
Connecting genotype and phenotype in minor spliceosome diseases
- Corresponding author: mikko.frilander{at}Helsinki.Fi
Abstract
Minor spliceosome is responsible for recognizing and excising a specific subset of divergent introns during the pre-mRNA splicing process. Mutations in the unique snRNA and protein components of the minor spliceosome are increasingly being associated with a variety of germline and somatic human disorders, collectively termed as minor spliceosomopathies. Understanding the mechanistic basis of these diseases has been challenging due to limited functional information on many minor spliceosome components. However, recently published cryo-electron microscopy (cryo-EM) structures of various minor spliceosome assembly intermediates have marked a significant advancement in elucidating the roles of these components during splicing. These structural breakthroughs have not only enhanced our comprehension of the minor spliceosome's functionality but also shed light on how disease-associated mutations disrupt its functions. Consequently, research focus is now shifting toward investigating how these splicing defects translate into broader pathological processes within gene expression pathways. Here we outline the current structural and functional knowledge of the minor spliceosome, explore the mechanistic consequences of its mutations, and discuss emerging challenges in connecting molecular dysfunctions to clinical phenotypes.
Keywords
INTRODUCTION
Increasing numbers of human diseases caused by mutations in the core components of the minor spliceosome, also called minor spliceosomopathies, have been identified within the last 10 years. Unlike the major spliceosome that carries out the bulk of cellular splicing and affects the expression of virtually all intron-containing genes, the minor spliceosome is functionally and structurally similar, but specialized to approximately 750 minor introns that constitute less than 0.5% of all introns in most organisms. In humans, minor introns reside in 700 genes often denoted as minor intron containing genes, or MIGs, each of which typically contains only one minor intron surrounded by multiple major introns (Turunen et al. 2013; Olthof et al. 2019; Moyer et al. 2020). For detailed information on the minor spliceosome, the reader should consult several excellent reviews on the topic (Turunen et al. 2013; Baumgartner et al. 2019; Olthof et al. 2022).
The minor spliceosome presents several challenges for understanding the etiology of the diseases associated with it. Similar to the major spliceosome, this cellular machinery is composed of five snRNAs, of which four are unique to the minor spliceosome. Like the major spliceosome, it is composed of a large number of protein components, a subset of which are unique to the minor spliceosome. It also follows a similar elaborate assembly/disassembly cycle as the major spliceosome (Patel and Steitz 2003; Turunen et al. 2013). These properties alone pose a significant challenge for understanding causes and consequences of disease mutations in any of the components. The additional difficulty with the minor spliceosome stems from its low cellular abundance, which is approximately 1% of that of the major spliceosome and has hampered the determination of its exact composition but also hindered many functional experiments in biochemical settings. Finally, while the ∼750 target introns in 700 genes are considerably less than the up to ∼200,000 intron targets for the major spliceosome, it is nevertheless a formidable group of genes to be connected in genotype–phenotype investigations. Despite these obstacles, recent years have witnessed several breakthroughs in the understanding of both the functions of the minor spliceosome and the associated diseases.
The discovery of unique components of the minor spliceosome and identification of novel minor spliceosomopathies are interlinked processes since all such diseases have been found to carry mutations in the unique snRNA and protein components. Originally, unique components of the minor spliceosome included only the four specific snRNA components, specifically U11, U12, U4atac, and U6atac snRNAs that substitute the respective major spliceosome U1, U2, U4, and U6 snRNAs, whereas the U5 snRNA was found to be shared between the spliceosomes (Hall and Padgett 1996; Tarn and Steitz 1996a,b). The list of unique components was subsequently expanded by the discovery of eight unique proteins in the U11/U12 di-snRNP (Will et al. 1999, 2004). Further expansion was on the waiting list for 15 years until genetic screens in maize by Bai et al. (2019) and Zuo et al. (2019) identified additional minor spliceosome protein components that set the stage for further expansions. Presently, 15 unique protein components are known, covering most of the steps in the minor spliceosome assembly/disassembly cycle (see Fig. 1; Bai et al. 2021; de Wolf et al. 2021; Siebert et al. 2022; Suzuki et al. 2022; Norppa et al. 2024). A parallel development has been taking place on the disease front following the identification of the first minor spliceosomopathy, MOPD I, caused by mutations in the RNU4ATAC gene that codes for the U4atac snRNA (Edery et al. 2011; He et al. 2011). Presently, mutations in two of the four snRNAs and five of the 15 known protein components have been associated with at least 11 minor spliceosomopathies (Table 1; Fig. 1).
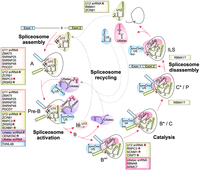
The minor spliceosome utilizes unique snRNA and protein components throughout the splicing cycle. Specific snRNA and protein components of various minor spliceosomal complexes are listed in text boxes colored according to the relevant snRNP. Factors with associated human disorders are shown in bold and marked with a red star.
Disorders caused by germline and somatic mutations in minor spliceosome-specific snRNAs and proteins
The growing catalogs of unique components and diseases have been instrumental in defining the compositional differences between the major and minor spliceosomes and mapping the introns and genes affected by the diseases. There is a wealth of phenotypic, genotypic, and transcriptome data for many of the minor spliceosomopathies revealing their shared general characteristics. For example, minor spliceosomopathies are predominantly developmental disorders, they often show striking tissue specificity, and at the molecular level the disease mutations are always hypomorphic. However, there are considerable challenges in connecting the known disease genotypes, that is, the mutations in the minor spliceosome components, to the observed clinical phenotypes. In this regard, recent developments in the field, particularly the high-resolution cryo-electron microscopy (cryo-EM) structures of several spliceosomal assembly intermediates, are now starting to fill one of the major gaps in our understanding by building solid foundations for interpreting the molecular phenotypes of these diseases. Here we will first discuss insights emerging from the structural data in explaining the molecular basis for the requirement of minor spliceosome-specific protein components, and how this can be linked to the observed molecular disease phenotypes. In the final section, we discuss the recent attempts in further progressing from molecular to clinical phenotypes.
STRUCTURAL FOUNDATIONS FOR THE MINOR SPLICEOSOME DISEASES
The recent high-resolution structures of the minor spliceosome pre-B and Bact complexes and the individual U11 snRNP (Bai et al. 2021, 2024; Zhao et al. 2025) are among the most transformative works for understanding the functions of the specific components of the minor spliceosome. These works provide the foundations for deciphering the consequences of disease mutations. The cryo-EM structures have provided a gratifying confirmation for the earlier conclusions based on biochemical and genetic analyses on the snRNA components, that the spliceosome catalytic core structures relevant for the catalytical steps are highly similar between the two spliceosomes (Tarn and Steitz 1996a; Shukla and Padgett 1999; Frilander and Steitz 2001; Turunen et al. 2013). Therefore, the extent of structural differences between the major and minor snRNPs and spliceosomal complexes observed in the high-resolution structures (described in more detail below) has been more extensive than anticipated. One of the major conclusions coming from the structural studies is that the need for specific proteins originates mostly from sequence-level differences between the minor and major snRNAs. The divergent regions in the minor snRNAs serve as interfaces for the binding of specific proteins in the minor spliceosome, and at the same time, they replace the corresponding RNA elements and their protein partners used by the major spliceosome. The key functions recognized for the unique proteins in the minor snRNPs and spliceosomal complexes can be classified to three general groups: (1) stabilization of the catalytic core architecture, (2) binding to the specific minor snRNA structures to mediate specific snRNP architecture, and (3) differential recognition of minor intron splice sites.
U11/U12 di-snRNP and U4atac/U6atac.U5 tri-snRNP
The U11/U12 di-snRNP provides an illuminating example of the role of specific proteins in shaping both the large-scale snRNP structures but also in providing specificity for minor intron recognition. The di-snRNP interacts with minor intron 5′SS, branch point sequence (BPS) and 3′SS elements both in the intron recognition step (A complex), but also in the pre-B complex that contains all five snRNAs (see Fig. 1). It has a highly divergent protein composition, featuring eight minor spliceosome-specific proteins (Will et al. 1999, 2004) of which five associate with the U11 snRNP and three with the U12 snRNP (Fig. 1). The recent structures of both the U11 mono-snRNP and the pre-B complex have now revealed that the protein diversity is specifically linked to the unique folding of U11 snRNA. Despite sharing a highly similar overall secondary structure with the U1 snRNA, U11 snRNA has been found to adopt a unique and compact 3D structure which serves as the recognition platform for the five U11 snRNP-specific proteins (Bai et al. 2024; Zhao et al. 2025). This affects both the overall architecture of the U11/U12 di-snRNP and specific molecular interactions during intron recognition.
At the architectural level, the rod-like PDCD7 protein is the most striking structural element and serves as a backbone for the U11 snRNP and, together with more compactedly folded U11 snRNA, gives it a unique shape (Bai et al. 2024; Zhao et al. 2025). PDCD7 also affects the higher-order di-snRNP formation by interacting with the RNPC3 protein bound to the U12 snRNA stem–loop III (see Fig. 2C). This interaction forms a molecular bridge essential for the cooperative recognition of the minor intron 5′SS and BPS sequences by the U11/U12 di-snRNP (Frilander and Steitz 1999; Benecke et al. 2005; Zhao et al. 2025). At the functional level, the specific proteins have a role in the recognition of each of the three intronic elements (5′SS, BPS, 3′SS). Of these, the initial 5′SS recognition of minor introns differs remarkably from that of major introns, where U1 snRNA base-pairs across the exon–intron junction and is stabilized by the U1-C protein (Kondo et al. 2015). In contrast, the recognition of the minor intron 5′SS involves a more intricate set of interactions. Positions −1 to +4 of the minor intron 5′SS are recognized by SNRNP48 and SNRNP35 proteins and also by the U11 stem–loop III that forms noncanonical contacts with this region (Turunen et al. 2008; Bai et al. 2024). In comparison, the downstream +5 to +10 positions adhere to a more conventional recognition mechanism. Here, the 5′-end of the U11 snRNA base-pairs with these nucleotides while ZMAT5, a paralog of U1-C, further stabilizes this interaction (Bai et al. 2024).

Mutational spectrum of minor spliceosomopathies. (A) Secondary structure of the U4atac/U6atac snRNA duplex, with mutations causing various RNUATAC-opathies indicated. The mutated nucleotides are colored according to the disorder they were first described in. If the same mutation has subsequently been reported in additional disorders, these are indicated by colored squares next to the mutation. The “Other” category includes phenotypes that fall in between or outside clinically defined disorders (McMillan et al. 2021; Tabib et al. 2023). (B) U4atac snRNA mutations in their structural context. The main image shows the cryo-EM structure of the U4atac/U6atac di-snRNP and U11 snRNP in the minor spliceosome pre-B complex (Protein Data Bank ID: 8Y6O). Insets show close-up views of the 5′ SL, stem II, Sm site, and 3′ SL regions. Mutated U4atac nucleotides are colored red and numbered by their position in the insets. Sm site mutations are shown for illustrative purposes as Sm-mutant snRNAs are not expected to assemble into snRNPs. Except for PRPF6, U5 snRNP components and tri-snRNP-specific proteins are grayed out for clarity. Structure visualization was carried out using PyMOL. (C) Secondary structure of the U12 snRNA showing mutations underlying EOCA and CDAGS. Binding sites of the RNPC3, RBM41, and ZCRB1 proteins are indicated. (D) Domain structure of minor-spliceosome-specific proteins implicated in human disease. Locations of germline mutations are shown at the bottom of the domain structure, and for ZRSR2, somatic mutations are shown on top of the domain structure. Somatic ZRSR2 mutations were downloaded from the COSMIC database and filtered for Primary Tissue (Hematopoietic and lymphoid) and Histology (Hematopoietic neoplasm).
Compared to the U11/U12 di-snRNP, the minor U4atac/U6atac.U5 tri-snRNP is more similar to its major spliceosome counterpart and shares most of the protein components with the major tri-snRNP. However, it is missing the RBM42 and SNRNP27 proteins present in the major tri-snRNP due to differences between minor and major tri-snRNP architectures (Charenton et al. 2019; Bai et al. 2021). The main unique features include, in addition to the distinct U4atac and U6atac snRNAs, two unique proteins: CENATAC and its interactor TXNL4B/Dim2 (de Wolf et al. 2021; Suzuki et al. 2022; Bai et al. 2024). Of these, CENATAC binds simultaneously to the unique structure composed of closely spaced trimethylguanosine cap of the U4atac snRNA and the central stem–loop of the U6atac snRNA (CSL, see Fig. 2A,B; Bai et al. 2024). CENATAC is present in both the U4atac/U6atac di-snRNP and the minor tri-snRNP (de Wolf et al. 2021). In contrast, TXNL4B binds to the U5 snRNP within the minor tri-snRNP, where it presumably functions as a CENATAC/TXNL4B heterodimer, replacing TNXL4A/Dim1 in the major spliceosome (de Wolf et al. 2021; Suzuki et al. 2022; Bai et al. 2024).
Spliceosomal complexes
During the complex transitions before the first catalytic step (A > pre-B > B > Bact complexes) several additional unique proteins, namely SCNM1, RMB48, ARMC7, and CRIPT enter the nascent spliceosome (Fig. 1). The precise timing of their entry is presently unclear due to lack of structural and interaction data for the A and B complexes. Of these proteins, the U12 snRNP-associated SCNM1 is a functional homolog of the SF3A2 and SF3A3 proteins in the major spliceosome. Unlike its major spliceosome counterparts that associate with the U2 snRNP, SCNM1 has not been detected in the purified U11/U12 di-snRNP (Will et al. 1999, 2004). Its presence in the minor pre-B and Bact complexes (Bai et al. 2021, 2024), however, suggests recruitment either at the A complex stage or during A to pre-B transition. In contrast, RBM48, ARMC7, and CRIPT are present only in the Bact complex, consistent with recruitment during the transitions from the prespliceosome to the activated spliceosome (pre-B > B > Bact). The entry of these proteins thus coincides with major structural transitions of the minor spliceosome, particularly the dissociation of U11 snRNP, the dissociation of U4atac snRNP and CENATAC and TXNL4B proteins, and the dissociation of all the U12 snRNP-specific proteins except RNPC3 (Bai et al. 2021).
In the Bact complex, the function of the minor spliceosome-specific proteins is to stabilize the precatalytic structures. Here, the SCNM1 protein plays the role of the SF3A2/A3 subunits of the SF3a complex in the major spliceosome, interacting with both the U12/BPS helix and the SF3b complex bound to U12 snRNA. It also extends into the active site, interacting with the 5′SS and U6atac snRNA, as well as RNF113A, which is a shared component of both spliceosomes. Together with CRIPT, which also interacts with the SF3b and RNF113A, SCNM1 spatially shields the 5′SS and BPS, which are located 50 Å apart in the precatalytic conformation. This structure is similar to that in the major Bact, but without the SF3a complex and with the SCNM1 and CRIPT proteins (Haselbach et al. 2018; Zhang et al. 2018). Incidentally, the major Bact structure is also compatible with CRIPT binding; however, it has not been observed in the present structures. The two other proteins in the minor Bact, RBM48, and ARMC7, form a heterodimer that apparently compensates for the structural differences between U6 and U6atac snRNAs. Specifically, U6atac snRNA is missing the 5′-stem–loop present in the U6 snRNA which in the major Bact binds the RBM22 and BUD31 proteins. In the minor spliceosome, these two proteins are replaced by the RBM48/ARMC7 dimer that binds to the 5′-monophosphate cap of the U6atac snRNA and potentially participate in similar postcatalytic processes as RBM22 in the major spliceosome (Zhang et al. 2017).
Due to the lack of detailed structural and interaction data, little is known about the composition of catalytic and postcatalytic minor spliceosome complexes. The only exception is the recently identified U12 mono-snRNP, which is likely a dissociation product of the postcatalytic minor spliceosome. It contains two unique proteins, of which ZCRB1 is also a component of the U11/U12 di-snRNP. However, it apparently dissociates from U12 snRNA during the B/Bact complex formation and then reassociates during or after the postsplicing transitions (Norppa et al. 2024). Interestingly, ZCRB1 is also known to interact with the 2′-O-methylated A residue within the UUAmA sequence at the 5′ end of U12 snRNA (Li et al. 2024). The other component, RBM41, is a recently identified paralog of RNPC3, that binds to the same stem–loop III of the U12 snRNA (Norppa et al. 2024). While the specific roles of these proteins remain unclear, proteomics and microscopy data suggest that they may escort the U12 snRNP to the Cajal body to initiate a new round of assembly for the U11/U12 di-snRNP.
FROM STRUCTURES TO DISEASES: INSIGHTS INTO MINOR SPLICEOSOMOPATHIES
Minor spliceosomopathies constitute a phenotypically heterogenous group of diseases caused by either germline or somatic mutations in any of the specific components of the minor spliceosome. Presently, this group includes ten recessive congenital disorders caused by loss-of-function mutations in two of the specific snRNAs (U12 and U4atac) and in four of the specific protein components (RNPC3, ZRSR2, SCNM1, CRIPT, and CENATAC). Additionally, somatic mutations in the ZRSR2 gene have been linked to an interconnected group of hematological malignancies (see Table 1). Each of these diseases is linked to minor spliceosome components and has been characterized at a phenotypical level, and in some cases also by RNA sequencing.
When evaluated collectively, the typical molecular outcome with the majority of mutations is a reduction in the levels of the minor spliceosome components. The component here can be an snRNA, a specific protein, an individual snRNP, or an snRNP complex. The mutations can be further classified into three categories depending on whether the underlying mechanisms: (a) can be explained by the present structural data; (b) require structural data that is not yet available; or (c) involve cases where structural data is not applicable.
“RNU4ATAC-opathies”: disrupting RNA backbone interactions and snRNP interfaces
Among the congenital minor spliceosomopathies, the RNU4ATAC locus encoding the U4atac snRNA features the highest number of disease-associated mutations. When viewed alongside the structural data, these mutations demonstrate the value of structural data in providing a detailed view on known disease mechanisms, but also in revealing novel disease mechanisms. The U4atac mutations concentrate on four separate regions (see Fig. 2A): 5′ stem–loop (5′SL), 3′ stem–loop (3′SL), Sm site and region forming U4atac/U6atac stem II. While a few U4atac mutations occur in homozygous form, most are found in combination with another mutation in the second RNU4ATAC allele. This allelic heterogeneity has resulted in phenotypically overlapping clinical conditions that are now collectively referred to as “RNU4ATAC-opathies” (Duker et al. 2023) with both shared and disorder-specific phenotypes (Table 1). This group includes the earlier discrete clinical conditions microcephalic osteodysplastic primordial dwarfism type I (MOPD I), Roifman syndrome (RS), and Lowry-Wood syndrome (LWS) (Edery et al. 2011; He et al. 2011; Merico et al. 2015; Farach et al. 2018). Furthermore, some individuals with RNU4ATAC mutations exhibit ciliopathy-related phenotypes characteristic of Joubert syndrome (JBTS) alongside phenotypes shared with other RNU4ATAC-opathies (Khatri et al. 2023; referred here to as JBTS-like). Of the individual disorders, MOPD I is associated with the most severe phenotype, commonly causing death in infancy or early childhood, with survival to adulthood reported only in a few cases.
According to the accepted model of the RNU4ATAC-opathy pathogenesis, the disease-causing mutations in U4atac snRNA impair minor spliceosome function by reducing the cellular levels of the U4atac/U6atac.U5 tri-snRNP by a combination of two mechanisms (Jafarifar et al. 2014; Almentina Ramos Shidi et al. 2023). Of these, the Sm site mutations have been shown to destabilize the U4atac snRNA, thereby reducing the cellular U4atac snRNP levels available for tri-snRNP formation (Jafarifar et al. 2014). In contrast, mutations in the U4atac 5′SL and stem II have been thought to reduce the minor tri-snRNP levels by disrupting the molecular interactions between the U4atac/U6atac di-snRNP and U5 snRNP that are thought to be mediated by SNU13, PRPF3, PRPF4, and PRPF31 proteins (Jafarifar et al. 2014; Merico et al. 2015; Khatri et al. 2023).
The cryo-EM structure of the minor pre-B complex (Bai et al. 2024) now refines this model by providing a detailed view on the RNA–protein contacts in 5′SL and stem II, showing that the majority of these are mediated though interactions with the RNA backbone. This offers a simple mechanistic model for the pathogenesis where the base-pairing interactions disrupted by disease mutations (see Fig. 2A) lead to a conformation change in the U4atac/U6atac stem II backbone and consequently weaken or disrupt the specific snRNA–protein interactions within 5′SL or stem II (Fig. 2B; Bai et al. 2024). The mutations disrupting the interactions between 5′SL and SNU13 are expected to be particularly deleterious because of the essential role of SNU13 in folding the 5′SL RNA to a kink-turn motif (Cojocaru et al. 2005; Woźniak et al. 2005) that serves as the starting point for the binding of the remaining U4atac/U6atac di-snRNP proteins (Nottrott et al. 2002; Liu et al. 2007, 2011; Jafarifar et al. 2014).
The structure also tentatively suggests a novel disease mechanism for the U4atac snRNA mutations located at the 3′SL (G111 > A and G114 > C, see Fig. 2A,B) that do not conform to the disease mechanisms identified earlier. In the minor pre-B structure, these two positions have been reported to be part of one of the four selective interaction interfaces between the U11/U12 di-snRNP and the U4atac/U6atac.U5 tri-snRNP that drive the incorporation of minor, but not major tri-snRNP to the minor spliceosome (Bai et al. 2024). Strikingly, the U4atac mutations in positions 111 and 114 are disrupting the base-pairing interactions on the proposed interface I, which can prevent the U4atac backbone interactions by the U11-specific PDCD7 and SNRNP25 proteins (see Fig. 2A,B). While this possibility is yet to be confirmed, it nevertheless exemplifies the utility of using structural data in interpreting the human disease data.
CH: disrupting the interaction points between the U11 and U12 snRNPs
Minor spliceosomopathy linked to the RNPC3 mutations P474T/R502X was originally described to cause isolated growth hormone deficiency and postnatal growth restriction, with pituitary hypoplasia (Argente et al. 2014). Since this original finding both the mutational and phenotypic spectrum has significantly expanded in the recent years and as a consequence, the disease condition has now been renamed as congenital hypopituitarism (CH) (see Table 1; Fig. 2D; Argente et al. 2014; Verberne et al. 2020; Yamada et al. 2021; Akin et al. 2022; Bezen et al. 2022; Yavas Abali et al. 2024). The affected RNPC3 protein has a domain structure containing N- and C-terminal RRMs separated by a proline-rich region. The majority of individuals with pathogenic RNPC3 mutations are compound heterozygous or homozygous for a missense mutation in the C-terminal RRM, with the heterozygotes carrying a frameshift, nonsense, or splice site mutation in the second allele (Fig. 2D).
The function of the RNPC3 protein is to form a molecular bridge between the individual U11 and U12 snRNPs within the U11/U12 di-snRNPs and therefore the disease-causing missense mutations in this protein are particularly interesting as they can delineate the structurally important regions of this protein. RNPC3 is poorly resolved in the present cryo-EM data sets, but there is limited structural information from NMR analyses of the C-terminal RRMs carrying P474T or R502X mutation. Additionally, AlphaFold predictions and model building onto the cryo-EM density map have been able to reconstruct a putative contact point between the U11 snRNP and RNPC3, consisting of fragments of the U11-specific PDCD7(172–453) and ZMAT5(136–156) and the U12-specific RNPC3 (143–192) (Zhao et al. 2025), which includes the positions for the G148A and P162S missense mutations. From this limited data, it is nevertheless possible to conclude that each of the missense mutations is likely to interfere with the RNPC3 bridging function and thus reduce the cellular levels of both the RNPC3 protein and the U11/U12 di-snRNP. The most detailed experimental evidence is available for the P474T mutation, where a combination of biochemistry and NMR analyses suggested that the affected C-terminal RRM is only partially folded, which reduces the affinity to U12 snRNA SL III, but is also linked to decreased stability of the protein. The neighboring R502X mutation, on the other hand, leads to RNPC3 mRNA decay by NMD, but the potential mutated protein is also essentially devoid of RNA-binding activity (Norppa et al. 2018). Additional biochemical evidence suggests that the other missense mutations in either the C-terminal RRM (Y443C, L483F) or its N-terminal extension (F410V) essential for the stability of the domain (Netter et al. 2009) are also likely to impair U12 snRNA binding. The two N-terminal missense mutations (G148A and P162S), in turn, are in the region that likely interacts with the PDCD7/ZMAT5 module in U11 snRNP (Zhao et al. 2025), suggesting a pathomechanism where these mutations weaken or sever the RNPC3/U11 snRNP interaction.
MVA: toward understanding the minor intron subtype selectivity by CENATAC
CENATAC is a recently identified minor spliceosome component and a novel disease gene for mosaic variegated aneuploidy (MVA), a rare disorder characterized by mitotic defects leading to chromosomal instability and mosaic aneuploidies in multiple tissues (de Wolf et al. 2021). The patients described thus far are compound heterozygous for two truncating mutations that both delete 64 amino acid residues from the C terminus of CENATAC (Fig. 2D). The most striking molecular outcome observed both in the patient cells and cultured cells depleted of CENATAC is a transcriptome-wide splicing defect that affects almost exclusively minor introns with AT-AN (“A-type”) terminal dinucleotides, while the introns belonging to GT-AN (“G-type”) subtype are mostly unaffected. The mechanistic basis of this selectivity is presently unknown due to the lack of suitable structural data, most likely minor B complex, while present data are limited to the preceding pre-B complex and the following Bact complex (Fig. 1). In the major B-complex the first 5′-G residue of the major introns is specifically recognized by the PRPF8 protein (Zhang et al. 2024) by a combination of stacking interactions and hydrogen bonding between the G residue and peptide backbone. The minor B complex structure is needed to resolve whether the CENATAC or its interacting protein TXNL4B can influence this process, particularly with the AT-AN minor intron subtype, and thus provide mechanistic insight to the observed intron subtype bias.
EOCA and CDAGS: decreased stability of the U12 snRNA
Biallelic U12 snRNA (RNU12 gene) mutations have been reported in two phenotypically distinct congenital disorders, early-onset cerebellar ataxia (EOCA) and CDAGS syndrome (Table 1; Elsaid et al. 2017; Xing et al. 2021). All EOCA patients reported to date are homozygous for an 84C > T mutation at the base of the U12 SL III (Fig. 2C; Elsaid et al. 2017). In contrast, CDAGS patients are compound heterozygous for a *3C > T mutation, located in the 3′-extension of U12 snRNA that is trimmed off during its maturation, and a mutation in either the Sm site (75A > G, 77T > A or 77T > G) or near the base of the SL III (86G > A) (Fig. 2C; Xing et al. 2021).
With these diseases, the causal mutations directly affect the cellular levels of U12 snRNA by decreasing its half-life. The process is best characterized with the 84C > U EOCA mutation, which leads to an “overtrimming” phenotype by the TOE1 exonuclease that is involved in natural 3′ end processing of the snRNAs, followed by further 3′ to 5′ decay by the nuclear exosome (Norppa and Frilander 2021). Interestingly, the EOCA study also identified additional destabilizing variants in the U12 SL III segregating in human population, including an 86G > U mutation that disrupts the same base pair as the CDAGS-causing 86G > A mutation, suggesting a potential shared disease mechanism between a subset of CDAGS mutations and those of EOCA. In contrast, the CDAGS Sm site mutation likely destabilizes the U12 snRNA similarly as the comparable U4atac mutations, while the *3C > T CDAGS mutation in the U12 snRNA 3′ extension has no clear precedent in the literature but could conceivably impact the 3′-end processing of the U12 snRNA primary transcript.
OFD and MDS: disruption of the initial 3′SS recognition by ZRSR2?
Both germline and somatic mutations in ZRSR2 have been associated with human disorders. Germline mutations in ZRSR2 cause orofaciodigital syndrome (OFD) (Hannes et al. 2024), while somatic ZRSR2 mutations have been described in various types of hematological malignancies, but have been most thoroughly characterized in myelodysplastic syndrome (MDS) (Table 1; Yoshida et al. 2011). From a structural point of view, ZRSR2 is challenging as it is not visible in the present cryo-EM structures even though it can be detected in the pre-B complex by mass spectrometry (Bai et al. 2024). RNA CLIP data indicates that ZRSR2 interacts with the stem III of U12 snRNA (Kwon et al. 2024). In vitro splicing studies have shown that it is involved in minor intron 3′SS recognition, consistent with its status as a paralog of U2AF1 that is involved in 3′SS recognition in the major spliceosome (Shen et al. 2010). Due to the X-chromosomal location of the ZRSR2 gene, all the reported ZRSR2-mutant OFD patients are males hemizygous for one of two different frameshift mutations in the last exon of ZRSR2, which likely lead to production of truncated proteins lacking the C-terminal RS domain (Fig. 2D; Hannes et al. 2024). In contrast, in MDS and other hematological malignancies, a large number of nonsense, missense, and frameshift mutations are relatively evenly distributed across the open reading frame without obvious hotspots (Fig. 2D). Noteworthy, a significant fraction of the nonsense and frameshift mutations are expected to completely disable ZRSR2 function.
Comparison of the RNA-seq data from OFD and MDS patients samples reveal that minor intron splicing defects are significantly weaker with the OFD (Madan et al. 2015; Inoue et al. 2021; Hannes et al. 2024). This is consistent with the notion that MDS mutations typically lead to ZRSR2 null allele, while with the OFD, ZRSR2 likely retains partial functionality. A possible pathomechanism could be a loss of either the RS domain-mediated exon definition interactions (Akinyi and Frilander 2021) or the interactions with the nearby positive splicing regulators. Alternatively, it is also possible that the C-terminal truncations affect the RNA-binding properties of the ZRSR2. In contrast, the majority of the MDS patient samples studied in detail thus far are likely carrying ZRSR2 null alleles (Madan et al. 2015; Inoue et al. 2021). Interestingly, RNA-seq data from these studies show that while missplicing can be detected with most minor introns, the majority of the respective transcripts appear to be correctly spliced (Madan et al. 2015; de Wolf et al. 2021), suggesting that ZRSR2 may not be strictly necessary for the correct 3′SS recognition. The observation that missplicing is enriched with introns featuring a short distance between BPS and 3′SS (Inoue et al. 2021) further suggests that the degree of the splicing defects may be linked to the architecture of 3′SS recognition. Another possibility is that ZRSR2 loss could be complemented by ZRSR1, an intronless pseudogene of ZRSR2 present both in mouse and humans. However, while ZRSR1 is known to complement ZRSR2 loss during mouse early development (Gómez-Redondo et al. 2020), it appears not to be functional in the human hematopoietic lineage (Madan et al. 2022).
OFD and RTF: destabilizing the Bact complex?
Two of the syndromes, OFD caused by germline mutations in SCNM1 (Iturrate et al. 2022) and Rothmund-Thomson syndrome (RTS) caused by mutations in CRIPT (Averdunk et al. 2023) are potentially affecting the stability of the minor Bact complex. The OFD patients carrying SCNM1 mutations are homozygous for any of the three mutations depicted in Figure 2D (Iturrate et al. 2022), while the CRIPT mutations are either frameshifts or missense mutations in the highly conserved CXXC motifs of the protein (Fig. 2D; Akalın et al. 2023; Averdunk et al. 2023). As described earlier, SCNM1, a functional analog of SF3a complex in major spliceosomes, has likely a vital role in stabilizing the precatalytic conformation of the minor Bact (Bai et al. 2021). The impact of SCNM1 mutations on minor intron splicing was originally demonstrated in mouse before the identification of SCNM1 as a minor spliceosome component (Buchner et al. 2003), and subsequently confirmed by RNA-seq and RT-PCR analysis of OFD patient cells (Iturrate et al. 2022).
In contrast, the role of CRIPT in the minor spliceosome is less clear. In minor Bact, it binds to the same overall region as SCNM1 suggesting that it may also influence the stability of the Bact precatalytic conformation. Mutations in the CXXC motifs are expected to disturb the Zn-finger motif, which likely affects the binding of CRIPT to Bact. Presently, the effect on splicing in the patient cells has not been reported, but it is worth noting that RTS shares several phenotypic characteristics with the other minor spliceosomopathies (Table 1). It is also possible that any effects on splicing are not limited to minor introns, because the major Bact architecture is also compatible with CRIPT binding (Bai et al. 2021), and CRIPT has been reported to copurify with the major Bact, BAqr, and C complexes at substoichiometric levels (Schmitzová et al. 2023; Zhang et al. 2024). It should also be noted that CRIPT appears to be a multifunctional protein as there is extensive literature on CRIPT functioning as a microtubule-binding protein in dendrites and mitotic spindle disassembly (Omelchenko et al. 2020; Xu et al. 2022). Therefore, the contributions of CRIPT mutations for synaptic, mitotic, and spliceosomal functions remain to be determined.
FROM MECHANISTIC DEFECTS TO CLINICAL PHENOTYPES
As our mechanistic understanding of the functions of minor spliceosome components and the molecular consequences of their mutations solidifies, the main challenge in deciphering the pathomechanisms is now shifting toward the subsequent steps in the gene expression pathway. At the mechanistic level, these include the mechanisms responsible for the mutation- and disease-specific defects during the processing of individual introns. At the gene expression level, these include the postsplicing effects at the levels of whole transcriptomes and proteomes, but also the further downstream consequences during development and differentiation that eventually lead to the observed clinical phenotypes.
Mechanistic defects and intron-level effects
Presently most attention on the molecular characterization of the minor spliceosomopathies has been devoted to documenting the minor intron splicing defects, either at the level of individual introns or at the transcriptome-wide scale. These studies have revealed that the typical molecular consequence of minor spliceosome dysfunction is a partial splicing defect shown as an increase in minor intron retention in RNA-seq and RT-PCR analyses. Interestingly, with all minor spliceosomopathies studied thus far the splicing defects have been detected only with a subset of the potential minor intron targets. Additionally, the splicing defect can also spread to the flanking exons and neighboring major introns, manifesting either as activation of cryptic or known alternative splice sites, exon skipping, or increased retention of major introns (Argente et al. 2014; Madan et al. 2015; Merico et al. 2015; Elsaid et al. 2017; Cologne et al. 2019; de Wolf et al. 2021; Inoue et al. 2021). Such a molecular phenotype, where only a subset of minor introns is responding to the minor spliceosome dysfunction, is consistent with a partial loss of the minor spliceosome functions but raises the question of the mechanism behind the differential response among the minor introns. Intronic features, such as the splice site strength, intron length, sequence features, and the location of minor introns in the transcripts are obvious candidates, but no such correlative interactions have been detected with most diseases. The known exceptions are MDS, where the ZRSR2 mutations causing intron retention have been preferentially associated with minor introns with short BPS to 3′SS distance (Inoue et al. 2021); MVA, where the CENATAC mutations lead to a splicing defect with introns carrying AT-AN terminal dinucleotides (de Wolf et al. 2021); and MOPD1, where the already poorly spliced introns in lowly expressed genes appear to be more affected (Cologne et al. 2019).
The variable disease phenotypes suggest that the identity and magnitude of the missplicing events in each minor spliceosomopathy has a unique mechanistic basis related to the specific mutations, but no systematic comparisons have yet been carried out. Also, the question of whether other factors besides the integral components of the minor spliceosome can influence the responsive introns is understudied, particularly in disease settings. Potential modifiers of splicing efficiency include the known interactors of the minor spliceosome, such as SR proteins (Hastings and Krainer 2001; Meinke et al. 2020) and the neighboring major spliceosomes via exon definition interactions (Wu and Krainer 1996, 1998; Akinyi and Frilander 2021). Supporting this possibility, data from the MOPD1 patients shows that the magnitude of minor intron splicing defects vary significantly between different cell types, and the strongest effects are seen with lymphoid cells, while patient fibroblasts show very weak responses to disease mutations (Cologne et al. 2019). These results are consistent with the observation that under normal physiological conditions outside of the disease context, minor intron splicing efficiency varies in a tissue-specific manner, suggesting that it is a more regulated event than earlier thought (Olthof et al. 2019). A possible link to tissue-specific expression comes from the work on SR protein family member SRSF10 that contains a minor intron. The expression of SRSF10 is regulated by the minor spliceosome, but at the same time, SRSF10 can influence the tissue-specific expression of the entire family of SR proteins through cross-regulation interactions (Meinke et al. 2020). Together, these observations argue that the cell- and tissue-specific factors not only influence minor intron splicing, but that the minor intron splicing has also an active role in their regulation.
Disease phenotypes from a cumulative subclinical effect?
RNA-seq analyses of the patient cells have confirmed that minor spliceosome mutations cause a significant number of aberrant splicing events, with anything from 30 and up to 300 minor introns being affected, depending on the disease and the cell type analyzed (Argente et al. 2014; Madan et al. 2015; Merico et al. 2015; Elsaid et al. 2017; Cologne et al. 2019; de Wolf et al. 2021; Inoue et al. 2021). These aberrant splicing events are predominantly intron retention events that typically incapacitate the affected transcripts as a consequence of NMD, nuclear retention or formation of truncated proteins (Verma et al. 2018). The large number of primary targets raises the question of whether the disease phenotypes are driven by individual misregulated MIGs or through a concerted action of multiple affected MIGs. This question is particularly pertinent for minor spliceosomopathies where the primary intron retention events are relatively mild. For example, both with MVA patient fibroblasts and MDS bone marrow cells the median ΔPSI values for intron retention are around 0.25 and only with a few transcripts do the individual ΔPSI values exceed 0.5 (Madan et al. 2015; de Wolf et al. 2021). Given that the fraction of nonfunctional transcripts for individual genes in minor spliceosomopathies is typically always less than 50%, it is conceivable that many of the phenotypic characteristics of the individual diseases are likely to be the outcome of the concerted action of multiple relatively mild splicing defects that alone are not sufficient to lead to pathological outcomes. We further argue that for probabilistic reasons, the cellular pathways containing large number of MIGs are likely to be more vulnerable for minor spliceosomopathy mutations than those containing individual MIGs.
Several cellular pathways and functional groups are particularly enriched for MIGs and can be envisioned to integrate the weak downregulation signals originating from minor intron splicing defects. The most prominent pathways or cellular functional groups, in terms of enrichment and number of affected genes, are those linked to the cell proliferation and mitotic functions, cilium assembly, and voltage-gated ion channel activity (Olthof et al. 2019; de Wolf et al. 2021; Khatri et al. 2023). Of these, the pathways linked to cell proliferation are particularly relevant as cellular growth defects are observed with many minor spliceosomopathies, manifesting, for example, as tissue hypoplasia or microcephaly in many germline diseases and as cytopenia in MDS (Table 1). The relevant pathways include, in addition to the Ras-Raf-MAPK pathway that shows highest pathway-level enrichment for MIGs, other growth-associated pathways such as the PI3K, WNT pathways, and various cell-cycle/apoptosis regulators (Fig. 3). Together, these growth-associated pathways include up to 150 of the 700 MIGs. They have been best characterized in the context of MDS (Madan et al. 2015; Nishimura et al. 2024) but have a central role in the developmental processes as well.
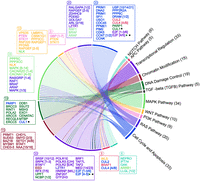
The distribution and network of 152 MIGs across growth-associated pathways. The MIG distribution across pathways was analyzed using DAVID (Sherman et al. 2022). The pathways are depicted on the right as arches and genes on the left side of the diagram. Each arc is color-coded, and its length is proportional to the number of MIGs associated with that pathway. Boxes on the left list the genes belonging to each pathway, their color matching the main pathway they are part of. The numbers above the boxes indicate the total number of genes. The genes inside the boxes are also colored according to their main pathways. Genes present in more than two pathways are indicated with color-coded star symbols (★). Numbers in brackets represent gene families.
The other two prominent functional groups containing an overrepresented number of MIGs are the genes related to ciliogenesis and genes coding for the voltage-gated ion channels. The enrichment on voltage-gated ion channels has been long noted (Wu and Krainer 1999; Olthof et al. 2019), and while not linked specifically to any of the minor spliceosomopathies, could be associated with the motoneuronal defects observed with a subset of the diseases. The other overrepresented group containing up to 86 MIGs has been associated with ciliogenesis and cilium assembly, and of these, 28 MIGs have been directly linked to ciliopathies. Furthermore, experimental evidence from both human cells and zebrafish supports the importance of the minor spliceosome for ciliogenesis. Cells derived from TALS and JBST-like syndrome patients (Khatri et al. 2023), as well as OFD patients with either SCNM1 or ZRSR2 mutations (Iturrate et al. 2022; Hannes et al. 2024), show alterations in primary cilia structure consistent with ciliopathy phenotype (Khatri et al. 2023). Interestingly, a recent preliminary work on the DDX59 helicase, the mutations of which lead to OFD syndrome (see Table 1) and ciliopathy, has also been reported to be essential for minor intron splicing (Che et al. 2024), thus providing another link between a minor intron splicing defect and ciliopathies. Therefore, defects in ciliogenesis, which has significant developmental functions (Anvarian et al. 2019) may be one of the notable common features of minor spliceosomopathies.
The proposed model of cumulative subclinical effects underlying the minor spliceosomopathy phenotypes is particularly compatible with the data from closely related diseases, such as the RNU4ATAC-opathy disease group. The partially overlapping phenotypic characteristics observed in this disease group (Table 1) may arise from the shared disease mutations in the heterozygous individuals, which give rise to a range of splicing defects of which a subset are shared between the diseases, as suggested by the comparison of RFMN and TALS RNA-seq data sets (Cologne et al. 2019), and thus contribute to the disease phenotype. However, it is also likely that a subset of MIGs have more weight on the phenotypical outcome. Here, MDS provides an illuminating example where the disease phenotype was initially associated with splicing defects in multiple proliferation-related MIGs, including several E2F transcription factors and many regulators of the MAPK pathway (Madan et al. 2015), but a later study revealed that most of the phenotypic features can in fact be explained by a minor intron splicing defect in LZTR1, a MIG in the affected Ras-Raf pathway (see Fig. 3) that codes for a cullin-3 adaptor for ubiquitination of RAS-related GTPases (Inoue et al. 2021). Furthermore, there are additional known effectors that can further influence the tissue specificity and the extent of the disease phenotypes, such as the tissue-specific regulation of MIGs (Olthof et al. 2019), feedback and cross-regulatory interactions among the components of the minor spliceosome (Verbeeren et al. 2010, 2017; de Wolf et al. 2021) but also extending to the major spliceosome components (Rogalska et al. 2024), and any other secondary gene expression changes of both MIGs and non-MIGs as observed both in the patient cells and in model organisms (Pessa et al. 2010; Baumgartner et al. 2018; Inoue et al. 2021; Gómez-Redondo et al. 2022). Together, we promote a model where the disease phenotypes in minor spliceosomopathies arise as a consequence of concerted action of subclinical splicing defects in multiple MIGs on the same pathways, combined with potential dominating effects from individual MIGs and further modified by the tissue-specific factors and secondary downstream effects or other genotype-specific modifiers.
PERSPECTIVES
There are two safe bets for the future development in the minor spliceosome and minor spliceosomopathy research. One of them is that there will be additional cryo-EM structures and additional detailed structures of minor spliceosome components to provide a refined view on the inner workings of the minor spliceosome, but also to refine the understanding of disease mechanisms. As a subcategory, it is also likely that the list of specific components for the minor spliceosome is not yet completed. In fact, in the present literature there are already two examples of potential novel minor spliceosome-specific proteins: WDR25, a putative paralog of the PRP17 protein in the major spliceosome that was identified in a phylogenetic co-occurrence analysis (Vosseberg et al. 2023), and DDX59, an RNA helicase that appears to be essential for minor intron splicing (Che et al. 2024). The other safe bet is that there will be additional minor spliceosomopathies, either associated with existing minor spliceosome components, or any of the yet-to-be discovered novel components. DDX59 mentioned above is a prime example of such disease, which in this case leads to OFD and ciliopathy.
As stated above, various splicing defects centered on the minor introns represent the hallmark feature shared by all known minor spliceosomopathies (reviewed by Akinyi and Frilander 2021). For this reason, transcriptional profiling of patient cells is expected to remain the primary approach for documenting the diagnostic intron retention and cryptic/alternative splice site activation events and connecting them to the disease phenotypes. At the methodological level, short-read RNA-seq is often sufficient for the identification of the primary splicing defects, assuming that the potential tissue-specific effects, described above, have been noted. For detailed analyses of transcript isoforms, particularly those investigating complex correlative events near in the minor introns and the surrounding major introns (Akinyi and Frilander 2021), long-read methods provide the best accuracy for the quantification of the co-occurring splicing events transcript isoforms (Inamo et al. 2024). In contrast, single-cell RNA-seq methods may not (yet) have a sufficient read depth and coverage for accurate quantification of the typically low-level minor intron splicing defects (Gupta et al. 2024) but could still be valuable in documenting the global gene expression level changes.
The inherent difficulty of most minor spliceosomopathies and their transcriptome-level characterization is that they are developmental diseases. Therefore, the correct sampling of the right tissues or cell populations at the right time is practically impossible. Presently, most of the RNA-seq data for minor spliceosomopathies is in fact derived from cells that are not related to the disease pathology, such as skin fibroblasts or blood lymphoblasts. Model organisms are the golden standard for overcoming such constraints, but at least in the case of a mouse CH model, the animals carrying RNPC3 disease mutations were found to be phenotypically normal (Akin et al. 2022). While the reasons for such species-specific differences are not known, it is tempting to speculate that if the human disease is caused by the cumulative effect of multiple subclinical splicing defects, such subtle changes in gene expression may not be readily transferable between the different organisms. An alternative may be to use patient-derived induced pluripotent stem cells (iPSCs) and for example organoid models to investigate the pathomechanisms in the actual cellular and genotypic context (Singh et al. 2022).
One of the exciting recent developments in the field does not directly relate to the minor spliceosomopathies, but rather the minor spliceosome is acting as a modifier for another disease, in this case to promote prostate cancer progression. In this work, Augspach et al. (2023) showed that the elevated levels of the U6atac snRNA, and the consequent increased activity of the minor spliceosome in prostate cancer cells is associated with the progression of prostate cancer in particular, but the results may be generalizable to other cancers as well. At the mechanistic level, it is likely that the normally highly unstable U6atac snRNA is being stabilized in cancer cells possibly through the action of a stress-activated p38MAPK pathway (Younis et al. 2013). This increases the normally inefficient splicing observed with a subset of minor introns (Patel et al. 2002; Younis et al. 2013; Oghabian et al. 2018) and promotes the expression of growth-associated genes (Augspach et al. 2023). As a consequence, this work suggests that minor spliceosomes may constitute a novel vulnerability factor for cancer cell growth, such that even a partial inhibition to bring the minor spliceosome activity down to, or below its normal parameters, while not completely inhibiting the splicing activity, can be used as a cancer therapy (Augspach et al. 2023; Juan-Mateu and Valcárcel 2023). In this context, the detailed structural and functional understanding of the unique factors of the minor spliceosome and the mechanism of their dysfunction in disease may be beneficial for the development of future cancer therapeutics. Additionally, it is possible that other minor spliceosome gain- or loss-of-function mutations or conditions could similarly act as the modifiers of other unrelated diseases.
ACKNOWLEDGMENTS
This work was supported by funding from the Academy of Finland (grant 341477) and the Sigrid Jusélius Foundation to M.J.F.
Footnotes
-
Article is online at http://www.rnajournal.org/cgi/doi/10.1261/rna.080337.124.
-
Freely available online through the RNA Open Access option.
This article, published in RNA, is available under a Creative Commons License (Attribution-NonCommercial 4.0 International), as described at http://creativecommons.org/licenses/by-nc/4.0/.








