suppression strategies to overcome the deleterious effects of PTC-causing mutations. Strategies to induce targeted mRNA decay in the case of dominantly inherited diseases are also shown.
Gene replacement or editing therapeutics
The past two decades have seen tremendous progress in the field of gene therapy with several successes in the clinic, including the development of Zynteglo for patients with transfusion-dependent β-thalassemia (Locatelli et al. 2022; Kohn et al. 2023). Gene replacement therapies involve viral-based gene delivery systems that rely on the ability of viruses to infect cells and deliver a functional version of a gene (either the full-length gene or a gene fragment, each of which would encode functional protein), thereby having the potential to circumvent the effects of the disease-causing nonsense codon (Fig. 1, first panel). In addition to directly replacing a nonsense- or frameshift-mutated gene with DNA that produces functional protein, this approach can also be used to deliver other therapeutic modalities. A splice-switching antisense oligonucleotide (ASO) (Kim et al. 2023) or trans-splicing components (Berger et al. 2016) would modulate splicing to overcome the effects of a disease-causing nonsense codon. Recombinant adeno-associated viral vectors (AAVs) are used due to their small size, high efficiency, and tunable tissue delivery or “tropism.” However, some issues around safety and manufacturing challenges, including cost, remain a concern for clinical development. High-dose AAV delivery has been shown to induce the presence of anti-AAV antibodies that pose significant problems (Smith et al. 2022), including the onset of thrombotic microangiopathy as observed in DMD clinical trials (Salabarria et al. 2024). Additional challenges include treatment duration to achieve a long-lasting therapeutic effect given that providing a second dose is not an option, and concern over the risk of hepatotoxicity or genotoxicity using lentiviral vectors (Kohn et al. 2023; Whiteley 2023).
Since the discovery of genome engineering by CRISPR–Cas9 (Doudna and Charpentier 2014), CRISPR–Cas9-mediated gene editing has become an invaluable tool for biomedical research and an approved treatment option for some diseases, including the use of Casgevy for sickle cell disease (Frangoul et al. 2021). The ability of this sophisticated RNA-guided methodology to modify a specific genomic locus, either at a distinct promoter region or to replace part or all of a gene, has revolutionized cell-based gene therapies. While still in the early days of therapeutic development, CRISPR–Cas9-mediated gene editing harnesses huge potential to replace or correct a mutated gene, including those with a frameshift or nonsense mutation. Several preclinical discoveries and strategies highlight the potential of CRISPR–Cas9 and related gene-editing approaches for the correction of homozygous and heterozygous nonsense variants in diseases such as cystic fibrosis or Leber congenital amaurosis (Afanasyeva et al. 2023; Vaidyanathan et al. 2024).
Additional advancements in this field have led to the development of CRISPR tools that efficiently correct point mutations by making a single-nucleotide change in a DNA or RNA sequence in a process called base-editing. This method uses components of the CRISPR system together with other enzymes to directly correct point mutations without making double-stranded DNA breaks (Komor et al. 2016; Gaudelli et al. 2017). Both cytidine deaminase-mediated base editors (CBEs) and adenine base editors (ABEs) have been described, with CBEs producing C-to-T or G-to-A substitutions and ABEs producing A-to-G or T-to-C substitutions. Base-editing therefore holds enormous potential to bypass the effects of a nonsense mutation by correcting the mutation, rescuing RNA and protein expression. This method has been successful in preclinical models of various inherited diseases, including DMD, cystic fibrosis, and dystrophic epidermolysis bullosa (Geurts et al. 2020; Sheriff et al. 2022; Jin et al. 2024).
While this field undoubtedly holds promise for correcting nonsense mutations and offering a durable therapeutic strategy, several hurdles remain, including tuning on-target specificity and improving delivery methods, especially to the central nervous system. Studies in several species highlight the presence of unintended mutations caused by CRISPR–Cas9 at locations in the genome other than in the targeted site (Tuladhar et al. 2019; Höijer et al. 2022). Another challenge that is particularly relevant to base-editing is overcoming targeting limitations imposed by the PAM sequence. The PAM sequence must be positioned relatively close to the targeted base to ensure efficient editing, thereby limiting this technology to <30% of known pathogenic single-nucleotide changes (Rees and Liu 2018). Taking all these factors into account, we advocate for thorough validation and characterization of such approaches in preclinical models both in vitro in cell culture and in vivo in model organisms to enhance the development of safe therapeutics. A thorough investigation of how gene editing impacts mRNA dynamics, the amino acid sequence, and function of the encoded protein in different cell and tissue types is warranted. Advances in RNA sequencing methods (including long-read and single-cell RNA-sequencing) and proteomics tools (such as single-molecule protein-sequencing using a nanopore [Lucas et al. 2021; Motone et al. 2024]) will prove instrumental to fully characterizing the molecular and cellular consequences of gene editing at PTC-containing alleles.
RNA replacement or RNA-editing therapeutics, including splicing modulation
The success of mRNA vaccines for COVID-19 and its subsequent iterations have magnified advancements in mRNA replacement therapy with the goal of delivering mRNA to restore protein expression that has been lost due to disease-causing mutations, including nonsense, frameshift, and splice-site mutations (Rohner et al. 2022; Khorkova et al. 2023). The delivery of linear, usually chemically modified, mRNA to cells is proving promising for several rare genetic disorders, including metabolic disorders such as phenylketonuria (PKU) and propionic acidaemia (PA) (Pérez-García et al. 2022; Koeberl et al. 2024). Such advances offer renewed hope for patients and families with other incurable diseases caused by PTC-containing alleles (Fig. 1, second panel). Indeed, promising results from a clinical trial for PA by Moderna, Inc. highlights the potential of mRNA replacement in diseases with unmet medical needs. Unfortunately for cystic fibrosis patients, an inhaled mRNA replacement therapy, MRT5005, although relatively safe and well-tolerated, failed to exhibit clear clinical benefits in patients (Rowe et al. 2023), highlighting room for improvement in this therapeutic area. Indeed, some of the biggest challenges in mRNA replacement therapy lies in optimizing the mRNA “cargo,” improving delivery systems for enhanced tissue tropism, and developing minimally invasive delivery methods to patients. Relating to mRNA cargo, several factors that are intrinsic to cells impact the relationship between mRNA levels and production of the encoded protein. mRNA features such as structure, stabilizing elements, and modifications all impact mRNA stability and translation efficiency, and they often lead to instability and immunogenicity issues (Metkar et al. 2024). Recent advances in this field have seen the development of circular RNA (circRNA)-based therapies that lead to enhanced mRNA half-life and translation efficiency of the introduced mRNA (Chen et al. 2023b; Sun and Yang 2023). In the coming years, important lessons are expected to emerge from the development of circRNA vaccines (Qu et al. 2022) that will be highly applicable for mRNA replacement therapy in the rare disease setting.
As mentioned above, the tight coupling of NMD and pre-mRNA splicing offers ample opportunities to develop innovative treatments for PTC-causing diseases by targeting splice-site selection (Fig. 1, second panel and Fig. 2; Supek et al. 2021; Neil et al. 2022). ASO and small-molecule approaches have been successfully developed to modulate the major spliceosome and overcome the devastating effects of a nonsense or frameshift mutation that results in a PTC. Several splice-switching ASOs have been developed and approved to treat DMD patients harboring a specific PTC in one of several exons (e.g., exon 45, 51, or 53) within dystrophin pre-mRNA (Servais et al. 2022; Clemens et al. 2023). ASO binding to specific regions within the pre-mRNA leads to skipping of the exon harboring the PTC, thereby allowing for production of an mRNA that is not targeted for NMD (Fig. 2, left panel). This strategy results in the production of a truncated but functional mini-dystrophin protein in patients suffering from DMD. Splice-switching using ABE has been described for DMD caused by a single base-pair substitution at a splice donor or acceptor site. This so-called “single-swap editing” approach leads to exon skipping of the PTC-containing exon and shows beneficial effects in preclinical models of DMD using AAV delivery methods (Chai et al. 2023).
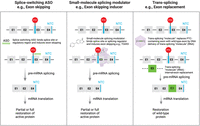
Therapeutic approaches targeting pre-mRNA splicing. Modalities targeting RNA splicing to circumvent the deleterious effects of PTC-causing mutations are shown with the position of the normal termination codon (NTC) indicated. Splice-site selection may be modulated by a splice-switching ASO (left) or a small molecule that manipulates protein–RNA interactions at or near exon–intron junctions (middle), both of which may induce exon skipping of a PTC-containing exon during pre-mRNA splicing and may lead to the restoration of partial or full protein activity. RNA trans-splicing may also be used to replace a PTC-containing exon (right), with internal exon replacement depicted in the diagram. Exon replacement using trans-splicing may lead to restoration of full-length wild-type protein. Notably, exon 3 in the two leftmost panels needs to consist of a multiple of three nucleotides. E, exon; lines between exons, introns.
Other strategies that use a splice-switching ASO have been developed to treat diseases caused by haploinsufficiency. In these strategies, the splicing of a fraction of pre-mRNA that is normally produced from the nonmutated allele but that generates an NMD-prone transcript is redirected to skip the poison exon, thereby upregulating the amount of cellular product from this allele to above the usual 50% of normal levels. In the case of Dravet syndrome, which manifests as a severe myoclonic form of epilepsy, a splice-switching ASO developed by Stoke Therapeutics is currently in phase 1/2 clinical trials and targets normal SCN1A pre-mRNA to exclude a poison exon during pre-mRNA splicing. The strategy upregulates SCN1A mRNA expression and the level of the encoded sodium channel Nav1.1 protein, offering the potential to treat this devastating disease (Lim et al. 2020). Modulating alternative splicing of pre-mRNA, produced by a normal allele, to circumvent mis-splicing that generates an NMD-prone transcript, may offer new hope for elevating the level of protein derived from the normal allele so as to obviate certain genetic diseases.
Several challenges exist with ASOs, including the need for repeated administration and their low bioavailability to, for example, muscle, spinal cord, and brain. This is particularly evident in the case of spinal muscular atrophy (SMA), where an orally available small-molecule splicing modulator called Evrysdi (Risdiplam) activates functional protein production from the normally inactive SMN2 gene either alone or in combination with a splice-switching ASO (Baranello et al. 2021; Kokaliaris et al. 2024). Given the success of Evrysdi, additional efforts should shift to the development of next-generation orally bioavailable small-molecule splicing modulators that induce exon skipping of PTC-containing exons without inducing a shift in the translational reading frame (Fig. 2, middle panel). Developments in this area include the discovery of a small-molecule CLK1 inhibitor, TG693, that promotes the skipping of exon 31 within the dystrophin pre-mRNA in both DMD patient cells and a mouse model of DMD (Sako et al. 2017). Orally bioavailable small molecules offer advantages over ASOs and gene therapy approaches that use viral delivery. First, they do not require invasive procedures for administration, thereby avoiding the risks associated with repeated intrathecal injections or other delivery methods. Second, any potential adverse effects of drug administration may be mitigated by dose titration or by stopping administration at any point. Ongoing and future efforts to identify small-molecule splicing modulators to help bypass the deleterious effects of PTC-causing mutations and to treat a broader range of genetic diseases are important, especially for neurological disorders where brain exposure remains a challenge with ASOs. Once the molecular cause of a disorder has been identified, then a search for an orally available splicing modulator can begin.
Another very promising area for therapeutic development of diseases associated with nonsense and frameshift mutations, including splice-site mutations, is trans-splicing (Berger et al. 2016). First observed in trypanosomes (Van der Ploeg et al. 1982), RNA trans-splicing involves the joining of one RNA to another to form a single chimeric RNA. This method can be used to repair mutated exons within pre-mRNA, in theory using either spliceosome-mediated or ribozyme-mediated trans-splicing (Sullenger and Cech 1994; Puttaraju et al. 1999; Berger et al. 2016). This posttranscriptional gene-editing approach employs engineered exons in proximity to a disease-causing mutated exon and is usually delivered as DNA using viral vectors (Fig. 2, right panel). The enormous potential of trans-splicing therapeutics is evident in preclinical studies to correct PTC-generating mutations in models of Sickle Cell Disease (Lan et al. 1998), DMD (Lorain et al. 2013), Frontotemporal dementia with parkinsonism linked to Chromosome 17 (FTDP-17) (Rodriguez-Martin et al. 2009), and HD (Rindt et al. 2012), and in an ongoing phase 1/2 clinical trial by Ascidian Therapeutics to correct disease-causing mutations in the ABCA4 gene that result in Stargardt disease. More recently, the utility of AAV-gene delivery, especially for longer genes, has been expanded by engineering the trans-splicing of two halves of an mRNA, each expressed from a different AAV, to reconstitute functional mRNA that encodes full-length protein (Yan et al. 2000; Riedmayr et al. 2023). While trans-splicing efficiency in vivo is usually relatively low, recent work to optimize trans-splicing via optimized ribozyme function has been used to restore levels of dystrophin and dysferlin protein in mouse models of DMD and a type of limb-girdle muscular dystrophy, respectively (Lindley et al., 2024). Thus, trans-splicing has the capacity to target a diverse spectrum of genetic diseases.
Translational readthrough or nonsense suppression therapeutics
As mentioned above, the dependency of NMD on translation, particularly translation termination, offers several opportunities to develop innovative treatments for PTC-associated diseases by targeting the process of protein synthesis, in particular decoding in the ribosome A-site (Fig. 1, third panel). For decades, substantial efforts have been placed, both at the discovery and clinical level, to advance therapeutics that allow for translation termination bypass, also called readthrough, at PTCs to restore protein expression (Supek et al. 2021; Spelier et al. 2023). The majority of these developments stem from findings that a class of antibiotics known as aminoglycosides, which bind to the ribosome A-site, modulate translation fidelity by promoting recognition of amino-acylated near-cognate transfer RNAs (tRNAs) at the A-site and lead to amino acid incorporation at a PTC, as demonstrated for a PTC within CFTR mRNA (Burke and Mogg 1985; Howard et al. 1996; Prokhorova et al. 2017). Readthrough-promoting small molecules, including the aminoglycoside-like molecules Gentamicin, G418, and ELX-02, and nonaminoglycoside compounds Amlexanox, and, reportedly, Translarna (Ataluren) have been shown to induce PTC readthrough. PTC readthrough then leads to synthesis of the full-length protein, and in several cases improve disease symptoms in patients and/or models of CF, DMD, Rett syndromes, dystrophic epidermolysis bullosa, and the peripheral neuropathy Charcot-Marie-Tooth disease (Wilschanski et al. 2003; Welch et al. 2007; Brendel et al. 2009; Benslimane et al. 2023; Chen et al. 2023a; Spelier et al. 2023; Woodley et al. 2024). However, Gentamicin and G418 are not suitable therapeutics in the long term due to nephrotoxicity and ototoxicity (Hayward et al. 2018), and overall disappointing clinical outcomes and failures of readthrough-promoting small molecules (including Translarna) (Spelier et al. 2023) have dampened enthusiasm for this approach and raised questions regarding the precise mechanism of action of drugs like Ataluren. Most recently, 2,6-diaminopurine, which posttranscriptionally methylates certain tRNAs, including the tRNA charged with tryptophan, has been identified as a strong suppressor of UGA PTCs in CFTR mRNA when administered orally or in utero in a mouse model of CF (Leroy et al. 2023).
To restore protein expression loss caused by nonsense mutations, a new direction is to inhibit a translation termination factor, such as eRF1 and eRF3, in the hope that their inhibition would increase readthrough by lowering translation termination at PTCs. So far, these efforts appear promising, and there are several new classes of molecules under development, including eRF1 degraders, that are showing therapeutic potential in models of CF, DMD, and Hurler disease (Baradaran-Heravi et al. 2021; Sharma et al. 2021; Lee et al. 2022; Gurzeler et al. 2023). While encouraging, attention should be paid to ensure that these molecules do not induce sufficient transcriptome-wide readthrough at native stop codons to cause side effects. While over 90% of native or annotated stop codons are followed by a downstream in-frame stop codon in the 3′ UTR (Wangen and Green 2020), readthrough into the 3′ UTR may be deleterious. A deeper understanding of the molecular and cellular basis of stop-codon readthrough (Loughran et al. 2014), and differences between premature translation termination and translation termination at native stop codons (Wangen and Green 2020), will greatly accelerate drug discovery efforts for nonsense suppression. Accelerating high-throughput screening approaches, particularly in a disease-relevant context, are warranted to aid in the discovery of additional classes of readthrough-promoting small molecules (Carrard et al. 2023).
Continuing to advance our understanding of how the NMD machinery itself is activated in response to distinct PTCs, how sequence and structural context impact NMD efficiencies (Khoroshkin et al. 2024), and the cross talk between the NMD pathway and the translation machinery (Zinshteyn et al. 2021), will significantly advance novel PTC-selective therapeutic developments. It is worth noting that NMD efficiency varies widely across the transcriptome, with a significant fraction of transcripts predicted to be sensitive to NMD escaping degradation by unknown mechanisms (Rivas et al. 2015; Sato and Singer 2021). Studies suggest that cell-to-cell variation in NMD efficiency may rely on the expression levels of surveillance factors, particularly SMG1 and phosphorylated UPF1. It is interesting to note that NMD efficiency may impact readthrough potential, and it has been proposed that damping NMD in combination with readthrough-promoting small molecules may be a new therapeutic avenue to consider for genetic diseases associated with NMD (Linde et al. 2007; Huang et al. 2018; Amar-Schwartz et al. 2023). Related to this observation, it seems feasible that stabilization or increasing the levels of an NMD-sensitive transcript may enhance or increase the efficacy of nonsense suppression and readthrough therapeutics.
The finding that the efficacy of readthrough-promoting small molecules can be increased by blocking components of the NMD pathway has led to the development of an innovative approach to induce stabilization of an NMD target by use of what are termed “EJC blockers.” This approach uses ASOs and relies on the fact that disrupting the binding of all EJCs that reside downstream from a PTC inhibits NMD of the host mRNA (Nomakuchi et al. 2016). For example, in a model of CF caused by CFTR-W1282X, a cocktail of ASOs targeting EJC-binding sites downstream from the PTC-containing exon disrupts the interaction between CFTR mRNA and EJC proteins, leading to increased expression of CFTR mRNA and, restoration of a truncated but partially functional protein (Kim et al. 2022). These findings hold promise for further therapeutic developments that attenuate NMD in a transcript-specific manner by blocking EJC deposition downstream from a disease-causing PTC. Due to the limitations of ASOs mentioned above, a new direction may be to focus on the discovery of orally bioavailable small molecules that act as EJC blockers in a transcript- and site-specific manner. Considering the importance of EJCs to mRNA metabolism, therapeutics would likely need to be tailored to target each site of PTC-distal EJC deposition.
Another emerging approach to suppress the deleterious effects of nonsense mutations involves nonsense suppressor tRNAs (sup-tRNAs) or anticodon-edited tRNAs (ACE-tRNAs) (Porter et al. 2021), which allow for insertion of an amino acid at a PTC, thereby restoring expression of full-length protein from a PTC-containing transcript. These engineered tRNAs are designed based on natural features of endogenous tRNAs, but harbor altered anticodons to allow for base-pairing with a PTC, and subsequent amino acid incorporation into the growing peptide chain during translation elongation. The premise for this mechanism is rooted in findings that, despite the extreme complexity and sophisticated nature of mRNA translation, incredible flexibility of the translation machinery exists as is evident from the discussions above regarding PTC readthrough and several regulatory mechanisms (Gesteland and Atkins 1996; Gebauer and Hentze 2004). While nonsense suppression using engineered tRNAs was first validated in Xenopus oocytes using a human beta-globin mRNA harboring a PTC (Temple et al. 1982), significant progress has recently been made to optimize not only the efficiency and specificity of suppression but also in vivo delivery of the suppressors, with clinical applications in mind. Several studies now demonstrate success in restoring the production and function of proteins for disease-relevant PTC-containing mRNAs, including CFTR, in both in vitro and in vivo models of the disease (Lueck et al. 2019; Ko et al. 2022; Wang et al. 2022; Albers et al. 2023), with startup companies such as Alltrna and Tevard Biosciences Inc. making progress toward the clinic. The efficiency of suppression using this approach is sufficiently high to inhibit NMD, allowing for more template mRNA availability and, therefore, likely more protein production. This is critical since PTCs that trigger NMD in an EJC-dependent mechanism target newly synthesized mRNAs: EJCs are largely absent from the bulk of cytoplasmic mRNAs, thereby rendering them immune to further decay by NMD (Kurosaki et al. 2019). Thus, PTC suppression during the window of time that a newly made cytoplasmic mRNA harbors EJCs, i.e., during the window of time that NMD reduces mRNA abundance, is critical to maintain mRNA levels as high as possible.
While there is a risk that engineered tRNAs may impact translation at native stop codons or other codons and may lead to transcriptome-wide errors in translation, ribosome profiling experiments suggest this is not necessarily the case upon sup-tRNA optimization. These studies reveal that the extent to which an engineered tRNA induces nonsense suppression is highly dependent on sequence context and translation speed upstream of the PTC (Bharti et al. 2024). Understanding translation elongation rate may accurately predict the suppression efficiency at a PTC and will be important to consider in sup-tRNA design. Engineered tRNAs may be delivered as DNA or RNA, with successes from both AAV-based delivery methods and lipid nanoparticles reported (Porter et al. 2021). Critical features of engineered tRNAs are sequences that dictate charging with the correct amino acid by endogenous aminoacyl-tRNA synthetases, and sequences that undergo posttranscriptional modifications to maintain stability and allow for optimal interactions with translation elongation factors, such as eukaryotic elongation factor 1A (eEF1A). Another important consideration when developing sup-tRNA therapeutics is how the engineered tRNAs are processed or degraded in cells. tRNA-derived fragments appear to have a significant role in protein synthesis and cellular processes (Guzzi and Bellodi 2020). It becomes important to fully characterize how the degradation products of exogenously introduced sup-tRNAs might alter cellular metabolism. While the strong therapeutic potential of engineered tRNAs is clear from preclinical studies, their journey to the clinic will likely face challenges, especially with regard to design, delivery, and safety. As sup-tRNAs are a suitable therapeutic for nonsense but not missense or frameshift mutations, a new area for development includes missense-correcting tRNAs (mc-tRNAs) as potential treatment for disease-causing missense mutations (Hou et al. 2024). And, at least in Saccharomyces cerevisiae, a tRNA harboring a 4 nt anticodon appears to restore a reading frame harboring a single-nucleotide insertion to normal (Gaber and Culbertson 1984), suggesting that such a therapeutic could be tailored for small frameshift mutations. Understanding the structural basis of nonsense suppression using engineered tRNAs at the ribosome will undoubtedly shed new insights into the design of next-generation sup-tRNAs and will help advance studies to patients with unmet medical needs.
Posttranscriptional modifications are widespread across the transcriptome, enhancing both chemical and functional diversity, and providing an additional layer of gene expression control (Roundtree et al. 2017; McCown et al. 2020). Pseudouridine (Ψ), an isomer of uridine, is found in most classes of RNA and is commonly employed in the field of RNA therapeutics to reduce RNA immunogenicity and increase RNA stability (Karikó et al. 2008). Hundreds of H/ACA small nucleolar RNAs (snoRNAs) modify site-specific uridines, largely in noncoding RNAs, and they have been well-characterized in terms of requirements and rules for conducting site-specific pseudouridylation (Watkins and Bohnsack 2012; McMahon et al. 2015). Recently, targeted pseudouridylation of PTCs (i.e., UAA, UAG, UGA, all of which contain uridine at the first position) using engineered H/ACA snoRNAs has been observed to suppress translation termination and promote readthrough to produce a full-length protein with an efficiency that also inhibits NMD (Fig. 3; Morais et al. 2020). While first described using yeast (Karijolich and Yu 2011), this approach, when applied to human cells, utilizes an engineered snoRNA to guide pseudouridylation of uridine within a specific PTC by the recruitment of an endogenous ribonucleoprotein complex containing the pseudouridine synthase Dyskerin (Adachi et al. 2023; Song et al. 2023). Replacing uridine within a PTC with pseudouridine leads to PTC recognition by an aminoacyl-tRNA in the ribosome A-site, thereby suppressing translation termination in human cell models of disease (Adachi et al. 2023; Song et al. 2023). It appears that ΨGA codons predominantly code for phenylalanine or tyrosine, with ΨAA and ΨAG codons coding for threonine or serine. While the precise mechanisms underlying this process remain poorly defined, structural studies support unusual codon–anticodon base-pairing at the ribosome decoding center that outcompetes release factor binding to the PTC. This approach holds high promise because it is tunable and can be made to target a specific PTC based on designing snoRNAs complementary to the sequences flanking a PTC of interest. Furthermore, the relatively small size of the snoRNA-derived guide RNAs makes them favorable for delivery using either lipid nanoparticle or AAV-delivery methods (Fig. 3). While it is important to test the feasibility of this method in vivo using disease models, and to fully understand incorporation and misincorporation efficiencies in distinct tissues, we are optimistic that this approach may open new avenues for therapeutic development.
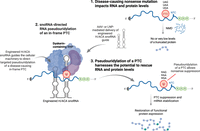
Nonsense suppression by snoRNA-directed RNA pseudouridylation. Depicted is the emerging potential of H/ACA snoRNA-directed RNA pseudouridylation to induce nonsense suppression of an in-frame PTC. (1) A PTC created by a nonsense mutation, which resides upstream of the normal termination codon (NTC), triggers NMD of the PTC-containing transcript. A small fraction of the PTC-containing mRNA may escape NMD and, when translated, may lead to production of a truncated, nonfunctional protein. (2) Delivery of a designer H/ACA snoRNA guides pseudouridine (Ψ) modification of a target uridine within a PTC by the cellular machinery. (3) Pseudouridine within the PTC leads to efficient translational readthrough of the modified PTC, NMD suppression, and increased expression of full-length protein.
Similarly to pseudouridine, adenosine-to-inosine (A-to-I) RNA editing by cellular double-stranded RNA-specific adenosine deaminases (ADARs) (Slotkin and Nishikura 2013) has emerged as an approach to induce nonsense suppression. This is mainly feasible based on the observations that inosine is recognized by the cellular machinery, including the ribosome, as guanosine, ultimately leading to an A-to-G base substitution in mRNA. Targeted ADAR-catalyzed editing has been shown to alter mutations and induce suppression at the PTC of a transcript in human cell lines and cultured neurons from mice (Montiel-Gonzalez et al. 2013; Sinnamon et al. 2017; Wettengel et al. 2017), opening up avenues for further therapeutic development. To exemplify a variation of this approach, A-to-I editing of a specific PTC has been achieved with a guide RNA that delivers the deaminase domain of the human ADAR2 enzyme, fused to the dCas13b protein, to a specific adenosine in a PTC-containing mRNA in a method called “RNA Editing for Programmable A to I Replacement, version 2” (REPAIRv2) (Melfi et al. 2020). One concern regarding all of the therapeutic approaches mentioned in this section is the potential of generating novel immunogenic epitopes in patients related to the restored target protein, as has been observed in DMD upon dystrophin reexpression (Mendell et al. 2010). Thorough characterization and monitoring of immune responses using these modalities should be closely monitored in preclinical models of disease, including human and animal models where T-cell responses can be easily assessed.
TARGETED mRNA DECAY AS POTENTIAL TREATMENTS FOR DOMINANTLY INHERITED DISORDERS
Several of the therapeutic modalities described above could be applied to induce the targeted decay of mRNA deriving from a disease-causing allele in dominantly inherited disorders irrespective of mutation type. Splice-switching ASOs or small-molecule splicing modulators could be developed to alter splicing in an allele-specific manner to induce NMD or other RNA decay mechanisms, thereby eliciting therapeutic benefit (Fig. 1, last panel). Similarly, RNA degradation by CRISPR–Cas13d has been shown to hold potential in preclinical models of HD by selectively targeting the CAG-expanded HTT transcript for decay (Morelli et al. 2023). Although in the early stages of development, RNATACs or RIBOTACs (Childs-Disney et al. 2022; Song et al. 2024) could also be further explored as an option to target disease-causing PTC mutations in a transcript-specific manner. While these approaches have reportedly been applied to target toxic RNA transcripts in repeat expansion disorders (Bush et al. 2021, 2022), significant efforts are warranted to develop RNA-binding small molecules that induce decay of PTC-harboring transcripts.
THERAPEUTIC APPROACHES THAT INHIBIT THE NMD PATHWAY
While initially discovered by studying human diseases, it is now clear that NMD has cellular roles beyond the quality control of aberrant RNAs by targeting ∼5%–10% of physiological mRNAs as a means of maintaining cellular homeostasis (Nasif et al. 2018; Kurosaki et al. 2019). Generally, the efficiency of NMD is inhibited during stress, such as during the unfolded protein response (Karam et al. 2015), or differentiation, such as during myogenesis (Gong et al. 2009). NMD can be viewed as preventing an inappropriate cellular response to stress until the stress is sufficiently large to warrant a response that is in part augmented by the cell inhibiting NMD. It follows that any disease-associated alterations to NMD efficiency may result in serious cellular imbalances that could facilitate or exacerbate, if not cause, disease. Several examples of diseases characterized by hyperactivated NMD are now known. These include Fragile X syndrome (FXS), which is the most common single-gene cause of intellectual disability and autism due to the absence of the ubiquitously expressed RNA-binding protein FMRP. In a mouse model of FXS, NMD hyperactivation begins in utero, by embryonic day 16, and extends postbirth until postnatal day 24 (Kurosaki et al. 2021b). Inhibiting NMD using either of two compounds (NMDI-1 and NMDI-14), each of which disrupts a different NMD factor interaction, normalizes the transcriptome dysregulation observed in neurons differentiated from FXS patient-derived induced pluripotent stem cells as well as the differentiation process itself (Fig. 4; Kurosaki et al. 2021a). Efforts to identify small molecules that could pass the blood–brain barrier and dampen the efficiency of NMD during the postnatal developmental window of NMD hyperactivity in FXS is an active area of current research.
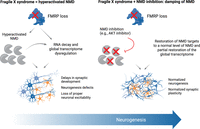
Proposed role of NMD in FXS and potential benefit of NMD inhibitor treatment in FXS. Schema highlighting the reported roles of NMD misregulation in FXS (left). Dampening NMD (either genetically or pharmacologically) leads to NMD target stabilization and rescue of transcriptome misregulation in FXS (right). The development of NMD inhibitors to dampen NMD hyperactivity in FXS is an attractive area of further therapeutic development.
Dampening the NMD pathway may also prove therapeutic for certain cancers, as has been extensively reviewed elsewhere (Supek et al. 2021; Tan et al. 2022). Pharmacological inhibitors of the NMD pathway have shown efficacy in some cancer models. Mechanistically, it appears that, while some cancers suppress NMD to promote oncogene expression, other tumor types are driven in part by the selection of driver mutations that result in PTC-containing transcripts that are subject to NMD, such as in tumor suppressor genes, including the TP53 gene (Chen et al. 2017; Gudikote et al. 2021). In addition, small-molecule NMD inhibition has been associated with triggering an antitumor immune response. This is likely due in part to findings that neoantigens derived from PTC-harboring mRNAs that are generated by somatic mutations can be highly immunogenic. Inhibiting NMD likely increases neoantigen presentation and is proposed to enhance the response to immunotherapy. In mouse models of metastasis, inhibiting NMD has been shown to slow tumor growth and increase T-cell infiltration, and mRNAs harboring frameshift mutations that escape NMD have proven to be immunogenic (Pastor et al. 2010; Litchfield et al. 2020).
Many cancers characterized by microsatellite instability often manifest hyperactivated NMD, which presumably eliminates new or altered transcripts that could block tumor progression (Bokhari et al. 2018). Inhibiting the NMD pathway may have clinical value in hypermutated cancers or tumors with DNA damage repair deficiencies, such as subtypes of breast cancer. In line with these findings, chemotherapeutic drugs such as doxorubicin, which work in part by eliciting DNA damage, naturally inhibit NMD (Popp and Maquat 2015). Inhibition contributes to apoptosis by allowing for the translation of NMD targets, including those that encode proapoptotic proteins. Such studies illuminate that supplementing DNA-damaging chemotherapeutics with an NMD inhibitor may be more beneficial than treatment with the DNA-damaging reagent alone. Additional research in this area, including the development and use of more drug-like NMD inhibitors in preclinical models, will further accelerate progress in this therapeutic area. There will undoubtedly be a new wave of modalities that target NMD in cancer and other diseases associated with NMD hyperactivation. New developments in protein modeling using artificial intelligence are envisioned to speed up structure-based drug discovery approaches and accelerate the generation of drugs that inhibit NMD factor interactions as a means to globally inhibit NMD.
FUTURE PERSPECTIVE AND OUTLOOK
A multitude of innovative approaches are in development that may lead to novel therapeutics for NMD-opathies, that is, diseases caused by the generation of one or more NMD targets or a cell-wide abnormality in NMD efficiency. We are optimistic that the future success of NMD therapeutics will be accelerated by extensive screening for novel therapeutics, coupled with a detailed molecular characterization of the precise role of NMD in specific diseases. Also in the therapeutic pipeline are NMD inhibitors that may have utility in cases where inhibiting NMD promotes cancer-cell killing. Carefully assessing and characterizing the efficiency of failed and ongoing strategies targeting NMD in clinical trials will undoubtedly speed up progress toward future drug development efforts, with the goal of providing safe and effective disease-modifying treatment options for patients with no or limited current treatment options.
COMPETING INTEREST STATEMENT
Mary McMahon is an employee at ReviR Therapeutics. The authors declare no other competing interests.
ACKNOWLEDGMENTS
The authors thank Dr. Paul August and Jawad Abid for critical feedback on this manuscript. We apologize to authors whose work could not be cited due to space limitations. Research in the Maquat laboratory is supported by the National Institutes of Health R35 GM149268, the FRAXA Research Foundation, and the New York State Empire Discovery Institute.
Footnotes
-
Article is online at http://www.rnajournal.org/cgi/doi/10.1261/rna.080334.124.
-
Freely available online through the RNA Open Access option.
This article, published in RNA, is available under a Creative Commons License (Attribution-NonCommercial 4.0 International), as described at http://creativecommons.org/licenses/by-nc/4.0/.








