
SF3B1: from core splicing factor to oncogenic driver
- Corresponding author: jlm2{at}columbia.edu
Abstract
Highly recurrent somatic mutations in the gene encoding the core splicing factor SF3B1 are drivers of multiple cancer types. SF3B1 is a scaffold protein that orchestrates multivalent protein–protein interactions within the spliceosome that are essential for recognizing the branchsite (BS) and selecting the 3′ splice site during the earliest stage of pre-mRNA splicing. In this review, we first describe the molecular mechanism by which multiple oncogenic SF3B1 mutations disrupt splicing. This involves perturbation of an early spliceosomal trimeric protein complex necessary for accurate BS recognition in a subset of introns, which leads to activation of upstream branchpoints and selection of cryptic 3′ splice sites. We next discuss how specific transcripts affected by aberrant splicing in SF3B1-mutant cells contribute to the initiation and progression of cancer. Finally, we highlight the prognostic value and disease phenotypes of different cancer-associated SF3B1 mutations, which is critical for developing new targeted therapeutics against SF3B1-mutant cancers still lacking in the clinic.
Keywords
INTRODUCTION
RNA splicing is a complex process conserved across eukaryotes by which introns are excised from precursor messenger RNAs (pre-mRNAs) and exons are ligated to form mature mRNA transcripts (Wilkinson et al. 2020; Rogalska et al. 2023). In multicellular organisms, the combinatorial selection of different intronic and exonic segments through the process of alternative splicing is critical to achieving tissue-specific gene expression that regulates cell differentiation, function, and homeostasis (Baralle and Giudice 2017). Early RNA-sequencing (RNA-seq) studies revealed that in humans, up to 95% of multiexon transcripts undergo alternative splicing (Pan et al. 2008; Wang et al. 2008), which can contribute to functional protein diversity and phenotypic complexity (Weatheritt et al. 2016; Blencowe 2017; Liu et al. 2017; Reixachs-Solé et al. 2020; Wright et al. 2022; Kjer-Hansen and Weatheritt 2023). The critical importance of pre-mRNA splicing in regulating cellular behavior has also been confirmed by the observation that splicing dysregulation contributes to human diseases, including cancer (Bonnal et al. 2020; Bradley and Anczuków 2023). In fact, tumors have up to 30% more alternative splicing than normal cells (Kahles et al. 2018), and 68% of cancer samples have at least one splicing-derived neoepitope (Frankiw et al. 2019). It is further estimated that up to 50% of all disease-causing mutations affect splicing (Cartegni et al. 2002; López-Bigas et al. 2005; Wang and Cooper 2007), underscoring the importance of understanding the molecular mechanisms of pathogenic RNA splicing to create opportunities for therapeutic correction (Desterro et al. 2020; Rogalska et al. 2023).
Alterations that affect splicing regulation in cancer can arise in different ways. The simplest type mechanistically is cis-acting mutations that occur in regulatory sequence elements of the pre-mRNA that disrupt normal splicing (Supek et al. 2014; Jung et al. 2015). Another type is trans-acting mutations in genes encoding spliceosomal proteins (Bejar 2016; Dvinge et al. 2016; Zhang et al. 2024a) or perhaps snRNAs (Shuai et al. 2019), which can also alter splicing patterns. And finally, expression changes of splicing regulators, such as SR or hnRNP proteins, can dysregulate alternative splicing patterns of target transcripts (Karni et al. 2007; David et al. 2010; Anczuków et al. 2012).
Highly recurrent somatic mutations in human genes encoding components of the splicing machinery were first described in patients with myelodysplastic syndromes (MDS) (Papaemmanuil et al. 2011; Yoshida et al. 2011; Graubert et al. 2012) and chronic lymphocytic leukemia (CLL) (Wang et al. 2011; Quesada et al. 2012), establishing a direct causal relationship between splicing dysregulation and hematological malignancies. Since then, mutations in human splicing factor-encoding genes have been identified in precancerous clonal hematopoiesis (Genovese et al. 2014; Jaiswal et al. 2014; Abelson et al. 2018; Desai et al. 2018; Fabre et al. 2022; Jaiswal and Bick 2022; Robertson et al. 2022), acute myeloid leukemia (AML) (Metzeler et al. 2016; Papaemmanuil et al. 2016), and solid tumors, such as uveal melanoma (Furney et al. 2013; Harbour et al. 2013; Martin et al. 2013), breast cancer (Ellis et al. 2012; Koboldt et al. 2012), lung adenocarcinoma (Imielinski et al. 2012; Esfahani et al. 2019), pancreatic ductal adenocarcinoma (Biankin et al. 2012), and bladder urothelial carcinomas (Seiler et al. 2018). Moreover, a comprehensive mutational analysis of the Tumor Cancer Genome Atlas revealed that 119 splicing factors are recurrently mutated across 33 different cancer subtypes (Seiler et al. 2018), suggesting that splicing dysregulation is a genetic driver of oncogenesis (Supek et al. 2014; Dvinge et al. 2016).
The most frequently mutated spliceosomal component in cancer is the core splicing factor 3b subunit 1 (SF3B1). SF3B1 is the largest subunit of the SF3b complex, a multiprotein component of the U2 small nuclear ribonucleoprotein particle (snRNP), which directly contacts the intronic branchsite (BS) and contributes to the selection of 3′ splice sites (3′ss) during the earliest steps of RNA splicing (Wilkinson et al. 2020). Most disease-causing mutations in SF3B1 alter hotspot amino acid residues that disrupt the interaction between SF3B1 and another splicing factor important for accurate recognition of branchpoint sequences (BPSs) called SUGP1 (Zhang et al. 2019), which is also mutated in cancers (Liu et al. 2020; Alsafadi et al. 2021). SF3B1 binding to SUGP1 allows this cofactor to engage and activate the RNA helicase DHX15 (Zhang et al. 2022), which is essential in a subset of introns for the correct selection of the 3′ss (Zhang et al. 2023). SF3B1 is mutated in ∼30% of all MDS patients, including up to 86% of cases of MDS with ring sideroblasts (MDS-RS) (Huber et al. 2022), which has prompted the designation of SF3B1-mutant MDS as a new disease subtype (Malcovati et al. 2020; Arber et al. 2022; Bernard et al. 2022). SF3B1 is also the most commonly mutated splicing factor gene in other cancers besides MDS, including ∼25% of uveal melanomas, 37% of vulvovaginal mucosal melanomas (Hintzsche et al. 2017), and up to 15% of CLL patients (Wang et al. 2011).
Several reviews have described how aberrant pre-mRNA splicing in general (Zhang and Manley 2013; Lee and Abdel-Wahab 2016; Agrawal et al. 2018; Bonnal et al. 2020; Desterro et al. 2020; Stanley and Abdel-Wahab 2022; Bradley and Anczuków 2023; Anczukow et al. 2024), and splicing factor mutations in particular (Bejar 2016; Dvinge et al. 2016; Chen et al. 2021; Bashari et al. 2023; Zhang et al. 2024a), contribute to the hallmarks of cancer. Here, we focus on the functional and pathological consequences of SF3B1 mutations in cancer. We first discuss SF3B1 function during early spliceosome assembly and describe how many of its cancer-associated mutations induce splicing errors by disrupting the SF3B1–SUGP1 interaction, which leads to aberrant BS recognition and activation of cryptic 3′ss. Moreover, we describe how decoding the molecular mechanisms of SF3B1-mutant spliceosomes established an important role for the RNA helicase DHX15 in early BS recognition. We then identify and discuss disease-relevant transcripts that are misspliced in SF3B1-mutated cells, and characterize the effects of these splicing alterations on cancer pathogenesis. Since not all SF3B1 mutations are of equal consequence, we examine the connection between different residue mutations, disease phenotype, and prognostic value. Finally, we pinpoint the challenges associated with studying the pleiotropic effects of SF3B1 mutations on the transcriptome and highlight how a mechanistic understanding of the molecular impact of these mutations can create novel therapeutic opportunities for SF3B1-mutant cancers.
MOLECULAR FUNCTIONS OF SF3B1 IN BRANCHSITE RECOGNITION
Early branchsite recognition is a highly dynamic process that happens in two steps
The removal of introns from pre-mRNA transcripts is orchestrated by the spliceosome, a megadalton macromolecular RNA-protein complex comprised of five small nuclear RNAs (snRNAs) and more than 100 proteins that dynamically assemble anew at each intron (Wahl et al. 2009). U1 and U2 snRNPs define exon–intron boundaries through the stepwise recognition of core consensus sequences in introns. Spliceosome assembly begins with the direct recognition of the GU dinucleotide at the 5′ss by U1snRNP via base-pairing of the U1snRNA to this motif. Simultaneously, splicing factor 1 (SF1), the heterodimeric U2snRNP auxiliary factors U2AF2 (U2AF65) and U2AF1 (U2AF35) recognize the BS, the polypyrimidine tract (PPT), and the YAG motif at the 3′ss, respectively.
Accurate selection of the 3′ss is predicated upon the recognition of the correct BS, given that the first AG motif downstream from the BS is usually selected as the 3′ss (Zhuang and Weiner 1990). Unlike the conserved dinucleotides at the 5′ss and 3′ss, the BS is composed of a highly degenerate intronic heptamer (YNCUGAC) called the BPS, which surrounds the branchpoint adenosine (BP-A) (Reed and Maniatis 1988; Taggart et al. 2017). This motif is usually located 18–37 nt upstream of the 3′ss, but its variable distance from the 3′ss, which can be up to 400 nt, and the number of BPS per 3′ss can greatly influence the fidelity of 3′ss selection and lead to alternative splicing (Xie et al. 2023). In the initial recognition of the BS during what is known as E complex formation, SF1 binds the BPS through its RNA-binding domain (Arning et al. 1996; Berglund et al. 1997; Peled-Zehavi et al. 2001), and U2AF2 flexibly adheres to the PPT through its two tandem RNA-recognition motifs (RRMs) while also stabilizing the BPS interaction through an interface comprising a U2AF homology motif (UHM) of U2AF2 bound to a U2AF ligand motif (ULM) of SF1 (Zamore and Green 1989; Zamore et al. 1992; Berglund et al. 1998). U2AF1 binds the 3′ss via its two zinc fingers (ZnF) (Ruskin et al. 1988; Valcárcel et al. 1996; Merendino et al. 1999), which contain hotspot residues for recurrent oncogenic mutations (Yoshida et al. 2011; Graubert et al. 2012) that alter splicing in a sequence-dependent manner (Okeyo-Owuor et al. 2015; Yoshida et al. 2020; Biancon et al. 2022) based on the different preferences of each ZnF for the nucleotides flanking the YAG motif (Yoshida et al. 2015). The initial binding of SF1 to the BPS in the E complex mainly serves a kinetic role for 3′ss recognition while ensuring that the BS is positioned for its subsequent identification by the U2snRNP in precatalytic (pre-A) spliceosome (Guth and Valcárcel 2000; Liu et al. 2001).
In the pre-A complex, the U2snRNP is recruited to the predefined BS through a highly dynamic assembly of splicing factors around the BPS and the 3′ss that are brought together in ATP-dependent reorganization of the spliceosome driven by RNA helicases (MacMillan et al. 1994). The critical molecular event in BS recognition is the “hand-over” of the BS from SF1 to the branchpoint-interacting stem–loop (BSL) of the U2snRNA, which “covers” the complementary BPS in the intron (Fig. 1). This process involves two sequential steps, which are (1) the loading of U2snRNP into the vicinity of the BS, and (2) the unwinding of the BSL to hybridize with the BPS, forming the pre-spliceosomal A complex (Wu and Manley 1989; Zhang et al. 2023). When U2snRNP has defined the correct BS, the BPS heptamer forms an RNA duplex with the 6 bp motif (GUAGUA) of the U2 snRNA through a base-pairing interaction that excludes the BP-A (Wu and Manley 1989). The unpaired BP-A thus “bulges” out from the duplex, and its 2′OH group carries out a nucleophilic attack on 5′ss during the first transesterification reaction of splicing, leading to 5′ cleavage and intron lariat formation (Berglund et al. 2001). The recruitment and positioning of the U2snRNP during early BS recognition in the pre-A complex involves multivalent and transient protein interactions between the U2snRNP complex, including SF3B1, and additional splicing factors that have evaded capture by recent cryo-electron microscopy (cryo-EM) snapshots of the spliceosome (Zhang et al. 2021; Tholen et al. 2022; Yang et al. 2023). For this reason, the molecular events leading to the stable association of the U2 snRNP with the intronic BS are not completely understood. Fortuitously, studying the molecular consequences of oncogenic SF3B1 mutations, which lead to altered BS usage and cryptic 3′ss, has provided multiple insights into the mechanisms promoting the first step of the BS hand-over in the pre-A complex, which we argue is the main determinant of BS selection in eukaryotic pre-mRNA splicing.
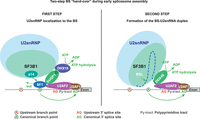
A mechanistic model for the two-step “hand-over” of the BS from SF1 to the U2snRNP. (Left) In the first step of the BS “hand-over” from SF1 to U2snRNP, SUGP1 is located near the BS through direct contacts with SF1 and U2AF2 and recruits the SF3B1-containing U2snRNP to the vicinity of the BS by directly contacting the hotspot region of the SF3B1 HD through two regions flanking its G-patch domain, which loops-out to activate DHX15. Then DHX15 “pulls” the pre-mRNA by helicase tracking into the RNA path of SF3B1 and displaces SF1 from the predefined BS, which becomes available for correct recognition by the p14 subunit of SF3B1 in the pre-A complex. (Right) In the second step of the hand-over, following the dissociation of SUGP1 from the complex, DDX46 is recruited to the U2snRNP by the same hotspot region previously occupied by SUGP1. DDX46 then unwinds the BSL of the U2snRNA to allow the formation of the U2-BS RNA duplex with a bulged BP-A, which is incorporated into the RNA channel of SF3B1 in the catalytically primed A-complex. (Figure modified from Zhang et al. 2019.)
Cancer-associated SF3B1 mutations affect hotspot residues in its HEAT domain
SF3B1 (SF3b155) is the largest subunit of the SF3b subcomplex, which together with SF3a and the U2snRNP core form the 17S U2snRNP complex (Will et al. 2002). SF3B1 comprises an intrinsically unstructured N-terminal domain (NTD) and a C-terminal domain containing 20 HEAT (Huntingtin, Elongation factor 3, PR65/A, TOR) repeats, which are tandem repeat structural motifs of two alpha helices linked by a short loop, forming a right-handed superhelix (Cretu et al. 2016). The NTD contains several ULMs that interact with the UHMs of U2AF2 (Spadaccini et al. 2006; Galardi et al. 2022), and an adjacent region that interacts with the SF3b subunit p14 (SF3b14a), which directly contacts the BP-A (Schellenberg et al. 2006). Since SF3B1 can be cross-linked with the nucleotides flanking the BPS near the binding sites for U2AF2 and p14 (Gozani et al. 1998), it has been suggested that SF3B1 acts in cooperation with its BS-binding partners to recruit the U2snRNP onto the correct BS during the formation of the pre-A complex (MacMillan et al. 1994; Query et al. 1996; Gozani et al. 1998). The crystal structure of an in vitro reconstituted human SF3b complex revealed that the HEAT-repeat domain (HD) of SF3B1 is a composite platform of protein–protein interactions that stabilize the recruitment of the U2snRNP particle onto the BPS. This structure revealed that, in addition to the UHs present in the NTD, the HD also contains regions where SF3B1 is directly cross-linked to p14 and U2AF2, in H3+H14–H17 and H14–15, respectively (Cretu et al. 2016). Thus, the superhelix structure was proposed to serve as a scaffold to define the positions of p14 and U2A65 on the intron and stabilize the U2snRNA/BPS RNA duplex. However, none of the cancer-mutated residues in SF3B1 are located in the NTD (where SF3B1 primarily contacts SF1 and U2AF2) or at the interface with other SF3b core proteins, which could irreparably destabilize the U2snRNP complex and lead to splicing arrest. An exception is the E592K mutation, which targets a residue at the interface with PHF5A (SF3B7), although the mutation does not appear to affect the interaction (Choi et al. 2024).
More than 40 residues in SF3B1 are known to be targeted by cancer-associated mutations, including the most frequent substitution of lysine by glutamic acid in residue 700 (K700E). Of these, 33 are in HEAT repeats 4–7, suggesting that these mutations affect functions of SF3B1 related to this specific domain and leave the remaining scaffold functionally intact. Critically, nine hotspot residues in H4–H8, including K700, are solvent-exposed residues that likely contribute to SF3B1 interaction with other spliceosomal proteins rather than subunits of the SF3b complex (Cretu et al. 2016). Cancer-associated SF3B1 mutations found in the HEAT domain do not prevent splicing but rather lead to alternative BS usage and recognition of cryptic 3′ss in downstream target mRNAs (Darman et al. 2015; Alsafadi et al. 2016). It was suggested early on that SF3B1 mutations alter splicing by perturbing the interaction with U2AF2 (DeBoever et al. 2015), possibly because these mutations affect hotspot residues in the vicinity of the region cross-linked to U2AF2, p14, and the mRNA (Cretu et al. 2016). However, the K700E mutation does not affect the stability of an in vitro SF3b-U2AF2 complex and its ability to bind RNA compared to WT (Cretu et al. 2016). This indicates that SF3B1 mutations lead to altered splicing by preventing interaction with other splicing factors necessary for correct BS recognition during the formation of the pre-A complex.
SF3B1 hotspot mutations disrupt SF3B1 interaction with SUGP1
One approach to elucidate the basis of mutant SF3B1 missplicing is to identify any protein(s) missing from mutant versus WT spliceosomes. Affinity purification of endogenous SF3B1-bound spliceosomes from SF3B1K700E-mutant and WT human K562 leukemia cells showed that this mutation impaired the association of SF3B1 with the splicing factor SUGP1 (SURP and G-patch domain-containing protein 1; previously known as SF4 [Sampson and Hewitt 2003]). No SF3b subunits were found to be missing from the SF3B1K700 spliceosome, further confirming that the SF3B1K700 mutation does not disrupt the interaction between SF3B1 and SF3b complex partners (Zhang et al. 2019). Importantly, SUGP1 knockdown (KD) completely recapitulated multiple cryptic splicing events observed in SF3B1K700E cells, as did expression of three SF3B1 derivatives with mutations affecting other hotspot residues in H4–H7 (E622D, R625C, and H662Q). These derivatives also disrupted the SF3B1–SUGP1 interaction, while a fourth, K666N, partially disrupted both SUGP1 interaction and splicing. Moreover, SUGP1 overexpression partially rescued aberrant splicing in SF3B1K700E cells, indicating that incorporation of SUGP1 into the mutant spliceosome is dose-dependent and that restoring the critical interaction between SF3B1 and SUGP1 during early spliceosome assembly can correct BS recognition and 3′ss selection (Zhang et al. 2019). SUGP1 contains two tandem SURP domains, which are exclusively found in splicing factors (Nameki et al. 2022) and mediate interactions between U2snRNP proteins and SF1 (Crisci et al. 2015; Nameki et al. 2022), a short ULM that is directly recognized by RRM3 of U2AF2 (Zhang et al. 2019), and a C-terminal flexible glycine-rich motif called a G-patch, which can bind and activate DEAH-box RNA helicases (DHXs) (Robert-Paganin et al. 2015; Studer et al. 2020; Bohnsack et al. 2021). Strikingly, expression of SUGP1 derivatives with mutations in the most conserved residues of the G-patch domain (G574A–G582A), but not in the SURP or ULM domains, reproduced usage of altered 3′ss of SF3B1-mutant cells, indicating that the G-patch domain of SUGP1, and likely its activation of an RNA helicase, is essential for correct 3′ss selection (Zhang et al. 2019).
The presence of multiple functional domains in SUGP1 highlights its possible role in loading the SF3b module of the U2snRNP onto the correct BPS during the first step of the “hand-over.” In this model (Fig. 2), U2snRNP recruits SUGP1 to the pre-A complex through the interaction between SUGP1 and the HEAT domain of SF3B1, and SUGP1, in turn, assists in positioning the U2snRNP in the vicinity of the canonical BPS and 3′ss by direct interactions with SF1 and U2AF1 via its SURP and ULM domains, respectively. Then, the stable recruitment of the U2snRNP onto the BPS, which is the first ATP-dependent step in the splicing cycle that leads to the formation of the A complex (Maul-Newby et al. 2022), is achieved by the SUGP1 G-patch, which associates and promotes the ATPase activity of a DHX helicase required for the displacement of SF1 by p14, allowing base-pairing between the canonical BPS and the U2snRNA and selection of the correct 3′ss. In patients carrying mutations in the SF3B1 HD that disrupt its interaction with SUGP1, the U2snRNP is unable to recruit SUGP1, which fails to activate its cognate DHX ATPase, and thus SF1 is not displaced through helicase tracking. The mutant spliceosome must then recognize an available, upstream BS, possibly through BS scanning by the p14 subunit (Perea et al. 2016).
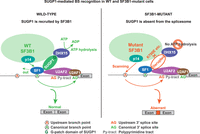
BS recognition in SF3B1 wild-type and SF3B1-mutant cells. (Left) Under normal conditions, the WT SF3B1 spliceosome is associated with SUGP1, which activates the helicase activity of DHX15 via its G-patch domain (G), thereby allowing the usage of the canonical BP and 3′ss for splicing. (Right) When specific residues of the SF3B1 HD are mutated in cancer, interaction of SF3B1 with SUGP1 is disrupted, leading to loss of SUGP1 during spliceosome assembly. In the absence of SUGP1, DHX15 is not activated, and SF1 is not efficiently displaced from the canonical BS thus blocking its recognition by the U2snRNP. The SF3B1 mutant spliceosome must then recognize an upstream BS and select a cryptic 3′ss. (Figure modified from Zhang et al. 2019.)
This mechanistic model (Fig. 2) argues that the loss of SUGP1 from the SF3B1-mutant spliceosome is solely responsible for the altered BP usage and cryptic 3′ss selection detected in cells harboring oncogenic SF3B1 mutations. Previous models, which stem from the comparison of RNA-seq data between WT and SF3B1-mutant cells and did not include structural analysis of the endogenous SF3B1-mutant spliceosome, suggested that SF3B1 mutations might (1) promote the selection of upstream 3′ss by causing a shift in the sterically protected region downstream from the BP, perhaps as noted above involving an altered interaction with U2AF2 (DeBoever et al. 2015), (2) enhance direct interactions of SF3B1 with specific nucleotides flanking the upstream BP (Darman et al. 2015), (3) induce conformational changes in the U2snRNP complex leading to the selection of a stronger upstream BP (Alsafadi et al. 2016), or (4) facilitate the recognition of inaccessible 3′ss buried in RNA secondary structures (Kesarwani et al. 2017). These previous models implied that the structural changes in SF3B1 induced by cancer-associated mutations would be considerable and refractory to rescue. However, as mentioned above, the splicing defects induced by SF3B1 mutations are partly reverted by overexpression of SUGP1. This suggests that restoring the SF3B1–SUGP1 interaction in SF3B1-mutant cells using proximity-inducing small molecules could be a therapeutic strategy for SF3B1-mutant cancers, which we discuss further below.
DHX15 is the RNA helicase activated by SUGP1 in early branchsite recognition
An important next question was to identify the putative DEAH-box helicase that is activated by the G-patch domain of SUGP1 during BS recognition. To this end, Zhang et al. (2022) performed affinity purification of an exogenously expressed SUGP1 C terminus followed by mass-spectrometry analysis, which revealed DHX15 as the RNA helicase bound to SUGP1. The SUGP1–DHX15 interaction is specifically mediated by the G-patch domain of SUGP1, and it is abolished by mutations affecting its most conserved glycine residues. KD of DHX15 or expression of several DHX15 derivatives containing mutations that disrupt the interaction between SUGP1 and DHX15, or the R222G hotspot mutation common in AML (Faber et al. 2016; Pan et al. 2017), recapitulate a subset of splicing defects found in SF3B1 mutant cells. Furthermore, the mutant SF3B1-specific splicing defects tested could all be rescued by expression of a DHX15–SUGP1 G-patch fusion protein (Zhang et al. 2022). The crystal structure of the DHX15–SUGP1 G-patch complex revealed that the G-patch contains an N-terminal helix that interacts with the DHX15 winged helix domain, and a C-terminal “brace loop” that contacts the DHX15 RecA2 domain (Zhang et al. 2022). Thus, the interaction between SUGP1 and DHX15 could tether these two domains together and facilitate the binding of DHX15 to its RNA substrates (Enders et al. 2023), thereby stimulating its ATPase and RNA helicase activity akin to the activation of DHX15 by the G-patch protein NRFK (Studer et al. 2020), whose structure is highly similar to that of the DHX15–SUGP1 G-patch complex. Notably, DHX15 is almost equally recruited to the WT and SUGP1-lacking SF3B1 mutant spliceosomes, which suggests that the SUGP1 G-patch is responsible for DHX15 activation but not its recruitment. Two additional studies, discussed further below, utilized in vivo assays to uncover the DHX15–SUGP1 interaction (Beusch et al. 2023; Feng et al. 2023).
Previous proteomic studies also indicated that DHX15 is present in U2snRNP and A-complex spliceosomes, along with G-patch proteins RBM5, RBM10, RBM17, CHERP, as well as SUGP1 (Agafonov et al. 2011; Hegele et al. 2012). Among the G-patch cofactors tested (RBM5, RBM17, CHERP, and SUGP1), only SUGP1 depletion was found to reproduce the splicing defects associated with SF3B1 mutations (Zhang et al. 2019), indicating that SUGP1 is the G-patch activator of DHX15 during early BS recognition (Zhang et al. 2022). Consistent with these results, Benbarche et al. (2024) found that another G-patch protein, GPATCH8, plays a role in mutant SF3B1 missplicing. Specifically, GPATCH8 was found to compete with SUGP1 for interaction with the same binding region of DHX15, and its loss was found to rescue missplicing mediated by SF3B1 mutations in a subset of introns enriched for GPATCH8-binding sites, perhaps reflecting an increased SUGP1–DHX15 interaction. However, whether GPATCH8 is present in SF3B1-mutant or WT spliceosomes is unclear, and the natural function of GPATCH8, including a possible role in RNA splicing, remains to be determined. Nonetheless, reducing GPATCH8 levels could offer therapeutic potential for SF3B1-mutant cancers, since GPATCH8 deletion normalized proliferation and erythroid differentiation of human hematopoietic progenitors and patient-derived AML cells heterozygous for the K700E and K666N hotspot mutations without affecting normal cells (Benbarche et al. 2024).
Possible roles for SUGP1 and DHX15 in disassembling unstable spliceosomes
DHX15, and its yeast homolog Prp43, are known for their canonical roles in facilitating the extraction of excised intron lariats during spliceosome disassembly (Arenas and Abelson 1997; Tanaka et al. 2007) and in ribosomal RNA (rRNA) biogenesis (Memet et al. 2017), which require different G-patch cofactors that direct the enzyme to the intron lariat or the pre-rRNA. During disassembly of intron lariat spliceosomes (ILS), DHX15 is activated by the G-patch protein TFIP11 to translocate on the 3′end of the U6snRNA (Arenas and Abelson 1997; Tanaka et al. 2007), whereas during rRNA biogenesis DHX15 is activated by G-patch protein NRFK to promote pre-rRNA cleavage at the A′ site (Memet et al. 2017). Recently, DHX15 has also been suggested to function in splicing quality control (QC) through a mechanism of kinetic proofreading (KP). For example, Maul-Newby et al. (2022) found that DHX15 destabilizes the interaction between U2snRNP and a minimal RNA substrate in an unproductive A complex in vitro, consistent with a role for DHX15 in QC of BS recognition by promoting dissociation of A complexes assembled on cryptic BS. This mechanism is supported by the fact that in splicing-proficient complexes extracted from HeLa cells, immunodepletion of DHX15 resulted in increased, rather than decreased, A complex formation and lower splicing efficiency (Maul-Newby et al. 2022). However, the reduced splicing efficiency could be an indirect effect from the loss of ILS disassembly upon DHX15 depletion rather than a direct inhibition of DHX15's putative QC function. A similar observation has been made in yeast, where Prp43 fused to the G-patch domain of its activator Ntr1 targets the U2snRNP–intron interaction to disassemble early spliceosomal complexes (Fourmann et al. 2016).
The notion that DHX15 functions in QC of BS recognition via proofreading has also been put forth in two other studies. Feng et al. (2023) found that KD of DHX15 or SUGP1 in 293T cells led to the activation of overlapping cryptic introns, suggesting that these proteins work together to repress suboptimal splicing. However, no significant overlap was observed between the altered splicing induced by DHX15 KD and that observed in cells expressing SF3B1K700E. DHX15 KD was previously shown to affect only 13% of SF3B1K700E-regulated introns (Zhang et al. 2022), which might contribute to this lack of overlap, as might the size and unspecified origin of the SF3B1K700E data set used by Feng et al. As expected, though, SUGP1 KD significantly recapitulated splicing defects observed in cells expressing SF3B1K700E, although to what extent was unclear from the data presented. In a related study, Beusch et al. (2023) performed base-editor mutagenesis of the human spliceosome and identified two mutations in the SUGP1 G-patch that weaken interaction with DHX15 and confer cellular resistance to pladienolide B (PB), an SF3b small molecule inhibitor. Since PB and related compounds reduce stable A complex formation (Folco et al. 2011; Cretu et al. 2018, 2021), the resistance-conferring mutations found in the SUGP1 G-patch could counteract the activity of PB by inhibiting a putative disassembly function of SUGP1 mediated by its interaction with DHX15, leading to increased A complex formation (Beusch et al. 2023).
Both the above studies confirmed the direct interaction between the SUGP1 G-patch and DHX15 but suggested that activation of DHX15 upon G-patch binding triggers rapid spliceosome disassembly, rather than displacing SF1 from the BPS through helicase tracking on the pre-mRNA. They further argued that DHX15 and SUGP1 share a QC mechanism of BS proofreading that is independent of SF3B1 and more general than the direct mechanism proposed by Zhang et al. (2019). When SUGP1 is recruited to an aberrantly assembled pre-spliceosome, for example at a cryptic BS, its G-patch could bind DHX15 and activate its helicase activity to disassemble the complex (Beusch et al. 2023; Feng et al. 2023). However, as discussed below, this model is difficult to reconcile with other recent studies.
SUGP1 and DHX15 can function directly in branchsite recognition
We have argued that SUGP1, through its multivalent interactions with the HEAT domain of SF3B1, and with SF1 and U2AF2, is responsible for localizing U2snRNP to the vicinity of the BPS during the first step of BS “hand-over” (Fig. 1). This novel mechanism is supported by structure-function studies that leveraged numerous cancer-associated mutations to elucidate the contribution of specific amino acid residues in SUGP1, SF3B1, and DHX15 in forming a splicing-proficient trimeric complex that leads to correct BS recognition and 3′ss selection. Notably, structural modeling of the SF3B1–SUGP1 heterodimer using AlphaFold-Multimer (AF-M) (Evans et al. 2022) revealed that SF3B1 H4–H7, which contain residues targeted by oncogenic SF3B1 hotspot mutations, directly contacts SUGP1 in two separate regions flanking, but not overlapping, its G-patch domain (Zhang et al. 2023). These two regions of SUGP1, which are not required for interaction with DHX15, both harbor cancer mutations that were experimentally validated to disrupt the SF3B1–SUGP1 interaction and recapitulate SF3B1-mutant splicing defects. In fact, the simultaneous interaction between the SF3B1 HD and SUGP1 at these two points of contact serves to “loop out” the G-patch domain and make it accessible to DHX15. The predicted SF3B1–SUGP1–DHX15 trimer bridges SF3B1 and DHX15 with SUGP1 in the middle, directly implicating DHX15 in early BS recognition in the pre-A complex (Fig. 3). A notable feature of the SUGP1–SF3B1 interface predicted by AF-M and confirmed experimentally is that one of the two SF3B1-interacting regions in SUGP1 consists of two conserved tyrosines at the very C terminus of SUGP1 that directly contact SF3B1 in a “mutational pocket” encompassing SF3B1 hotspot residues, including K700 (Fig. 3; Zhang et al. 2023).
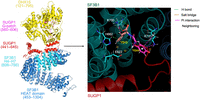
AlphaFold-Multimer predicted structures of the SF3B1–SUGP1–DHX15 heterotrimer and SF3B1–SUGP1 interface. (Left) Schematic drawing of the AF-M predicted structure of the SF3B1–SUGP1–DHX15 heterotrimer. The SF3B1 HEAT domain (453–1304) is shown in blue, with SF3B1 H4–H7 (606–756) shown in cyan. SUGP1 (441–645) is indicated in red, with the G-patch (560–606) shown in purple. DHX15 (121–795) is yellow. (Right) AlphaFold-Multimer–predicted interactions between the C-terminal residues of SUGP1 and SF3B1 hotspot residues mutated in cancer. (Figure adapted with modifications from Zhang et al. 2023.)
Mutant SF3B1-containing spliceosomes in which SF3B1 loses interaction with SUGP1 still in theory allow SUGP1 to interact with SF1 and U2AF2 via its SURP and ULM domains, respectively, and thus localize to the BS region. However, such complexes would be defective in recruiting the SF3B1-containing U2snRNP, thereby preventing utilization of the canonical BS. The U2snRNP will then recognize an available upstream BS and ultimately use a cryptic 3′ss, as proposed (Zhang et al. 2019, 2022, 2023). Thus, it is difficult to conceive a scenario where SUGP1 would function in QC proofreading later in splicing after its association with SF3B1, given that SUGP1 mutants that cannot bind SF3B1 but possess all the necessary domains to localize to the BS region nonetheless lead to altered splicing. If indeed SUGP1 functioned in QC by activating the DHX15-mediated disassembly of defectively assembled spliceosomes, then the endogenous SUGP1 and DHX15 proteins, whose levels are not decreased in heterozygous SF3B1-mutant cells, would still be able to interact with the WT spliceosome and prevent the cryptic splicing ubiquitously observed in these cells. Thus, we propose that SUGP1–DHX15 functions during the earliest step of BS hand-over from SF1 to the SF3B1-containing U2snRNP, which is localized to the vicinity of the BPS by direct contacts with SUGP1 and the ATPase function of DHX15, activated by the looped out SUGP1 G-patch domain. In this model, DHX15 translocates along the pre-mRNA via ATP-dependent helicase tracking until the BPS is matched to the BSL of the U2snRNA, thereby facilitating the recognition of the canonical BS (Fig. 4).
Another recent study also provides additional support, albeit indirect, for SUGP1/DHX15 functioning early in BS recognition. Specifically, Damianov et al. (2024a) localized U2snRNP binding across the transcriptome in WT and SF3B1K700E K562 cells, and unexpectedly detected mutant U2snRNP binding to thousands of cryptic BSs, the large majority of which are not associated with missplicing. These findings, which likely reflect the loss of SUGP1 from mutant SF3B1, seem most consistent with a positive role for SUGP1/DHX15 in facilitating accurate U2snRNP recognition of the correct BS, rather than with a role in disassembling thousands of apparently mislocalized complexes, often found in close juxtaposition with the authentic BS. These results indicate that SF3B1K700E- containing U2snRNPs bind BSs with considerably reduced precision relative to their WT counterparts.
DDX46 mediates U2snRNP-branchsite interactions but does not contribute to mutant SF3B1-induced missplicing
In addition to DHX15, two other RNA-dependent ATPases, DDX42 and DDX46 (or PRP5), are associated with the U2snRNP and contribute to its remodeling during the transition between pre-A and A complexes. DDX42 has been implicated in integrating the SF3b module into the U2snRNP, as it associates both with a free SF3b complex and U2snRNP particles that lack SF3b (Will et al. 2002; Yang et al. 2023), and is, therefore, unlikely to be involved directly in BS recognition in the pre-A complex. Upon binding to the U2snRNA, DDX42 is replaced by DDX46 resulting in the mature 17S U2snRNP, which is already near the BPS before any ATP-mediated rearrangements by this helicase (Yang et al. 2023). In the pre-A complex, DDX46 is located near the BSL of the U2snRNA, which covers the complementary sequence in the intronic BPS (Zhang et al. 2020). Subsequent ATP hydrolysis by DDX46 leads to a conformational change of the BSL, allowing base-pairing between the unwound U2snRNA and the BPS, thus promoting the formation of a preliminary U2-BS RNA duplex (Zhang et al. 2021). This duplex is further stabilized when the bulged adenosine of the BPS is recognized in a hinged pocket created by SF3B1 and PHF5A, a subunit of the SF3b complex. Strikingly, this BP-A pocket contains a cysteine residue in the nucleophilic ZnF of PHF5A that is covalently bound by several small molecule splicing inhibitors that compete with the pre-mRNA for access to the SF3B1 RNA channel and lead to altered BS usage (Folco et al. 2011; Cretu et al. 2018, 2021). Once the BS is tightly bound, the SF3B1 HD closes around the BP-A pocket forcing the ejection of DDX46 and directly engaging the newly formed branch helix (Zhang et al. 2021; Tholen et al. 2022).
Recent cryo-EM structures of the SF3b-DDX42 complex and DDX46-bound U2snRNP showed that the NTDs of these two helicases sequentially engage the RNA path of the SF3B1 HD in a mutually exclusive manner. Both the so-called N-plug of DDX42 and the N-terminal acidic loop of DDX46 occupy an overlapping binding site at the interface of the SF3B1 HD that is replaced by the intron region corresponding to the PPT upon formation of the A-complex (Yang et al. 2023). These structures suggest that after DDX46 unwinds the BSL of the U2snRNA for base-pairing with the pre-mRNA BPS, its N-terminal loop is only removed from the SF3B1 HD if the hinge pocket tightly binds the bulged BP-A upon correct BS recognition (Yang et al. 2023). Hence, the SF3B1-mediated release of the N-terminal loop of DDX46 from the hinge pocket is suggested to be an indirect proofreading mechanism for BS fidelity (Zhang et al. 2021, 2024b).
In this model, if the BP-A is tightly bound by the hinge pocket after U2-BS RNA duplex formation, the SF3B1 HD closes, thereby forcing the removal of the N terminus of DDX46 from the RNA channel. This, in turn, allows the U4/U6.U5 tri-snRNP to bind the A complex and splicing to proceed. Conversely, if for some reason (i.e., BPS mutations) the BP-A is not well placed in the hinge pocket, SF3B1 HD does not close, or closes at a slower rate, and the N terminus of DDX46 is not promptly removed from the SF3B1 HD, thereby preventing or delaying association with the tri-snRNP and arresting the splicing cycle, which could lead to disassembly of unproductive A-complexes stalled at cryptic BPS. Depending on the strength of the BP-A-hinge pocket interaction, (1) the SF3B1 HD could gradually close upon DDX46 and enable the graduated incorporation of the tri-snRNP, which would allow for the time-dependent recognition of suboptimal BS and selection of 3′ss, or (2) the SF3B1 HD could not close at all, thereby leaving DDX46 unremoved and preventing the incorporation of the tri-snRNP, which would lead to spliceosome disassembly and rejection of the pre-mRNA substrate. This could contribute to lower splicing efficiency, but not missplicing through altered BS usage.
The DDX46–SF3B1 HD interface that mediates this putative QC proofreading mechanism includes the hotspot area targeted by cancer-associated SF3B1 mutations, including K700E, which reduce binding of DDX46 to SF3B1 in humans and yeast (Tang et al. 2016; Zhang et al. 2024b). Hence, somatic mutations in SF3B1 that affect the DDX46–SF3B1 interaction were proposed to prevent the KP function of DDX46, by tapering off its SF3B1-mediated release from the RNA channel thus preventing closure of the hinge pocket around the BP-A and slowing the recruitment of the tri-snRNP, which would lead to activation of cryptic BS and alternative 3′ss (Zhang et al. 2024b). This model, however, is contradicted by the fact that DDX46 KD does not induce cryptic 3′ss usage in any of the top targets of mutant SF3B1 (Zhang et al. 2023). Furthermore, the N-terminal acidic loop of DDX46 binds the SF3B1 hotspot area, including K700, in an identical region to SUGP1, indicating that DDX46 can only be recruited to the spliceosome after SUGP1 is discharged from the HD. It is also notable that there are no known cancer mutations in DDX46, unlike both SUGP1 (Liu et al. 2020; Alsafadi et al. 2021) and DHX15 (Faber et al. 2016; Pan et al. 2017).
Limitations of structural studies of pre-spliceosomes
As we highlighted thus far, the multistep process of early BS recognition, from the initial identification of the BPS by SF1 in the E complex, through hand-over to the prelocalized U2snRNP in the pre-A complex and finally stable binding of the BS by the SF3B1 HD hinge pocket in the A-complex, is exceptionally dynamic. The intermediate states between conformational rearrangements in the precatalytic spliceosome are highly transient and unstable, which limits the ability of structural studies to resolve the detailed interactions that enhance the speed and fidelity of BS recognition. To overcome these technical constraints and enrich extremely short-lived transitional complexes, RNA structural biologists have leveraged multiple technologies, such as producing in vitro splicing complexes with BP-A-mutated or highly complementary BS sequences (Tholen et al. 2022; Zhang et al. 2024b) and affinity purification of endogenously tagged proteins coupled to the molecular effects of splicing inhibitors that stall splicing before formation of the A complex (Zhang et al. 2024b). Although encouraging, these spliceosome-trapping mechanisms could also be conducive to the artificial increase of “off-path” complexes that do not faithfully reproduce the natural order of splicing, especially in the convoluted landscape of BS recognition. Additionally, the highly degenerate sequence of the BPS in higher eukaryotes (Xie et al. 2023) further complicates a full understanding of BS-U2snRNA pairing.
A landmark example of the limitations associated with using cryo-EM of target-biased spliceosomes is the fact that despite multiple lines of evidence supporting a role for SUGP1 in the earliest step of BS recognition during assembly of the pre-A complex, no purified 17S U2snRNP has been found to contain SUGP1 (Zhang et al. 2020, 2021, 2024b; Tholen et al. 2022). This is consistent with the notion that the SF3B1–SUGP1 interaction occurs in an earlier step of splicing and could be unstable or transient. The recent cryo-EM structures that attempted to provide the first structural evidence in support of an early splicing QC control mechanism for DDX46, and possibly DDX42, in SF3B1-mutant cancers, were obtained by stalling the DDX46-bound U2snRNP immediately before assembly of the A complex, which happens later during BS recognition (Zhang et al. 2021, 2024b) and would omit SUGP1 since this factor needs to be discharged before DDX42 and DDX46 can bind SF3B1 (Zhang et al. 2023).
PHENOTYPIC CONSEQUENCES OF SF3B1 HOTSPOT MUTATIONS
SF3B1 mutations lead to pleiotropic cellular perturbations in a cancer-specific manner
Somatic missense mutations in SF3B1 are universally heterozygous and almost always mutually exclusive with hotspot mutations affecting other core splicing factors, indicating the low cellular tolerance for splicing dysregulation (Papaemmanuil et al. 2011; Yoshida et al. 2011; Graubert et al. 2012). While the SF3B1 K700E mutation is the most common mutation across multiple cancers (Yoshida and Ogawa 2014), other mutated residues are enriched in certain cancer types in an allele-specific manner that is still poorly understood. For example, E902K mutations are exclusively found in bladder cancer, R625 are preferentially enriched in melanomas, and G742 mutations almost always occur in CLL (Seiler et al. 2018).
SF3B1 mutations are found in ∼30% of all MDS patients (Papaemmanuil et al. 2011; Yoshida et al. 2011; Visconte et al. 2012) and have a striking frequency of up to 86% in a specific subtype of low-risk MDS in which erythrocyte progenitor cells in the bone marrow have iron-laden mitochondrial deposits around their nucleus called ringed sideroblasts (MDS-RS) (Huber et al. 2022). Patients with MDS-RS are characterized by ineffective erythropoiesis, multi- or single-lineage dysplasia, low blast count, and high transfusion burden (Adès et al. 2014; Cazzola 2022). Due to this clear genotype–phenotype relationship, SF3B1 mutations were included in the diagnostic criteria for MDS-RS, and more recently, SF3B1-MDS has been classified as an independent disease subtype associated with a relatively favorable prognosis. However, only the K700E substitution, which accounts for ∼60% of SF3B1 mutations in MDS-RS, independently predicts improved overall survival in MDS (Kanagal-Shamanna et al. 2021). In contrast, other recurrent non-K700E mutations rarely found in MDS-RS, such as E592K and K666*, are distinctly associated with high-risk forms of MDS with poor prognosis and progression to secondary AML (Dalton et al. 2020; O Sullivan et al. 2023; Choi et al. 2024). The molecular basis for this allele-specific association between different mutated residues in the C-terminal HEAT domain of SF3B1 and their prognostic significance in SF3B1-mutant MDS remains elusive. In the following section, we argue that the loss of interaction between SF3B1 and SUGP1 due to specific mutated residues and its effects on pre-mRNA splicing could determine the disease phenotype associated with different SF3B1-mutant MDS.
Oncogenic mutations in SF3B1 primarily affect the transcriptomes of multiple cancers by activating the recognition of aberrant BS, which most commonly leads to the selection of intron-proximal cryptic 3′ss upstream of the canonical 3′ss (Darman et al. 2015; DeBoever et al. 2015; Alsafadi et al. 2016; Zhang et al. 2019). Fifty percent or more of the hundreds of misspliced transcripts generated by this mechanism contain premature termination codons predicted to trigger nonsense-mediated decay of the mRNA and reduced protein expression, resulting in pleiotropic effects on cellular homeostasis that contribute to disease (Darman et al. 2015; DeBoever et al. 2015). For example, SF3B1 mutations have been repeatedly shown to reduce the levels of the MAP kinase MAP3K7, which can have multiple phenotypic effects on oncogenesis depending on the cancer type (Lee et al. 2018; Lieu et al. 2022). In mouse models of pancreatic cancer, loss of Map3k7 in Sf3b1K700E cells confers resistance to the apoptotic effects of the transforming growth factor beta, thereby promoting the progression of early-stage tumors, thus suggesting that SF3B1K700E acts as an oncogenic driver (Simmler et al. 2023). In the context of human hematopoiesis, SF3B1K700E-mediated loss of MAP3K7 disrupts p38 MAPK signaling, which acutely reduces GATA1 expression and leads to apoptosis and premature differentiation of erythrocyte progenitors, likely contributing to the severe anemia observed in MDS patients (Lieu et al. 2022). The heme biosynthesis and iron metabolism pathways have also been implicated in SF3B1-mutant cancers, which might explain the distinctive association between these mutations and MDS-RS (Bondu et al. 2019). Notably, the coordinated missplicing and reduced protein expression of the mitochondrial transporters TMEM14C and ABCB7 in SF3B1-mutant cells results in iron sequestration within the mitochondria and formation of ringed sideroblasts in MDS-RS (Dolatshad et al. 2015, 2016; Clough et al. 2022; Ochi et al. 2022). Moreover, the cooperative effects of the missplicing of these two transcripts are a prominent example of how not all SF3B1 mutations invariably result in the same missplicing pattern of disease-relevant transcripts, which could explain the varying prognostic outcomes associated with specific mutations, as explained below.
Clonal dominance of different SF3B1 mutations during hematopoiesis
SF3B1 mutations have a very high allelic burden with a variant allele frequency of up to 43% (Damm et al. 2012), indicating that these mutations arise early during CH and are likely to be initial genetic drivers of MDS and secondary AML. Indeed, a sequential sample analysis of MDS-initiating hematopoietic stem cells (HSCs) showed that SF3B1-mutant HSCs can propagate to the myeloid progeny and be an initiating event in MDS that drives malignant clone expansion to AML transformation (Mian et al. 2015). However, the clonal expansion necessary for AML transformation might be restricted to high-risk SF3B1 mutations since longitudinal tracking of CH in 385 individuals through phylogenetic and expansion analysis of mutant clones in peripheral blood found that K666N-mutant clones grow faster and exhibit higher clonal dominance than K700E clones (Fabre et al. 2022). This is further supported by another study that tracked the clonal competition between independent HSCs with different SF3B1 mutations from the same MDS-RS patients and found that K666N clones exhibit clonal dominance over K700E and N626D clones and also display mutation-specific RNA missplicing profiles (Moura et al. 2024). This suggests that the K700E mutation preferentially associated with MDS-RS and favorable prognosis (Kanagal-Shamanna et al. 2021) does not expand as fast as high-risk SF3B1 mutations, which drive clonal progression by acquiring subsequent genetic lesions.
Although SF3B1 mutations may occur alone and be the initiating oncogenic event (Mian et al. 2015), they can also be secondary to cooperative genetic lesions that can partially explain the clonal dynamics and disease phenotype induced by different mutated residues. Indeed, SF3B1 co-mutations are more frequent in MDS-RS with multilineage dysplasia than with single-lineage dysplasia, indicating that co-occuring genetic lesions contribute to increased failure of the bone marrow (Janusz et al. 2021). K700E mutations are preferentially associated with age-related epigenetic mutations in TET2, DNMT3A, and ASXL1 in MDS, which generally do not affect prognosis (Mian et al. 2013; Malcovati et al. 2014, 2015; Song et al. 2019; Dukenik et al. 2023; O Sullivan et al. 2023; Moura et al. 2024). In contrast, K666N and E592K mutations, which are rare in MDS-RS (∼0.1% and ∼1.5%, respectively), frequently co-occur with adverse prognostic mutations in RUNX1 and FLT3 (Dalton et al. 2020; Malcovati et al. 2020; Choi et al. 2024), which could explain their preferential selection during the progression of MDS to MDS with excess blasts and secondary AML (Dalton et al. 2020; Choi et al. 2024). Importantly, K666 residue mutations besides K666N co-occur with similar genetic lesions as K666N but are not enriched during disease progression from MDS to AML, which suggests that the distinct clinicopathologic features of SF3B1K666N MDS/AML are not solely driven by co-mutational differences (Dalton et al. 2020).
Loss of the SF3B1–SUGP1 interaction determines disease phenotype of SF3B1-mutant MDS
Besides the contribution of cooperative mutations, the distinctive missplicing induced by different mutated residues may also explain the specific prognostic effect of each mutation (Table 1). For example, the K700E mutation, which is associated with MDS-RS and severe anemia but not with progression to AML, leads to missplicing of both ABCB7 and TMEM14C, thus disrupting iron homeostasis. Contrarily, the E592K substitution in HEAT repeat 2, which does not disrupt the interaction between SF3B1 and SUGP1 or perturb the splicing of TMEM14C or ABCB7, is rare in MDS-RS and is exclusively associated with high-risk MDS and progression to AML (Choi et al. 2024). The K666N mutation in HEAT repeat 4, which is also associated with high-risk MDS and progression to AML (Dalton et al. 2020; O Sullivan et al. 2023), only slightly disrupts TMEM14 and ABCB7 to a much lesser extent than K700E and E622D (Choi et al. 2024). Interestingly, of the five mutations initially shown to disrupt the SF3B1–SUGP1 interaction (K700E, E622D, R625C, H662Q, and K666N), the K666N mutant disrupted the interaction only partially and did not show the same missplicing as the other four mutants (Zhang et al. 2019). Thus, the AML-enriched E592K and K666N mutations in SF3B1 that are specifically associated with poor prognosis and progression from MDS to secondary AML fully or partially maintain the SF3B1–SUGP1 interaction. Notably, the K700E and H662* mutations are significantly associated with a decrease in hemoglobin typical of anemia, whereas K666* mutations are not (Venable et al. 2021). This reinforces the notion that disease-specific clinicopathological traits depend on the molecular mechanism of each mutation.
Molecular, phenotypic, and clinical features of different cancer-associated SF3B1 mutations
These observations highlight the loss of the SF3B1–SUGP1 interaction as a disease-specific feature of SF3B1-MDS-RS and as a potential molecular biomarker to distinguish between the typical phenotype of K700E-mutant MDS-RS and the high-risk MDS/AML progression observed in patients carrying E592K or K666N mutations. Hence, the new SF3B1-MDS disease entity that is predicated upon the presence of RS and a favorable prognosis is more accurately defined at the molecular level by loss of the SF3B1–SUGP1 interaction rather than solely by the presence of genomic alterations in the SF3B1 allele.
Conclusions and outstanding questions
In this review, we have described the contribution of biochemical, phenotypic, and structural assays of oncogenic hotspot mutations in SF3B1, SUGP1, and DHX15 to understanding the fleeting interactions that mediate early BS recognition by the spliceosome (Zhang et al. 2019, 2022, 2023). Such studies have not only revealed the molecular consequences of these mutations in altered BS recognition and 3′ss selection observed in multiple cancer types, but also led to a new mechanism for the cooperative interaction of these three proteins in the hand-over of the BS during the assembly of the pre-A complex. Importantly, these studies also harnessed the recently developed power of artificial intelligence applied to biology, by using AF-M computational modeling to accurately predict the structure of a splicing-related multiprotein complex, the SF3B1–SUGP1–DHX15 protein trimer, which can be assembled in vitro but continues to evade cryo-EM visualization (Zhang et al. 2023).
In our current mechanistic model, in the first step of the BS hand-over, SUGP1 recruits U2snRNP to the vicinity of the BPS by directly contacting the hotspot region of the SF3B1 HD through two regions flanking its G-patch domain, which loops-out to activate DHX15. Then DHX15 “pulls” the pre-mRNA by helicase tracking into the RNA path of SF3B1 and displaces SF1 from the predefined BS, which becomes available for correct recognition by the p14 subunit of SF3B1 in the pre-A complex. In the second step of the hand-over, following the dissociation of SUGP1 from the complex, DDX42 and/or DDX46 are recruited to the U2snRNP by the same hotspot region previously occupied by SUGP1. DDX46 then unwinds the BSL in the U2snRNA to allow the formation of the U2-BS RNA duplex with a bulged BP-A, which is incorporated into the RNA channel of SF3B1 for the establishment of the branch helix in the catalytically primed A-complex. When specific residues of the SF3B1 HD are mutated in cancer, its interaction with SUGP1 is disrupted, leading to loss of SUGP1 during spliceosome assembly (Zhang et al. 2019). In the absence of SUGP1, DHX15 is not activated, and SF1 is not efficiently displaced from the canonical BS thus blocking its recognition by the U2snRNP. The SF3B1 mutant spliceosome must then recognize an upstream BS, and in case there is an appropriately spaced cryptic 3′ss, that 3′ss is selected and missplicing occurs.
This model does not rule out the possibility that DHX15, although not SUGP1, may have an additional function in splicing QC at a later step. This is consistent with the fact that DHX15 associates with spliceosomes independently of SUGP1 (Zhang et al. 2022), that DHX15 can be activated by multiple G-patch proteins (Hegele et al. 2012; Robert-Paganin et al. 2015; Bohnsack et al. 2021, 2022), and with the work of Maul-Newby et al. (2022) mentioned above. Interestingly, by leveraging the nucleotide preferences of DHX- and DDX-helicases, these authors also found that a DHX enzyme, through GTP hydrolysis, can not only promote an initial rearrangement that allows U2snRNP to engage an intron and support robust A-complex assembly, but also mediate the availability of the U2snRNA for base-pairing, either by unwinding an RNA structure (i.e., the BSL), or by dislodging a protein that protects the RNA.
Since DHX15 is the only DHX enzyme known to function in the U2snRNP (Maul-Newby et al. 2022), it is tempting to speculate that DHX15 could have an additional role in early BS recognition, to remove SUGP1 or another interacting G-patch protein from covering the U2snRNA, thereby supporting branch helix formation. This hypothesis is supported by the fact that DHX helicases in general, and possibly DHX15, can dislodge proteins without performing RNA duplex unwinding (Fairman et al. 2004). Thus, DHX15 could first contribute its unwinding activity to the direct recognition of the BS in the SF3B1–SUGP1–DHX15 splicing complex, and subsequently function to remove another ribonucleoprotein substrate to expose the U2snRNA. Notably, Damianov et al. (2024b) isolated parts of chromatin-associated spliceosomal A complexes bound by the G-patch proteins RBM5 and RBM10, which also contained DHX15 but limited amounts of SUGP1. RBM5 and RBM10 are alternative splicing factors that have been implicated in cancer which contain N-terminal RRM1-ZnF-RRM2 domains that interact with repressive cis-acting elements in target introns to promote exon exclusion (Collins et al. 2017; Inoue 2021; Soni et al. 2023, Damianov et al. 2024b) and have been previously reported to interact with DHX15 (Hegele et al. 2012). RBM5 in particular has been shown to interact with and stimulate the RNA helicase activity of DHX15 in a G-patch-dependent manner (Niu et al. 2012), which is reminiscent of the SUGP1–DHX15 mechanism. Nonetheless, RBM5 depletion did not recapitulate SF3B1-mutant splicing events (Zhang et al. 2019). Further studies will be valuable in elucidating how multiple G-patch proteins might function in BS recognition, and how mutations in such proteins can contribute to cancer pathogenesis.
Phenotypically, recurrent hotspot mutations in SF3B1 HEAT repeats H4–H7 that disrupt its interaction with SUGP1 and fail to activate DHX15, including K700E, lead to missplicing of specific genes associated with hematopoiesis and heme biosynthesis. These mRNA defects primarily drive the occurrence of MDS-RS, which is associated with severe anemia and high transfusion burden but not with clonal dominance and progression to secondary AML. Conversely, the E592K mutation rarely found in MDS-RS does not disrupt the SF3B1–SUGP1 heterodimer creating a unique RNA missplicing profile that could drive its selection during the progression of MDS to AML. Additionally, the K666N mutation, which partially maintains the interaction with SUGP1, is also associated with CH, high-risk MDS, and progression to AML. The striking disparity between the prognostic outcomes of SF3B1 mutations according to their effect on the SF3B1–SUGP1 interaction prompts us to suggest that the newly described disease entity known as SF3B1-MDS is actually molecularly defined by disruption of the SF3B1–SUGP1 interaction, which could thus be used as a biomarker to predict the prognostic effects of different SF3B1 mutations. Importantly, since restoring the SF3B1–SUGP1 interaction by overexpressing SUGP1 rescues missplicing in mutant cells, we posit that using proximity-inducing small molecules that can bridge critical interacting residues and restore the SF3B1–SUGP1 heterodimer could be a therapeutic strategy for MDS-RS and other SF3B1-mutant cancers (Zarzycka et al. 2016; Chen and Zacharias 2023; Lucero et al. 2023). It is important to highlight that although the K666 residue of the SF3B1 HD does not directly contact SUGP1 in the AF-M model, the K666N mutation could still perturb the local structure of the SF3B1–SUGP1 interface and thereby dislodge SUGP1 due to its proximity to the mutational pocket formed by other H4–H7 hotspot mutations. Moreover, other K666 mutations other than K666N, such as K666R, display a missplicing profile similar to K700E, which underscores the molecular and phenotypic singularity of the K666N mutation (Table 1).
A major goal for future studies of SF3B1-driven oncogenesis will be to elucidate the molecular mechanisms by which hotspot mutations associated with other cancers besides MDS-RS, such as E592K, K666N, and E902K, disrupt splicing. The E592K and E902K mutations do not neighbor the SF3B1–SUGP1 interface and likely perturb splicing through alternative mechanisms that remain to be described. The K666N mutation might perturb splicing in a manner related to SF3B1–SUGP1 disruption, albeit leading to a unique missplicing profile possibly due to the neomorphic contacts specifically made by the substitution of lysine by asparagine at residue 666, as opposed to other amino acids (Canbezdi et al. 2021). Unfortunately, AF-M is not trained to predict the effects of point mutations on protein structures (Evans et al. 2022), and thus it will be difficult to evaluate the specific structural consequences of these idiosyncratic mutations. Notwithstanding, these studies will not only advance our understanding of the elusive allele-specific relationships between different SF3B1 mutations and multiple cancer types but also provide novel insights into the earliest steps of splicing, which could be therapeutically targeted in SF3B1-mutant cancers.
ACKNOWLEDGMENTS
We thank Jian Zhang for insightful discussions and critical reading of the manuscript. Work from the authors’ laboratory was supported by National Institutes of Health (NIH) grant R35 GM118136 and an EvansMDS discovery research grant from the Edward P. Evans Foundation.
Footnotes
-
Article is online at http://www.rnajournal.org/cgi/doi/10.1261/rna.080368.124.
-
Freely available online through the RNA Open Access option.
This article, published in RNA, is available under a Creative Commons License (Attribution-NonCommercial 4.0 International), as described at http://creativecommons.org/licenses/by-nc/4.0/.
REFERENCES
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵








