
Accurate in silico predictions of modified RNA interactions to a prototypical RNA-binding protein with λ-dynamics
- Murphy Angelo1,
- Yash Bhargava1,
- Elzbieta Kierzek2,
- Ryszard Kierzek2,
- Ryan L. Hayes3,4,
- Wen Zhang1,5,
- Jonah Z. Vilseck1,6 and
- Scott Takeo Aoki1,5
- 1Department of Biochemistry and Molecular Biology, Indiana University School of Medicine, Indianapolis, Indiana 46202, USA
- 2Institute of Bioorganic Chemistry, Polish Academy of Sciences, Poznan 61-704, Poland
- 3Department of Chemical and Biomolecular Engineering, University of California Irvine, Irvine, California 92697, USA
- 4Department of Pharmaceutical Sciences, University of California Irvine, Irvine, California 92697, USA
- 5Melvin and Bren Simon Cancer Center, Indiana University School of Medicine, Indianapolis, Indiana 46202, USA
- 6Center for Computational Biology and Bioinformatics, Indiana University School of Medicine, Indianapolis, Indiana 46202, USA
- Corresponding authors: wz15{at}iu.edu, jvilseck{at}iu.edu, staoki{at}iu.edu
-
Handling editor: Eric Westhof
Abstract
RNA-binding proteins shape biology through their widespread functions in RNA biochemistry. Their function requires the recognition of specific RNA motifs for targeted binding. These RNA-binding elements can be composed of both unmodified and chemically modified RNAs, of which over 170 chemical modifications have been identified in biology. Unmodified RNA sequence preferences for RNA-binding proteins have been widely studied, with numerous methods available to identify their preferred sequence motifs. However, only a few techniques can detect preferred RNA modifications, and no current method can comprehensively screen the vast array of hundreds of natural RNA modifications. Prior work demonstrated that λ-dynamics is an accurate in silico method to predict RNA base binding preferences of an RNA-binding antibody. This work extends that effort by using λ-dynamics to predict unmodified and modified RNA-binding preferences of human Pumilio, a prototypical RNA-binding protein. A library of RNA modifications was screened at eight nucleotide positions along the RNA to identify modifications predicted to affect Pumilio binding. Computed binding affinities were compared with experimental data to reveal high predictive accuracy. In silico force field accuracies were also evaluated between CHARMM36 and Amber RNA force fields to determine the best parameter set to use in binding calculations. This work demonstrates that λ-dynamics can predict RNA interactions to a bona fide RNA-binding protein without the requirements of chemical reagents or new methods to experimentally test binding at the bench. Advancing in silico methods like λ-dynamics will unlock new frontiers in understanding how RNA modifications shape RNA biochemistry.
Keywords
INTRODUCTION
Modified RNAs have far-reaching impacts on disease and cellular functions. Over 170 RNA modifications have been identified in biology (McCown et al. 2020). Many modifications are proposed to be necessary for proper folding of RNA, but some modifications function directly in gene regulation. A classic example is N6-methyladenosine (m6A). A single methyl group added to the adenosine N6 nitrogen leads to the recruitment of the YTH family of RNA-binding proteins, promoting RNA destabilization and turnover (Roundtree et al. 2017; Patil et al. 2018). As a result, the m6A modification is proposed to be the single largest determinant of mRNA stability (Uzonyi et al. 2023) and has disease implications in many types of human cancer and viral pathogenesis (Horner and Reaves 2024; Koch and Lyko 2024). Thus, RNA modifications can play key roles in biology and pathology through changing how RNA-binding proteins interact with RNAs.
RNA-binding proteins interact with mRNA targets to affect mRNA stability and expression. They can bind to any region of an mRNA (Singh et al. 2015), but RNA binding is critical to exert their functions in transcript regulation (Coller et al. 1998; De Gregorio et al. 2001; Coller and Wickens 2007). For example, Pumilio is a prototypical RNA-binding protein and a member of the Pumilio and FBF (PUF) protein family required for embryonic and germ cell development, neuronal differentiation, some human cancers, and other biological functions (Wickens et al. 2002; Goldstrohm et al. 2018). Binding of mRNA by Pumilio is required to recruit other proteins for transcript repression via turnover (Wickens et al. 2002; Goldstrohm et al. 2018). Pumilio binds a conserved UGUAHAUA 8-mer RNA sequence motif typically found in mRNA 3′ UTRs, where H represents A, C, or U (Fig. 1A; White et al. 2001; Galgano et al. 2008; Morris et al. 2008; Lu and Hall 2011; Bohn et al. 2018; Jarmoskaite et al. 2019). RNA targets are recognized by a conserved RNA-binding domain consisting of helical PUF repeats (Wang et al. 2002; Gupta et al. 2008; Dong et al. 2011; Lu and Hall 2011). Each PUF repeat discerns a specific RNA base through a three amino acid code (Fig. 1B; Opperman et al. 2005; Cheong and Hall 2006; Lu et al. 2009; Goldstrohm et al. 2018; Zhou et al. 2021). Hence, RNA sequence variations would be expected to affect Pumilio binding, subsequently affecting transcript stability. More recently, two RNA modifications were also noted to change Pumilio-RNA-binding affinity in vitro (Vaidyanathan et al. 2017), demonstrating that noncanonical modifications can also affect RNA–protein interactions. The impact of the many other known RNA modifications on the binding profiles of RNA-binding proteins is still an unresolved question.
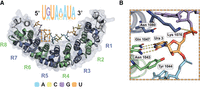
The structure of RNA bound to human Pumilio, a prototypical RNA-binding protein. (A) Crystal structure of human Pumilio 1 (PDB ID: 3Q0P) bound to a consensus RNA sequence. The Pumilio RNA-binding domain consists of eight helical PUF repeats forming a comma-shaped structure that binds RNA on its interior. Each PUF repeat (R1–R8) interacts with a single nucleobase. Binding is antiparallel, with the C terminus of Pumilio interacting with the 5′ end of the RNA sequence motif. Human Pumilio consensus motif presented, with letter size indicating in vivo sequence preference. Dashed box region enlarged. (A) Adenine, (G) guanine, (C) cytidine, (U) uridine. (B) Molecular detail of a single PUF repeat (R6) binding to a single nucleobase (uridine). A PUF repeat amino acid triplet binds to preferred bases through hydrogen bonding and stacking. Images by PyMOL.
Many in vitro and in vivo methods have been developed to identify the preferred binding motifs of RNA-binding proteins. In vitro methods like RNA systematic evolution of ligands by exponential enrichment (SELEX) use recombinant RNA-binding proteins and in vitro selection to identify preferred RNA substrates of a target protein (Dasti et al. 2020). In vivo methods like cross-linking and immunoprecipitation and sequencing (CLIP-seq) enrich for RNA-binding proteins from cell culture or in vivo sources and sequence the associated RNAs to determine the binding protein's RNA target and footprint (Xiang et al. 2024). Many other strategies and variations of these methods have been reported and are successful in identifying preferred, unmodified RNA sequence motifs. However, none to date can fully identify the effects of the numerous RNA modifications on protein binding. SELEX can screen for preferred interactions with specific RNA modifications (Vaidyanathan et al. 2017), but this strategy cannot currently differentiate between modified and unmodified RNA. Mass spectrometry can identify a diversity of RNA modifications (Herbert et al. 2024), but it still relies on RNA sequencing to determine their precise locations within the sequence. Of most concern, a majority of RNA modifications cannot currently be synthesized in vitro. There is a lack of reagents for solid phase chemical RNA synthesis, an inability to incorporate RNA modifications via in vitro transcription, and a lack of methods to add the RNA modifications post synthesis (National Academies of Sciences Engineering and Medicine 2024). Due to these limitations, current experimental methodologies can study the RNA–protein binding profiles for only a small subset of the 170+ known modifications (National Academies of Sciences Engineering and Medicine 2024).
Molecular dynamics and free energy calculations are accurate computational methods to model structural complexes and predict the binding affinities of nucleic acid to protein (Reyes and Kollman 2000; Gapsys and de Groot 2017; Kappel et al. 2019). Molecular dynamics-based simulations, though time consuming, can illustrate how chemical perturbations affect molecular interactions on an atomistic scale. λ-Dynamics (λD) is an efficient, high-throughput free energy method that can calculate free energy differences corresponding to changes in chemical structure (Kong and Brooks 1996; Knight and Brooks 2011). As an alchemical free energy method, it uses a sliding λ variable to investigate several structural and chemical modifications simultaneously during a single simulation. After ensuring that all chemical states are sampled equally, λD can calculate changes in binding free energy (ΔΔGbind) relative to an original reference molecule. The sign and magnitude of the ΔΔGbind results indicate if a structural modification is favorable or not and by how much. This method can accurately and efficiently compute both protein–ligand and protein–protein binding affinities. Excellent agreement has been observed in benchmark comparisons to conventional thermodynamic integration (TI) calculations. Notably, when exploring many alchemical perturbations simultaneously, λD is as accurate yet seven to 20 times more efficient than TI (Hayes et al. 2017, 2018; Vilseck et al. 2018, 2019; Barron and Vilseck 2024; Liesen et al. 2025). Prior work also established λD adept at predicting relative binding affinities of a library of RNA modifications to modified RNA-targeting antibodies (Angelo et al. 2025b). The antibodies in that work were designed to bind singly modified purine RNA bases, and λD accurately predicted off-target antibody binding of other modified bases. Thus, λD demonstrated promise as a practical way to predict the effect of chemical modifications on RNA–protein interactions. That work was limited, however, in that it did not test the accuracy of λD predictions with biologically relevant RNA-binding proteins that bind larger nucleic acid. Establishing such accuracy will be critical to enable comprehensive predictions of RNA-binding protein interactions with λD.
The following study extends prior work testing λD as a viable method to study modified RNA–protein interactions. Pumilio is used as a prototypical RNA-binding protein due to its biological and medical relevance, the wide breadth of structural and binding studies published, and its relatively large RNA-binding footprint. The work further establishes λ-dynamics as a capable method for predicting RNA–protein binding to modified RNAs and identifies computational practices for improved accuracy.
RESULTS
The goal of this study was to evaluate the accuracy of λD as an in silico strategy for exploring canonical and modified RNA binding to RNA-binding proteins. Pumilio (PUM) was chosen as a prototypical model RNA-binding protein because of its human medical relevance, available high-resolution structures of PUM bound to RNA targets (Opperman et al. 2005; Cheong and Hall 2006; Lu et al. 2009; Goldstrohm et al. 2018; Zhou et al. 2021), and extensive studies of its RNA-binding specificity (White et al. 2001; Galgano et al. 2008; Morris et al. 2008; Lu and Hall 2011; Bohn et al. 2018; Jarmoskaite et al. 2019). Like all Pumilio and FBF (PUF) family proteins, human PUM1 (hPUM1) and PUM2 (hPUM2) have a modular architecture of eight conserved α-helical PUF repeats that facilitates targeting specific RNA sequences (Fig. 1A; Opperman et al. 2005; Cheong and Hall 2006; Lu et al. 2009; Goldstrohm et al. 2018; Zhou et al. 2021). Each repeat binds a single base of its RNA target sequence (Fig. 1B), with a preferred RNA sequence motif of UGUAHAUA, where H is A, C, or U (Fig. 1; White et al. 2001; Galgano et al. 2008; Morris et al. 2008; Lu and Hall 2011; Bohn et al. 2018; Jarmoskaite et al. 2019). Prior work determined in vitro protein–RNA-binding affinities for a majority of the 48 possible canonical RNA base sequences (Jarmoskaite et al. 2019) and for several modified RNAs (Vaidyanathan et al. 2017). Thus, hPUM served as an excellent benchmark system for gauging λD's ability to model protein interactions with both modified and unmodified RNA.
λ-Dynamics accurately modeled Pumilio binding to canonical RNA mutants
λD was first used to explore how canonical base mutations were predicted to affect RNA binding to Pumilio. Each of the eight nucleobase sites along the RNA bound to hPUM1 and hPUM2 were mutated to a different canonical base, effectively modeling all possible single-nucleotide polymorphisms of the PUM consensus motif. λD calculations were performed with the CHARMM molecular modeling package and the BLaDE GPU accelerated engine in the isothermal–isobaric ensemble (see Materials and Methods; Brooks et al. 1983, 2009; Hayes et al. 2021). This work expanded upon the procedure used previously to establish λD's efficacy for modeling antibody binding to ribonucleosides (Angelo et al. 2025b). That system used CHARMM36 force field parameters, descriptions of the interatomic force potentials used to model molecules, for the protein and RNA bases (Denning et al. 2011; Best et al. 2012a,b; Hart et al. 2012; Xu et al. 2016; Huang et al. 2017). Other force fields for RNA, like Amber (Aduri et al. 2007; Zgarbova et al. 2011; Maier et al. 2015), have been used in molecular dynamic simulations but have not yet been tested with λD. To evaluate the effect of force field accuracy, both CHARMM36 and Amber-based force field parameters were used in parallel with λD in this work. Specifically, the Amber ff14SB protein, OL3 RNA, and modRNA08 force field parameters were used (Aduri et al. 2007; Zgarbova et al. 2011; Maier et al. 2015). For simplicity, this specific set of parameters is referred to as the “Amber” force field in this work. Computed binding results were then compared to in vitro binding affinities (ΔGbind) from existing literature (Fig. 2; Jarmoskaite et al. 2019).
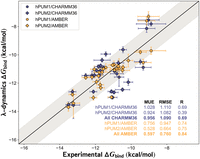
Pumilio-RNA sequence preferences predicted with λ-dynamics. Comparison of in silico λ-dynamics and in vitro measurements of Pumilio binding to a variety of RNA sequences with canonical RNA. Experimental binding affinity results (x-axis) from a previous publication (Jarmoskaite et al. 2019). These results were compared against the predicted λ-dynamics ΔGbind (y-axis) generated in this study. Both variables reported as kcal/mol. See Materials and Methods for details regarding the calculations and RNAs selected. Shaded area, root mean square error (RMSE) of 1.0 kcal/mol. Error bars report standard deviation from both data sets. Mean unsigned errors (MUEs) and RMSEs reported for human Pumilio 1 (hPUM1) and human Pumilio 2 (hPUM2) results computed with CHARMM36 (blue) and Amber (gold) force fields.
All λD results exhibited strong agreement with in vitro measurements (Fig. 2). Mean unsigned errors (MUEs) and root mean square errors (RMSE) between the λD predicted free energies differences (ΔGbind) results and previously published experiments (Jarmoskaite et al. 2019) were within or below the accepted gold standard of ∼1.0 kcal/mol agreement (Fig. 2; Supplemental Table S1; Wang et al. 2015a; Gapsys et al. 2019; Schindler et al. 2020; Ross et al. 2023). As a control, each native RNA base was perturbed to an identical but physically distinct copy of itself. These relative binding free energy differences (ΔΔGbind) were near zero, as expected of a base replacing itself, indicating that the λD calculations were functioning properly (Supplemental Table S1). Pearson R values indicated strong predictive trends for CHARMM36 (R = 0.69) and Amber (R = 0.84) force fields (Fig. 2). An improvement in accuracy was noted with the Amber force field. Relative to CHARMM36, MUEs improved by 0.36 kcal/mol, RMSE by 0.33 kcal/mol, and the Pearson R by 0.15 (Fig. 2). Terminal RNA bases represented the greatest challenge for λD, with sites 1, 7, and 8 containing predictive outliers exceeding ±1.0 kcal/mol in each screening (Supplemental Table S1). This may be due to the terminal RNA bases’ ability to rotate out of the PUF pocket and therefore unbind mid-λD simulation. The relatively weaker predictions of the CHARMM36 versus Amber force field suggested an area of future parameter refinement for modeling RNA interactions with CHARMM36. Other factors like slow conformational changes may also have important energetic contributions (see Discussion). Regardless, λD was able to accurately estimate hPUM1 and hPUM2 binding trends to canonical RNA. These results also suggest that λD had the potential to predict protein–RNA interactions of expanded chemical complexity.
λ-Dynamics predicted Pumilio binding to modified RNA
The accuracy of λD was further tested by probing how a library of RNA modifications affected hPUM1 binding. Of the 170 plus RNA modifications currently identified, many do not have commercially available reagents or lack protocols for in vitro syntheses. Following previous work (Angelo et al. 2025b), a library of 44 modified nucleobases was selected for λD screening based on the availability of commercial reagents and force field parameters. Using CHARMM36 force fields (Xu et al. 2016), λD screened 352 single-site mutants created by applying these 44 modifications at each position along the hPUM1 8-mer RNA consensus motif (see Materials and Methods; Fig. 3). The full results are reported in the Supplement (Supplemental Table S2). The computed ΔΔGbind results of the modified RNAs ranged from positive to negative, designating worsened to enhanced binding, respectively, relative to the wild-type RNA sequence. Using a conservative cutoff of ΔΔGbind ≤ –1.0 kcal/mol, approximately a fivefold or greater enhancement of binding, six modifications showed markedly enhanced binding to hPUM1 at three separate RNA sites (Supplemental Table S2). Overall, the λD predictions suggested that most modifications negatively impact RNA binding, yet some had little effect and others enhanced it.
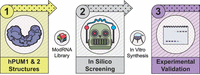
Strategy to test the predictive value of λ-dynamics for modified RNA and RNA-binding protein interactions. (1) λ-Dynamics and a modified RNA library were used with high-resolution crystal structures of human Pumilio 1 (hPUM1) and human Pumilio 2 (hPUM2) to predict modified RNA–protein binding. (2) Each RNA position in hPUM1 or hPUM2 was changed to a different RNA, and the change in free energy binding was calculated compared to the original RNA–protein structure. See Materials and Methods for more details. Some of the modified RNA modifications tested in silico could be synthesized in vitro. (3) These modified RNAs were tested for RNA binding to probe λ-dynamics prediction accuracy.
To further interrogate the λD predictions, 12 modified RNAs with worsened, neutral, and enhanced affinities were selected for extended follow-up 50 nsec λD simulations with both CHARMM36 and Amber force fields (see Materials and Methods; Fig. 4; Supplemental Table S3; Aduri et al. 2007; Xu et al. 2016). The modified RNA selection was based on the availability of published Ψ and m6A in vitro data (Vaidyanathan et al. 2017) or commercial reagents, which could be synthesized and tested in-house, referred to as the “PUM Library.” The extended CHARMM36 simulations displayed good agreement with the initial shorter screening, suggesting good convergence and reproducibility in the λD calculations. However, the Amber results were noticeably different from CHARMM36 for some of the PUM Library data points. Three of the Amber predictions differed from their CHARMM36 counterparts by more than 1.0 kcal/mol (Fig. 4). At site 4, 2-methylthio-N6-methyladenosine (ms2m6A) was predicted to have moderately worsened binding with Amber rather than strongly favorable binding affinity with CHARMM36. The Amber versus CHARMM36 ΔΔGbind differed by more than 3.0 kcal/mol. At site 5, 4-thio-uridine (s4U) worsened binding with Amber rather than having no significant change with CHARMM36. The Amber versus CHARMM36 ΔΔGbind differed by ∼1.3 kcal/mol. These energetic discrepancies between sulfur-containing bases may reflect differences in how each force field treats sulfur-containing moieties, a traditionally difficult element to parameterize accurately with molecular mechanics force fields (Mobley et al. 2009; Vilseck et al. 2014). Finally, at site 6, N6-isopentenyladenosine (i6A) saw an Amber versus CHARMM36 ΔΔGbind of ∼2.4 kcal/mol (Fig. 4). In summary, λD predicted that several RNA modifications would site-specifically alter hPUM1 RNA-binding affinity. Moreover, different RNA force fields yielded different predictions, and thus, alternative methods were warranted to determine which computational strategy was more accurate.
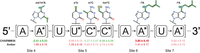
Example λ-dynamics predictions of how RNA modifications may affect binding to human Pumilio 1. The original RNA base is reported, along with the base change (*, RNA name and chemical structure above) and noted relative free energy binding change measured (ΔΔGbind, kcal/mol, 50 nsec simulations) with CHARMM36 and Amber force fields. Predicted decrease in binding affinity, positive ΔΔGbind, in red. Predicted increase in binding affinity, negative ΔΔGbind, in green. Results highlight modifications tested in vitro. Sites refer to RNA positions described in Figure 1. See Supplemental Table S2 for complete screening results.
λ-Dynamics’ predictions with Amber force fields closely matched Pumilio binding to modified RNA in vitro
To evaluate the accuracy of the λD predictions, computed ΔΔGbind were compared to experimentally determined modified RNA binding by Pumilio, measured here and in prior publications. First, electrophoretic mobility shift assays (EMSAs) were used to measure the in vitro binding affinity of select modified RNA to hPUM1 and hPUM2. Seven RNA modifications were selected based on commercial availability and the extended screening results (Fig. 4; Supplemental Table S3). These RNA modifications and paired, unmodified RNA controls were synthesized as 5′-fluorescein-capped RNA oligos through solid-state chemistry (see Materials and Methods). The modified RNA oligos changed a single site in the UGUACAAU hPUM recognition motif, with unmodified RNA serving as a control (Fig. 5). The 5-hydroxymethylcytidine (hm5C) building block was only available as DNA and therefore compared to an equivalent DNA–RNA chimera control (Fig. 5). Fluorescein-labeled RNAs were incubated with increasing concentrations of hPUM1 or hPUM2, then run on a nondenaturing PAGE gel under low voltage to separate unbound versus PUM-bound RNA. The lower and upper bands, corresponding to unbound and bound RNA, respectively, were quantified, and dissociation constants (Kd) were estimated based on the final curve results (see Materials and Methods; Fig. 5B; statistics in Supplemental Fig. S1).
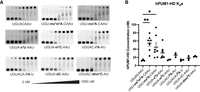
Human Pumilio 1 binding to modified RNAs in vitro was consistent with λ-dynamics predictions. (A) Electrophoretic mobility shift assays (EMSAs) were used to estimate the binding affinity in vitro. RNA oligos without or with the designated RNA modifications were incubated with increasing concentrations (0–1000 nM) of recombinant human Pumilio 1 (hPUM1) RNA-binding domain, run on a polyacrylamide gel, and imaged for carboxyfluorescein (FAM) fluorescence. A sample without protein served as an unbound RNA control. The lower band corresponds to unbound RNA. The upper band corresponds to RNA bound to the recombinant protein. The binding affinity can be estimated by calculating the protein concentration at the binding inflection point. Shown are representative gels from hPUM1 EMSA binding experiments. (B) EMSAs were performed at least three times, and calculated binding dissociation constants (Kd) were reported with their mean and standard deviation. (*) P < 0.05; (**) P < 0.01. See Supplemental Figure S1 for full statistical analyses and Materials and Methods for more experimental details.
The in vitro findings supported the λD predictions and demonstrated that the tested RNA modifications all had negligible to unfavorable effects on hPUM1 or hPUM2 binding (Fig. 5; Supplemental Fig. S2). With hPUM1, three of the seven PUM Library RNA oligos (hm5C and m5C at site 5, along with i6A at site 7) showed affinities comparable to their respective wild type. The remaining four (ms2m6A and ms2A at site 4, s4U at site 5, and i6A at site 6) significantly weakened binding (Fig. 5B). Similar results were also observed with hPUM2 (Supplemental Fig. S2). Next, the λD screening results were compared to a prior study measuring Pumilio binding to select RNA modifications. Vaidyanathan et al. measured in vitro binding affinities of hPUM2 to RNA containing pseudouridine (Ψ) or m6A RNA modifications (Vaidyanathan et al. 2017). That study and λD (Supplemental Table S3) noted that both modifications generally weakened RNA binding to Pumilio in vitro as more RNA sites incorporated the modifications. Thus, in vitro binding data support the binding trends predicted by λD (Fig. 6). While no distinctly favorable RNA modifications were identified in this study, several promising candidates remain untested due to lack of chemical reagents, underscoring opportunities for future exploration (see Discussion).
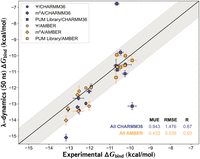
Pumilio-modified RNA interactions can be accurately predicted with λ-dynamics and Amber force fields. Comparison between λ-dynamics and in vitro ΔGbind measurements of human Pumilio 2 (hPUM2) binding to pseudouridine (Ψ) or N6-methyladenosine (m6A) modified RNA sequences performed previously (Vaidyanathan et al. 2017) or of binding data generated in this study (“PUM Library”). The experimental binding data (x-axis) are plotted against the extended 50 nsec λ-dynamics simulation results (y-axis) in kcal/mol. See Materials and Methods for details regarding the calculations performed. Shaded area, root mean square error (RMSE) of 1.0 kcal/mol. Error bars report the standard deviation from experimental sets. Mean unsigned errors (MUEs) and RMSEs reported for hPUM2 results computed with CHARMM36 (blue) and Amber (gold) force fields.
The in silico predictions and binding results were analyzed further to gauge the accuracy of the CHARMM36 versus Amber force fields (Fig. 6). Overall MUEs and RMSEs between λD and experiment were 0.95 and 1.48 kcal/mol for CHARMM36 and 0.43 and 0.54 kcal/mol for Amber, respectively (Fig. 6; Supplemental Table S3). The CHARMM36 predictions achieved a Pearson R of 0.67, indicating moderate predictive accuracy, but the Amber predictions had a significantly improved Pearson R of 0.93, indicating very strong correlation (Fig. 6). The visibly tighter fit of the Amber force field results reflects the improved RMSE compared to CHARMM36. This improvement in Amber could partially arise from selecting PUM Library sequences based on the larger CHARMM36 RNA modification screen, without subsequently repeating the computationally expensive screen with Amber. The RMSEs for Amber and CHARMM36 were 0.61 and 0.84 kcal/mol, respectively, for the Ψ and m6A published data only (Vaidyanathan et al. 2017). While both force fields demonstrated relatively good predictive accuracy, the CHARMM36 results were negatively affected by three large outliers, resulting in higher average errors (Fig. 6). The CHARMM36 ΔGbind for ms2m6A at site 4 and i6A at site 6 deviated from experiment by over 3.0 kcal/mol (Supplemental Table S3). Overall, Amber force fields performed more accurately than CHARMM36 force fields for this prototypical protein–RNA complex. These results collectively demonstrate that the λD in silico method can accurately predict in vitro RNA-binding protein interactions with both unmodified and modified RNAs.
DISCUSSION
Nearly 200 RNA modifications have been identified in biology, yet there are only a small number of methods to determine how they affect RNA–protein interactions. In vitro methods are limited by the lack of reagents for modified RNA synthesis, and in vivo methods are limited by their inability to detect all modifications within RNA. This work used λD free energy calculations to test how a library of RNA modifications could affect single-stranded RNA–protein interactions. Using human Pumilio as a prototypical RNA-binding protein, these results demonstrated that λD could accurately predict both unmodified and modified RNA binding without the dependence of in vitro chemical reagents to probe such interactions.
Simulation of biological macromolecules requires an accurate description of their physical properties, such as the force required to stretch or rotate a phosphodiester bond or nonbonded electrostatic and van der Waals interactions. These terms are collectively encoded as force field parameters in molecular mechanics simulations. Accuracy in free energy calculations, including λD, also depends upon proper sampling of all relevant biological and alchemical motions (Mobley 2012). This work demonstrated that the λD free energy results are consistent and reproducible over a range of 25–100 nsec of molecular dynamics sampling, suggesting convergence. Therefore, major differences with experiment likely stem from differences in force field parameterization. Both Amber and CHARMM36 RNA force fields have been developed to include modified RNA parameters, which have been optimized to accurately model RNA stability and conformational dynamics (Aduri et al. 2007; Denning et al. 2011; Zgarbova et al. 2011; Maier et al. 2015; Xu et al. 2016; Huang et al. 2017). However, pairing current Amber or CHARMM36 modified RNA force fields with protein force field counterparts has received limited investigation with commonly studied m6A and Ψ bases (Dutta et al. 2022; Piomponi et al. 2024). In this work, protein and nucleic acid force fields from Amber and CHARMM36 were paired to predict changes in binding free energies associated with nucleobase perturbations to a large library of both canonical and modified RNAs. Notable improvements in predictive accuracy were observed with Amber compared to CHARMM36. Their performance differences further highlight the critical influence of force field selection for free energy calculations. Two CHARMM36 outliers (Fig. 6) contained sulfur atoms. Parameter refinement or the incorporation of additional lone pair sites on sulfur atoms may improve these predictions, as performed previously (Cole et al. 2016; Zhang et al. 2021). Regardless, the RNA-PUM results herein suggest that either Amber alone or a CHARMM36 with Amber consensus approach should be used to maximize predictive accuracy for prospective analysis of RNA–protein binding interactions. Work is ongoing to identify if inaccuracies also exist in Amber RNA force fields for specific chemical moieties not explicitly evaluated in this work. This additional work will be valuable for minimizing false negative and false positive binding predictions with λD.
Understanding how modified RNAs interact with RNA-binding proteins is an active area of investigation. This includes investigating sequence preferences of modified RNA-binding proteins and how different RNA modifications affect canonical RNA-binding protein interactions. For example, in vitro SELEX and in vivo CLIP-seq methods have determined that the YTH family of RNA-binding proteins have a sequence preference for G(m6A)CH (Zhang et al. 2010; Wang et al. 2014, 2015b), while a wide variety of methods have determined RNA sequence preferences for human PUM (e.g., White et al. 2001; Campbell et al. 2012; Vaidyanathan et al. 2017; Van Nostrand et al. 2020). In vitro RNA-binding assays showed how Ψ and m6A modifications have a cumulative, negative effect on human PUM-RNA binding (Vaidyanathan et al. 2017). In this study, in silico λD correctly predicted many RNA modifications to have negligible or modest effects on binding, consistent with trends observed in vitro. The findings underscore the potentially nuanced and site-dependent effects of RNA modifications on protein binding, a conclusion reached in prior studies (Vaidyanathan et al. 2017). Comprehensive screening across all RNA sequence positions and with various RNA modifications is needed to fully understand how RNA modifications influence RNA–protein interactions.
In silico methodologies enable new opportunities to study molecular interactions without the limitations that chemical synthesis or molecular biology requirements place on in vitro or in vivo strategies. This work demonstrates how λD can be used to accurately screen preferred sequence binding motifs for unmodified RNA. More excitingly, the results also support that RNA-binding protein interactions with noncanonical RNAs can be predicted in silico with λD. While most RNA modifications will likely disrupt RNA–protein interactions, a few may enhance binding, leading to breakthroughs in epitranscriptomics and toward the rational design of unnatural, RNA-based therapeutics. With λD and other in silico strategies, probing any RNA–protein interaction is possible.
MATERIALS AND METHODS
Recombinant protein expression
Human PUM1 (hPUM1) and PUM2 (hPUM2) RNA-binding homology domain (HD) coding regions were codon optimized for E. coli, synthesized, and cloned into pET21a or pET28b expression vectors with a His6::V5::PreScissionC protease cleavage site or a His6::SUMO tag, respectively. The plasmids were expressed in LOBSTR cells (Kerafast EC1002), grown in Luria Broth (LB) media until 0.6–0.8 OD (600 nm absorbance), and then induced with 0.1 mM IPTG and cultured at 16°C and 160 rpm overnight. Bacteria were pelleted and frozen until use. Pellets were resuspended in 50 mL lysis buffer (20 mM Tris pH 8.0, 300 mM NaCl, 14.3 mM β-mercaptoethanol [BME], 1 mM EDTA pH 8.0, 5% [v/v] glycerol, 0.1% [v/v] Tween-20, and 20 mM imidazole pH 8.0) with a Pierce protease inhibitor tablet (Fisher PIA32955). The cells were lysed at 14,500 PSI by high-pressure homogenization with a microfluidizer (LM20 Microfluidizer, Microfluidics). Phenylmethylsulfonyl fluoride (PMSF) at a final concentration of 1 µM was added immediately after lysis. Tagged hPUM1-HD and hPUM2-HD were isolated from the soluble fraction using NiNTA resin (Fisher PI88222). The collected resin was subsequently washed with 100 mL lysis buffer at a flow rate of 1 mL/min. Resin with tagged protein was reconstituted in 10 mL lysis buffer and then cleaved using 0.04% (v/v) 3C protease (Neta GSCRPT-Z03092-500) or ULP1 protease overnight. Recombinant protein was eluted by flowthrough and then dialyzed into storage buffer (20 mM Tris pH 8.0, 100 mM NaCl, 1 mM DTT, 1 mM EDTA pH 8.0, 5% [v/v] glycerol, and 0.1% [v/v] Tween-20). hPUM1-HD and hPUM2-HD were further purified on a UNOsphere S column (Bio-Rad) followed by a SEC70 column (Bio-Rad) preequilibrated with storage buffer. Purified protein was then concentrated to 2–3 mg/mL.
Molecular modeling system setup
In silico simulations were performed on published RNA–protein complex structures selected to match the in vitro screens performed: (a) hPUM1-HD bound to UGUACAUC RNA for the canonical hPUM1-HD and RNA modification library screening (PDB ID: 3Q0P [Lu and Hall 2011]); (b) hPUM1-HD bound to UGUAUAUA RNA for the canonical hPUM1-HD screening at site 8 (PDB ID: 3Q0N [Lu and Hall 2011]); and (c) hPUM2-HD bound to UGUAUAUA RNA for m6A screening (PDB ID: 3Q0Q [Lu and Hall 2011]). For the ψ screening, the 3Q0Q RNA structure was mutated from U to A at site 5 using Chimera (Pettersen et al. 2004) and energy minimized to acquire a structural model of hPUM2-HD bound to UGUAAAUA. A similar strategy was applied to 3Q0P to acquire a structural model of hPUM1-HD bound to UGUACAAU for the extended λD modified RNA screening. All hPUM1-HD and hPUM2-HD structures were optimized by PDB-REDO prior to use (Joosten et al. 2014). Separate systems of unbound RNA oligos were created by extracting RNA coordinates from each optimized RNA–protein complex. Each system was then set up similarly as in previous work (Angelo et al. 2025b). Briefly, the protein–nucleotide complexes and unbound RNA oligos were solvated in cubic boxes of explicit TIP3P water (Jorgensen et al. 1983) using the CHARMM-GUI solution builder (Jo et al. 2008). Solvent padding of 10 Å was performed, and a final ionic strength of 150 mM NaCl or KCl was used to match experimental conditions. Preliminary 25 nsec screenings also incorporated 0–5 mM MgCl2. This was found to negligibly contribute to computed ΔΔGbind and was left out of subsequent simulations. Protonation states were applied based on PROPKA predictions at pH 7.0 (Sondergaard et al. 2011). All systems were energy minimized with a minimum of 250 steps of steepest descent minimization to remove potential steric clashes prior to molecular dynamics.
As mentioned in the Results, both CHARMM36 and Amber force fields were used to represent protein and nucleic acid components. CHARMM36 protein, nucleic acid, and modified RNA bases were used (Denning et al. 2011; Best et al. 2012a,b; Hart et al. 2012; Xu et al. 2016; Huang et al. 2017). Amber ff14SB (Maier et al. 2015), OL3 (Zgarbova et al. 2011), and modRNA08 (Aduri et al. 2007) force fields, referred to as “Amber” in this work, were ported into the CHARMM simulation package using an open-source script developed in-house (https://github.com/murfalo/chamberr). This script prioritizes the use of newer Amber parameters over older ones. For example, for N-glycosidic potentials, OL3 parameters were used except when OL3 was missing an atom type, such as for modified nucleotide R-groups. In such cases, modRNA08 parameters were used. This work was largely made possible by the open-source community's work on the ParmEd parameter and topology editor (https://github.com/ParmEd/ParmEd). Previous work, which manually converted Amber ff14SB topologies and parameters into CHARMM-formatted files (Maier et al. 2015), was used as an initial test suite to validate the script-converted force fields.
λ-Dynamics calculations
A library of 48 bases, comprising 44 modified and four unmodified RNA candidates, was selected for screening with λD (Angelo et al. 2025b). Simulations were conducted using the CHARMM molecular simulation package (version c48b2) with the Basic λ-Dynamics Engine (BLaDE) for GPU accelerated modeling (Brooks et al. 1983, 2009; Hayes et al. 2021). Simulations were run at 25°C and 1 atm in the isothermal–isobaric ensemble, as performed in published binding experiments (Jarmoskaite et al. 2019) and other λD studies (Hayes et al. 2017, 2018; Vilseck et al. 2018, 2019; Barron and Vilseck 2024; Angelo et al. 2025b; Liesen et al. 2025). BLaDE uses a Langevin thermostat, a Monte Carlo barostat, and periodic boundary conditions to maintain this ensemble (Chow and Ferguson 1995; Åqvist et al. 2004; Hayes et al. 2021). A Langevin coefficient of 0.1 psec−1 was used, and volume moves were attempted every 100 steps (Hayes et al. 2021). Long-range electrostatic interactions were modeled with particle mesh Ewald, and Lennard–Jones interactions were truncated with force switching. Cutoffs for all nonbonded interactions were set at 10 Å, with force switching beginning at 9 Å (Darden et al. 1993; Steinbach and Brooks 1994; Huang et al. 2016). SHAKE was used to constrain all bonds to hydrogen, facilitating the use of a 2 fsec MD timestep (Ryckaert et al. 1977). For purine-to-purine or pyrimidine-to-pyrimidine mutations, analogous atoms in the shared core were harmonically restrained to one another with the scaling of constrained atoms (scat) utility, developed for restraining alchemical substituents and protein backbones with λD (Hayes and Brooks 2021; Liesen and Vilseck 2024). Relative binding free energies, ΔΔGbind, were calculated using a conventional thermodynamic cycle, in which alchemical perturbations were performed in both protein-bound and solvated-unbound states of the RNA (Supplemental Fig. S3).
Prior to production λD sampling, the adaptive landscape flattening (ALF) algorithm (Hayes et al. 2017, 2018) was used to identify optimal biasing potentials, which help facilitate frequent and even alchemical transitions between all perturbed molecules. Optimal biases were identified after sampling ALF for a cumulative 48–55 nsec, after which five independent production simulations were conducted for each group of modified RNA bases. Canonical base production simulations of hPUM1-HD and hPUM2-HD were run for 25–100 nsec each. Convergence was confirmed by ensuring λD predictions remained unchanged within statistical noise as simulation times increased. For computing free energy differences, an initial 20% of the production data was excluded as equilibration. For the initial modified RNA base screen with CHARMM36 force fields, four mutations per simulation per RNA site were performed, and mutations were grouped based on structural similarity (see Supplemental Table S4). Additional extended modified RNA screenings were then conducted in a pairwise manner with λD using both CHARMM36 and Amber force fields (Brooks et al. 1983; Aduri et al. 2007; Denning et al. 2011; Zgarbova et al. 2011; Best et al. 2012a,b; Hart et al. 2012; Maier et al. 2015; Xu et al. 2016; Huang et al. 2017). In these head-to-head comparison simulations, production simulations were run for 50 nsec each. Final free energy differences were then calculated by Boltzmann reweighting end state populations with WHAM (Kumar et al. 1992) and computing statistical uncertainties with bootstrapping. As shown in Supplemental Figure S3, ΔΔGbind were calculated as a difference between bound and unbound λD results. The “Data Collection and Analysis” section describes how relative binding free energies were converted into absolute values for comparison to experiment. λD input files, topology and parameter files, and initial starting structures needed to reproduce this work have been uploaded to Zenodo (Angelo et al. 2025a).
RNA oligonucleotide preparation
RNA oligonucleotides used for binding affinity experiments were synthesized on an ABI 394 DNA/RNA synthesizer (Applied Biosystems [ABI]) as performed previously (Angelo et al. 2025b). m6A (Glen Research, 10-3005-90), i6A (Chemgene, ANP-8615), dhm5C (Glen Research, 10-1510-95), m5C (Glen Research, 10-3064-90), and s4U (Glen Research, 10-3052-90) (Chemgene, ANP-8626,) modified RNA phosphoramidites; fluorescein phosphoramidite (Lumiprobe, F5160), canonical DNA C phosphoramidite (Chemgene, ANP-5560), and canonical RNA (Chemgene, A, ANP-5671; U, ANP-5674; C, ANP-6676) phosphoramidites were purchased from commercial sources. Lacking commercial phosphoramidite reagents, ms2m6A-containing oligonucleotides were synthesized following previously established protocols (Kierzek and Kierzek 2003a,b; Kierzek et al. 2022). All synthesized oligos were gel extracted, lyophilized, and redissolved in water for downstream experiments. Concentrations of the aqueous RNA samples were determined by 260 nm UV absorption, using a Thermo Scientific Nanodrop One Spectrophotometer and theoretical 260 nm molar extinction coefficients provided by Integrated DNA Technologies.
Electrophoretic mobility shift assay (EMSA)
Binding affinities of the synthesized RNA oligos to hPUM1-HD and hPUM2-HD were determined using EMSAs. Fluorescein-labeled RNA oligos were diluted in 1× EMSA binding buffer (10 mM HEPES pH 7.5, 50 mM KCl, 1 mM EDTA, 0.1% [v/v] Tween-20, 1 mM DTT, 0.01 mg/mL BSA [Fisher Scientific, BP9706100), to a 10× concentration of 20 and 50 nM for hPUM1 and hPUM2 experiments, respectively. Serial protein dilutions were prepared for a final volume of 10 μL as follows. In 1× EMSA binding buffer, proteins were diluted to an initial 1.1× concentration of 1111 nM and threefold serial diluted. In tubes, 9 μL protein and 1 μL RNA were mixed together for final protein concentrations of 1000, 333, 111, 37, 12.3, 4.1, and 1.4 nM with 2 and 5 nM RNA for hPUM1 and hPUM2 experiments, respectively. An eighth lane with no protein was prepared using EMSA binding buffer alone. After incubating the protein×RNA sample at 4°C for 30 min, 2 μL of 6× EMSA loading buffer (15% [w/v] Ficoll 400, 0.01% [w/v] bromophenol blue) was added to each 10 μL dilution for gel loading. Nondenaturing, polyacrylamide gels (0.5× TBE [Tris/Borate/EDTA buffer; 45 mM Tris, 45 mM boric acid, and 1 mM EDTA], 5% [v/v] acrylamide/Bis 19:1; catalyzed with 0.1% [w/v] ammonium persulfate [APS], and 0.1% [v/v] N,N,N′,N′-tetramethylethylenediamine [TEMED]) were preequilibrated in 0.5× TBE using a Mini-PROTEAN vertical electrophoresis cell (Bio-Rad, 1658005) for 30 min at 4°C. Samples were loaded and run at 75 V for 45 min at 4°C. The gels were imaged with a Bio-Rad ChemiDoc imager on a U/V tray capable of measuring flourescein 528/532 nm absorption. Flourescein absorption intensities of the lower (unbound) and upper (bound) RNA bands were quantified using FIJI ImageJ (Schindelin et al. 2012). Kd values were calculated with a nonlinear fit in GraphPad Prism version 10.1.1 for MacOS (GraphPad Software). Averages, standard deviations, and graphs were also performed and made in GraphPad Prism. All EMSA experiments were performed in a minimum of three replicates, with additional replicates conducted as necessary to ensure that the standard error of the mean (SEM) for all estimated Kd values remained below 10. Statistical significance of differences in Kd between oligos was obtained using one-way ANOVA.
Data collection and analysis
To complement experimental EMSA binding data, additional hPUM1 and hPUM2 RNA in vitro binding data were collected from the literature (Vaidyanathan et al. 2017; Jarmoskaite et al. 2019). For unmodified RNAs, matching sequences and their variations to this study's binding data were selected from previously published data (Jarmoskaite et al. 2019). Duplicate entries were averaged into a single experimental ΔG for each sequence. In total, experimental ΔGs were obtained for 21 of 24 possible single-site mutants of UGUAUAUA bound to hPUM2. For hPUM1, experimental ΔGs were obtained for eight mutants of UGUACAUC. Binding data for 6 Ψ and m6A mutants to hPUM2 were obtained from Vaidyanathan et al. (2017).
For comparison of λD predictions with experimental values, experimental Kds were first converted to absolute ΔGs using the relationship ΔG = −RTlnKd at the experimental temperature. Next, λD predicted ΔΔGs were converted to absolute ΔGs following established methods (Keranen et al. 2017). Briefly, the data were first divided into groups based on the consensus oligo and protein target. For each data set, the
mean experimental ΔG (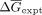 ) and predicted in silico ΔΔG (
) and predicted in silico ΔΔG (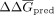 ) were calculated. Finally, the in silico ΔΔGpred were converted to absolute ΔGpred by recentering values relative to the experimental mean
) were calculated. Finally, the in silico ΔΔGpred were converted to absolute ΔGpred by recentering values relative to the experimental mean 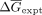 . In sum,
. In sum, 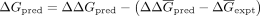 for each ΔΔGpred in the data set (Keranen et al. 2017). The λD predicted ΔGs (ΔGpred) and experimental ΔGs (ΔGexpt) were then directly compared.
for each ΔΔGpred in the data set (Keranen et al. 2017). The λD predicted ΔGs (ΔGpred) and experimental ΔGs (ΔGexpt) were then directly compared.
SUPPLEMENTAL MATERIAL
Supplemental material is available for this article.
ACKNOWLEDGMENTS
The authors thank members of the Aoki, Vilseck, and Zhang Laboratories for their helpful discussion and acknowledge the Indiana University Pervasive Technology Institute for providing supercomputing and storage resources that have contributed to the research results reported within this paper. S.T.A., W.Z., and J.Z.V. received startup funds from the Indiana University School of Medicine and its Precision Health Initiative (PHI). J.Z.V. is funded by the National Institute of General Medical Sciences (NIGMS) of the National Institutes of Health (NIH) (R35GM146888). S.T.A. is funded by the NIH/NIGMS (R35GM142691) and an Indiana University Research Support Funds Grant (RSFG). E.K. is funded by UMO-2020/39/B/NZ1/03054 and R.K. is funded by UMO-2022/45/B/ST4/03586.
Footnotes
-
Article is online at http://www.rnajournal.org/cgi/doi/10.1261/rna.080367.124.
-
Freely available online through the RNA Open Access option.
- Received December 23, 2024.
- Accepted July 14, 2025.
This article, published in RNA, is available under a Creative Commons License (Attribution-NonCommercial 4.0 International), as described at http://creativecommons.org/licenses/by-nc/4.0/.








