
From activator to suppressor: PACT is joining the company of PKR negative regulators
- 1Virology and Gene Therapy Track, Mayo Clinic Graduate School of Biomedical Sciences, Rochester, Minnesota 55901, USA
- 2Department of Molecular Medicine, Mayo Clinic, Rochester, Minnesota 55901, USA
- 3Division of Veterinary Medicine, Paul-Ehrlich-Institute, Langen 63225, Germany
- Corresponding author: pfaller.christian{at}mayo.edu
In this issue of RNA, Young et al. identify a new regulatory role of the protein activator of the interferon-induced protein kinase (PACT, PRKRA gene) on the activation of protein kinase R (PKR) (Young et al. 2025). PKR is one of four cellular stress-activated kinases inducing global protein translation shutdown through phosphorylation of their natural substrate, the eukaryotic initiation factor 2-α (eIF2α). PKR activation occurs through binding to double-stranded RNA (dsRNA), a hallmark of many viral infections. As such, PKR is an integral part of the cell-intrinsic innate immune response system against viral pathogens (Pfaller et al. 2011). However, tight regulation of PKR activation is essential for maintaining cell viability and health. A large body of evidence published in the past decade has revealed that endogenous dsRNA structures can activate PKR as well as the dsRNA sensors melanoma differentiation-associated gene 5 (MDA-5) and retinoic acid–inducible gene I (RIG-I), thereby causing aberrant inflammatory responses. Hyperactive MDA-5 is associated with type-I interferonopathies such as Aicardi–Goutières syndrome (AGS) (Crow and Manel 2015). Likewise, a growing number of studies have linked self dsRNA-mediated PKR activation to the onset of neurological disorders such as Parkinson's disease, Alzheimer's disease, and Huntington's disease (Mohan et al. 2025). In cancer cells, PKR activation can lead to autoinflammation and cell death; however, this is efficiently suppressed in many tumors. Cells have evolved multiple redundant and synergistic mechanisms to prevent these autoinflammatory responses. One central regulatory enzyme, the IFN-inducible 150 kDa isoform of adenosine deaminase acting on RNA 1 (ADAR1 p150), suppresses both MDA-5 and PKR activation by competitively binding to dsRNA molecules, as well as by editing of dsRNAs, which permanently alters dsRNA secondary structures and removes immunostimulatory capacities (Fig. 1A; Li and Walkley 2025).
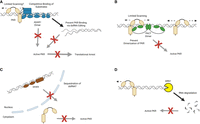
Mechanisms of PKR suppression. (A) ADAR1 p150 (blue) uses editing-dependent and editing-independent mechanisms to prevent PKR activation. (B) PACT dimers (green) occupy dsRNA molecules and prevent PKR scanning and dimerization. In addition, PACT dsRBD2 can directly bind to the PKR kinase domain and block its activity. (C) DHX9 (brown) likely sequesters dsRNA in the nucleus preventing its detection by PKR in the cytoplasm. (D) The exoribonuclease XRN1 (yellow) likely degrades dsRNA. Created in BioRender (https://BioRender.com/ta5mgpx).
In their study, Young et al. find that many triple-negative breast cancer (TNBC) cell lines express high levels of another dsRNA binding protein, PACT (Young et al. 2025). PACT depletion in a subset of TNBC cell lines led to PKR activation and induced cell death, indicating that PACT acted as a PKR suppressor. However, a different subset of TNBC cell lines required simultaneous depletion of both PACT and ADAR1 for efficient PKR activation, suggesting that both mechanisms to suppress PKR are redundant in these cells. PACT possesses two functional dsRNA-binding domains (dsRBD1/2) and dsRBD3 that does not bind dsRNA but allows PACT to form homodimers. The authors show through mutational analyses that dsRNA binding and dimer formation are required for the suppressive activity of PACT on PKR under physiological conditions.
In two other simultaneous studies, PACT was identified as a suppressor of PKR activation against self-RNAs (Ahmad et al. 2025; Manjunath et al. 2025). Manjunath et al. used a CRISPR-based screening method to identify genes necessary to activate PKR in the context of a viral infection. PACT was identified as a suppressor of PKR activation, and, remarkably, depletion of both PACT and ADAR1 led to synthetic lethality in uninfected cells dependent on PKR activation (Manjunath et al. 2025). Ahmad et al. provide detailed mechanistic insights into the mode of PKR suppression by PACT. While PACT does not prevent PKR binding to endogenous dsRNA substrates such as inverted Alu repeats, it prevents efficient scanning of PKR along longer dsRNA elements, a prerequisite for PKR activation (Ahmad et al. 2025). In addition, PACT dsRBD1/2 directly and independently of dsRNA interacted with the PKR kinase domain and served as a decoy substrate for phosphorylation, preventing PKR autophosphorylation.
Together, the findings of these three studies provide strong evidence for the suppressive activity of PACT on PKR activation. This is in stark contrast to PACT being originally described as activator of PKR (Patel and Sen 1998) but is in line with the observed rescue of embryonic lethality of Prkra knockout in mice by additional depletion of PKR (Dickerman et al. 2015). Key to these observed opposing functions seems to be the availability of dsRNA, as well as the concentration of PACT. In the absence of dsRNA, low amounts of PACT can directly interact with PKR, causing its autophosphorylation and activation; however, when dsRNA is present, or in the absence of dsRNA but high PACT concentrations, PACT suppresses PKR activation. Through the combined findings of all three studies, we are now able to establish a clear molecular mechanism of the suppressive function of PACT (Fig. 1B): PACT dimers occupy dsRNA molecules; while this does not per se prevent PKR binding to the same dsRNA molecules, it prevents sliding of PKR along the dsRNA scaffold, which is required for PKR dimerization, autophosphorylation, and hence activation. It remains an open question whether other dsRNA sensors such as MDA-5 are directly affected by PACT. It is well established that MDA-5 needs to form oligomeric filaments on long dsRNA structures for proper activation (Dias Junior et al. 2019); this could also be affected by PACT occupying the dsRNA.
PKR's central role in antiviral immunity and cancer cell death makes this dsRNA sensor a desirable therapeutic target. However, as we are gaining a deeper understanding of the consequences of PKR overactivation for the development of autoinflammation, it becomes clear why cells have evolved multiple redundant mechanisms to keep PKR in check. Notably, besides ADAR1 p150, several other factors have been recently discovered to antagonize PKR activation in cancer cells or during viral infection. These include the DExH-box RNA helicase DHX9 (Fig. 1C; Cottrell et al. 2024b), the exoribonuclease XRN1 (Fig. 1D; BenDavid et al. 2022), as well as the histone modifier WDR5 (BenDavid et al. 2024). PACT is now joining this increasing list of PKR-controlling factors (Cottrell et al. 2024a). It is now clear that regulating PKR is a main priority for the cell. Understanding how these mechanisms function across different tissue types will provide significant insights into strategies aimed at manipulating these regulatory effectors for therapeutic development against cancers, such as TNBC, viral infection, and neurological disorders. However, the goal to manipulate PKR activity may not be easily achievable, as we may have to target multiple redundant controlling mechanisms in parallel.
Footnotes
-
Article is online at http://www.rnajournal.org/cgi/doi/10.1261/rna.080743.125.
-
Freely available online through the RNA Open Access option.
This article, published in RNA, is available under a Creative Commons License (Attribution-NonCommercial 4.0 International), as described at http://creativecommons.org/licenses/by-nc/4.0/.








