
Conserved role for spliceosomal component PRPF40A in microexon splicing
- Corresponding author: adamn{at}ucr.edu
Abstract
Microexons (exons ≤30 nt) are important features of neuronal transcriptomes, but pose mechanistic challenges to the splicing machinery. We previously showed that PRP-40, a component of the U1 spliceosome, is globally required for microexon splicing in Caenorhabditis elegans. Here we show that the homologous PRPF40A is also globally required for microexon splicing in mouse neuroblastoma cells. We find that PRPF40A coregulates microexons along with SRRM4, a neuron-specific regulator of microexon splicing. The relationship between exon size and dependence on PRPF40A/SRRM4 is distinct, with SRRM4-dependence exhibiting a size threshold (∼30 nt) and PRPF40A-dependence exhibiting a graded decrease as exon size increases. Finally, we show that PRPF40A knockdown causes an increase in productive splicing of its spliceosomal binding partner Luc7l by the skipping of a small “poison exon.” Similar homeostatic cross-regulation is often observed across paralogous RNA-binding proteins. Here we find this concept likewise applies across evolutionarily unrelated but functionally and physically coupled spliceosomal components.
Keywords
INTRODUCTION
Many eukaryotic genes are split into protein-coding exons interrupted by intervening introns. These introns are removed by the spliceosome to generate mature mRNAs. While intron sizes vary substantially across genes and species, exon size is more constrained, with a median size of ∼120 nucleotides (nt) across various animal species (Ramírez-Sánchez et al. 2016). Indeed, exons below ∼51 nt are spliced inefficiently, partly due to physical constraints impeding spliceosomal assembly flanking small exons (Dominski and Kole 1991; Berget 1995).
Nevertheless, a subset of unusually small exons referred to as microexons, often defined as exons ≤30 nt, has recently emerged as a distinct functional class. Microexons are enriched in neuronal genes, and tend to be alternatively spliced such that the microexon is included in neurons but skipped in other cells (Irimia et al. 2014; Li et al. 2015; Gonatopoulos-Pournatzis and Blencowe 2020). Moreover, dysregulated microexon splicing is as a common molecular feature in autism spectrum disorder (Irimia et al. 2014). Thus, in spite of mechanistic hurdles complicating the splicing of exons below a certain size, microexons constitute an important feature the neuronal transcriptome.
Studies on the regulation of microexon splicing have identified a handful of required factors. The splicing factors RBFOX and PTBP1 were shown to regulate a number of microexons in mouse cells (Li et al. 2015), and the neuronal splicing factor SRRM4 (aka nSR100) was identified as a factor required for global microexon splicing in mice and human cells (Calarco et al. 2009; Irimia et al. 2014; Quesnel-Vallières et al. 2015). Likewise, we recently found in Caenorhabditis elegans that PRP-40, a component of the U1 snRNP, is globally required for microexon splicing (Choudhary et al. 2021).
The potential mechanistic relationships among these observations remain to be determined. In particular, we wanted to investigate the nature of global microexon regulators SRRM4 and PRP-40: Is PRP-40 required globally for microexon splicing in mouse as it is in C. elegans? If so, are the attributes of PRP-40-dependent exons and SRRM4-dependent exons identical? The possibility of different microexon regulatory strategies between mouse and worm is raised by evidence that mouse SRRM4 stimulates microexon splicing via its C-terminal domain, while C. elegans does not encode such a domain in its closest protein homolog (RSR-2) (Torres-Méndez et al. 2019). We therefore set out to determine in mouse cells the relationship between PRPF40A and SRRM4 in microexon splicing.
We find that knockdown of either PRPF40A or SRRM4 in mouse neuroblastoma cells causes a global decrease in microexon inclusion, thus the activity of PRPF40A in mouse is similar to PRP-40 in worm. Moreover, mouse microexons require both PRPF40A and SSRM4 simultaneously for efficient splicing. However, PRPF40A-dependent exons demonstrate a graded decrease in PRPF40A dependence as size increases, while SRRM4-dependent exons display size threshold, with exons above a ∼30 nt threshold not requiring SRRM4. Additionally, we reveal that PRPF40A knockdown causes a large increase of productive splicing of its spliceosomal binding partner Luc7l by skipping a small poison exon. We propose that this is similar to cases of homeostatic compensatory regulation across paralagous RNA-binding proteins, thus extending the concept to compensatory regulation across two evolutionarily unrelated but structurally neighboring factors (PRPF40A and LUC7L).
RESULTS
PRPF40A knockdown causes global loss of microexon splicing
PRP40 is a component of the U1 snRNP (Li et al. 2019). We previously showed that loss of C. elegans prp-40 does not result in systemic splicing defects, as might be expected for a spliceosomal component, but rather results specifically in loss of microexon and small-exon splicing (Choudhary et al. 2021). Yeast and worm genomes encode a single PRP40 gene, while mouse and human genomes encode two paralagous genes, PRPF40A and PRPF40B, with PRPF40A most resembling the PRP40 genes of yeast and worms (Fig. 1A). prp-40 homologs encode two N-terminal WW domains and a number of C-terminal FF domains. Biochemical experiments show that the FF domains interact with Luc7 and U1-70K, components of the U1 snRNP that binds to the 5′ splice site (Ester and Uetz 2008; Li et al. 2019). The WW domains interact with BBP (homolog of SF1), which binds to the branchpoint near the 3′ end of the intron (Abovich and Rosbash 1997; Li et al. 2019). Thus, PRP-40 is proposed to act as an intron-spanning bridge (Supplemental Fig. S1), and to be particularly important for the “intron definition” mode of splicing. Indeed, this intron definition mediated by PRP-40 appears to be essential for its role in facilitating efficient microexon splicing, which is too small to be spliced by the “exon definition” mode of splicing (Robberson et al. 1990; Black 1991).
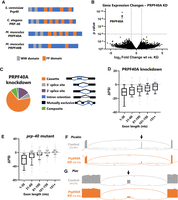
. PRPF40A knockdown causes global loss of microexon and small exon splicing. (A) Protein domain structure of PRP-40 homologs, modeled after TreeFam database (Ruan et al. 2008). (B) Volcano plot showing changes in gene expression as determined by DESeq2 upon PRPF40A siRNA knockdown. (C) The effect of PRPF40A knockdown on alternative splicing is largely restricted to changes in cassette exon inclusion (67%). (D) Effects of PRPF40A knockdown on cassette exon inclusion binned by exon size, including microexons (1–30 nt). ΔPSI = change in percent spliced in from control to PRPF40A conditions. (E) Same as in D, except for C. elegans prp-40 loss-of-function mutants. (F,G) Sashimi plots showing that PRPF40A knockdown results in dramatic decreases in microexon inclusion, but does not affect splicing of upstream or downstream exons, or lead to increased retention of the flanking introns. Numbers in parentheses represent PSI values for the replicate in question; numbers over splice junctions indicate number of reads for that splice junction in the replicate in question.
We showed that, as in worms, knockdown of PRPF40A in mouse neuroblastoma cells results in loss of microexon splicing for the handful of exons we tested (Choudhary et al. 2021). Here we wanted to extend these findings to a global analysis of splicing controlled by PRPF40A. To this end, we performed PRPF40A knockdown in mouse neuroblastoma N2a cells followed by RNA-seq.
Knockdown of PRPF40A causes little change in global gene expression levels except for, as expected, a strong decrease in PRPF40A levels (Fig. 1B). PRPF40A knockdown has specific effects on alternative splicing, largely limited to dysregulated cassette exon splicing (aka exon skipping). Other types of alternative splicing, for example, intron retention or 3′ splice site selection, are largely unaffected (Fig. 1C). Moreover, the effect of PRPF40A is directional, as knockdown consistently results in negative changes in Percent Spliced In (ΔPSI, Fig. 1D), indicating that the role of PRPF40A is to facilitate cassette exon inclusion.
The effect of PRPF40A on cassette exon inclusion depends on exon length. Microexons (≤30 nt) are most strongly affected, and as exon size increases, the dependence on PRPF40A decreases (Fig. 1D; Supplemental Fig. S2A–C). This pattern of size dependence is similar to our previous observations in C. elegans prp-40 mutants (Fig. 1E). Therefore, mouse PRPF40A, like C. elegans PRP-40, is required specifically for the inclusion of microexons and small exons. Furthermore, PRPF40A knockdown does not lead to an increase in retention of the introns flanking microexons (examples in Fig. 1F,G). This indicates that PRPF40A is not required generally for splicing fidelity, but is required specifically for the splicing decision of microexon inclusion versus skipping.
SRRM4 and PRPF40A are both globally required for microexons, but with different regulatory features
In parallel with our PRPF40A knockdown, we also knocked down two related factors of interest: PRPF40B, the paralog of PRPF40A; and SRRM4, a neuronal splicing factor required for the inclusion of many microexons (Supplemental Fig. S2D; Quesnel-Vallières et al. 2015). The results of PRPF40B knockdown are in striking contrast to those of PRPF40A, showing no global relationship between exon size and PRPF40B requirement (Fig. 2A). Nor does PRPF40B knockdown affect Prpf40a levels or exacerbate microexon dysregulation in conjunction with PRPF40A knockdown (Supplemental Fig. S2E,F). This indicates that PRPF40A, but not PRPF40B, acts in a similar manner to C. elegans PRP-40 in stimulating microexon inclusion.
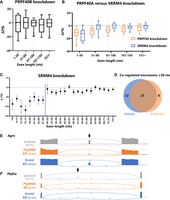
SRRM4 and PRPF40A coregulate microexons. (A) In contrast with PRPF40A knockdown, PRPF40B knockdown does not result in global splicing defects with respect to exon length. (B) SRRM4 knockdown results in loss of microexon inclusion. Both PRPF40A and SRRM4 are required for microexon splicing, but only PRPF40A is required for other small exon splicing (e.g., the 31–60 nt bin). (C) Finer-grained binning of SRRM4-dependent exons reveals a threshold of 30 nt beyond which SRRM4 is not globally required for exon inclusion. Note that these ΔPSIs are averages, and thus some exons of larger size are inevitably regulated by Srrm4. (D) Venn diagram showing microexons that were detected in both the Srrm4 and Prpf40A knockdowns (minimum 15 junctions in all six replicates plus all three control replicates), coregulated as defined by |ΔPSI}>5% in both conditions. (E) Example of a microexon (in Agrn) strongly dependent on PRPF40A but only mildly regulated by SRRM4. (F) Example of a microexon (in Mef2a) coregulated by SRRM4 and PRPF40A.
In contrast, knockdown of SRRM4 results in widespread loss of microexon inclusion, as expected (Fig. 2B). The magnitude of microexon dysregulation is similar between SRRM4 and PRPF40A knockdown (Fig. 2B). However, the global relationship between exon length and ΔPSI is different for the two factors. PRPF40A-dependent exons show a graded decrease in PRPF40A dependence as their size increases (Fig. 2B). But SRRM4-dependent exons display a size threshold, with microexons ≤30 nt strongly dependent on SRRM4, and exons above this threshold largely unaffected (Fig. 2C). As such, the global effects of PRPF40A and SRRM4 are similar for microexons, but diverge for small exons above the 30 nt threshold (Fig. 2B).
Most PRPF40A-dependent microexons are also SRRM4-dependent, and vice versa (Fig. 2D), although we did detect a few notable exceptions. For example, a 12 nt microexon in the Agrn gene is strongly dependent on PRPF40A, but not on SRRM4 (Fig. 2E). It may be that in cases such as this, the paralogous factor SRRM3 provides redundant splicing regulation (Nakano et al. 2019). In any event, the most common scenario, as in the 24 nt microexon in Mef2a (Fig. 2F), is that both factors are required for microexon inclusion, indicating that most microexons are coregulated by PRPF40A and SRRM4.
PRPF40A knockdown increases productive alternative splicing of its spliceosomal binding partner LUC7L
We noted a particularly strong effect of PRPF40A, but not SRRM4 or PRPF40B, on the splicing of a small 71 nt alternatively spliced exon in the Luc7l gene. Luc7l encodes a U1 spliceosomal component that in yeast physically interacts with Prp40 (Fig. 3A; Ester and Uetz 2008). Under normal conditions, the exon is included in more than half of Luc7l transcripts (53.3%). Inclusion of this exon causes a frameshift and is predicted to encode a truncated LUC7L protein lacking both conserved zinc finger motifs (Fig. 3B), and the mRNA is likely to be an NMD substrate (Jourdain et al. 2021). Therefore, the most common splicing outcome in normal conditions leads to nonfunctional LUC7L due to the inclusion of a “poison exon.” A second common splicing outcome is retention of the downstream and/or upstream introns (12% intron retention). The result of this splicing choice is also predicted to be NMD sensitive and encode a truncated, nonfunctional protein (Fig. 3B). Only the exon-skipped product, which constitutes a mere 35% of the spliced output in control N2a cells, is predicted to be functional.
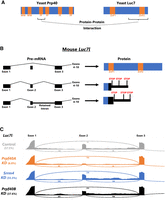
PRPF40A knockdown increases productive splicing of its spliceosomal binding partner LUC7L. (A) Summary of protein–protein interaction data (Ester and Uetz 2008) showing that yeast Prp40 directly interacts with yeast Luc7 via its first FF domain. (B) Mouse Luc7l gene model showing that if exon 2 is included, or if introns 1 or 2 are retained, then unproductive LUC7L is produced, lacking the region that interacts with Prp40, and containing premature stop codons likely to subject the transcript to NMD. (C) In control N2a cells, the majority of Luc7l is spliced into unproductive isoforms. (This is likely an underestimate, given that some unproductive mRNAs are likely destroyed by NMD.) Knockdown of PRPF40A, but not PRPF40B or SRRM4, results in strong increase of the productive isoform (exon 2 skipped and introns 1 and 2 spliced out). Numbers in parentheses represent PSI values for the replicate in question, and numbers over splice junctions indicate number of reads for that splice junction in the replicate in question.
The consequence of PRPF40A knockdown is a dramatic shift in Luc7l splicing from unproductive isoforms (exon included and/or intron retained) to the productive exon-skipped isoform: from 35% productive isoform in control to 80% in PRPF40A knockdown (Fig. 3C; Supplemental Fig. S2G). In the yeast U1 spliceosome, Prp40 and Luc7 are direct protein–protein interactors (Ester and Uetz 2008) and are juxtaposed next to each other in cryoEM structures (Li et al. 2019). As such, a major molecular response to the loss of PRPF40A is an increase in its spliceosomal partner LUC7L mediated by productive alternative splicing. Two paralogous genes, Luc7l2 and Luc7l3, also encode splicing-mediated nonproductive isoforms. However, the effect of Prpf40A is specific to Luc7l splicing (ΔPSI productive isoform = + 45%), as no large changes are seen for Luc7l2 or Luc7l3 productive isoforms (ΔPSI = −5.4% and +0.9%, respectively). We also tested whether PRPF40A-sensitive exons harbor 5′ splice site sequences diagnostic of regulation by Luc7l/Luc7l2 versus Luc7l3 (Kenny et al. 2022) and found both classes equally associated with PRPF40A-sensitive microexons (Supplemental Fig. S2G).
A feedback splicing response similar to the Prpf40a/Luc7l response described here has also been observed in human cells upon knockdown of the Luc7l paralog Luc7l2 (Jourdain et al. 2021; Kenny et al. 2022). Knockdown of Luc7l2 causes an increase in Luc7l exon skipping and a decrease in both intron retention and exon inclusion. This response appears to be a compensatory mechanism of the type often seen among paralogous RNA-binding proteins (Ni et al. 2007; Spellman et al. 2007; Lareau and Brenner 2015). Here we extend this phenomenon of compensatory splicing changes to include evolutionarily unrelated but physically associated components of the U1 snRNP. We speculate this might be a mechanism for homeostatic control of spliceosome formation and that abundant LUC7L might partially compensate for the loss of its binding partner PRPF40A.
DISCUSSION
Here we show that the spliceosomal component PRPF40A is globally required for the splicing of microexons and small exons, but not for other types of alternative or constitutive splicing. This role for PRPF40A is shared between mouse and C. elegans and thus the specific activity of this spliceosomal component in facilitating microexon inclusion appears to be a deeply conserved phenomenon.
We find that PRPF40A and SRRM4 coregulate microexon splicing, while small exons (∼31–60 nt) require PRPF40A but not SRRM4. We speculate that for microexons, SRRM4 might directly recruit PRPF40A to facilitate exon inclusion. Affinity-purification mass spectrometry experiments in mouse N2a cells revealed protein–protein interactions between SRRM4 and PRPF40A. (Gonatopoulos-Pournatzis et al. 2018) We propose a model (Fig. 4) in which SRRM4 binds to microexon-flanking introns via UGC-containing motifs (Gonatopoulos-Pournatzis et al. 2018) and interacts with PRPF40A to ensure microexon splicing. We previously implicated C. elegans PRP-40 in an “intron definition” splicing mechanism for microexons whose small size physically precludes them from the typical “exon definition” splicing mechanism favored in many metazoa (Berget 1995; Choudhary et al. 2021). An alternative mechanism described in mammalian cells involves a unique exon-definition process mediated by SRRM4 (Gonatopoulos-Pournatzis et al. 2018). The data here are consistent with either model, with the main commonalities being that SRRM4 binds to the flanking intron(s), interacts with PRPF40A, and both factors facilitate the splicing of microexons that would otherwise be skipped by standard splicing mechanisms. (Fig. 4).

Model for coregulation of microexons by SRRM4 and PRPF40A. For conventional-sized exons, splicing proceeds via exon definition, in which the unit of recognition is the exon. This is not physically feasible for microexons, which cannot accommodate the binding of both the U1 and U2 snRNPs on such a small sequence space. However, in our model, this physical constraint can be overcome when SRRM4, binding to UGC-containing motifs in the flanking intron, interacts with PRPF40A, which then facilitates splicing either through a novel exon-definition mechanism, or alternatively by an intron-spanning intron definition process. Either mechanism overcomes the failure to splice according to standard exon definition mechanisms, and the result is microexon inclusion in the mature mRNA.
While the activities of PRPF40A and PRP-40 appear to be similar between mouse and worm, there is reason to believe that this is not the case for SRRM4 and its worm ortholog RSR-2. The C terminus of SRRM4 appears to be essential for its microexon-stimulating role, and this domain is not present in worm RSR-2 (Torres-Méndez et al. 2019). We therefore speculate that stimulation of microexons in the C. elegans nervous system is accomplished by other sequence-specific RNA-binding proteins that in turn recruit PRP-40. For example, the splicing factor RBFOX, which regulates some mammalian microexons (Li et al. 2015), or other neuron-specific splicing factors (Norris et al. 2017; Taylor et al. 2023; Wolfe et al. 2024), might facilitate neuronal microexon splicing.
One of the strongest splicing changes upon PRPF40A knockdown is the loss of a poison exon in the Luc7l gene. We propose that this constitutes a compensatory homeostatic mechanism by which loss of PRPF40A leads to an increase in its spliceosomal binding partner LUC7L. Such compensatory mechanisms are often observed between paralagous RNA-binding proteins, and indeed such a compensatory mechanism is observed between Luc7l and its paralog Luc7l2 (Jourdain et al. 2021; Kenny et al. 2022). Here we extend this phenomenon to also apply to functionally related but evolutionarily unrelated factors. It should be noted that we have yet to test whether this compensation translates to major differences in protein abundance, and in the future, it will be interesting to test whether concomitant loss of Prpf40a and Luc7l results in a negative genetic interaction. Another interesting link between PRPF40A and LUC7L is that they are both U1 snRNP-associated proteins, but have restricted effects on subclasses of alternative splicing events. For PRPF40A, regulation is specific to microexons and small exons. For LUC7L, regulation is specific to certain classes of 5′ splice sites (Puig et al. 2007; Kenny et al. 2022). This suggests a degree of specialization within individual snRNPs such that different components are responsible for regulating different aspects of mRNA splicing.
MATERIALS AND METHODS
N2a cell maintenance and harvesting
N2a (Neuro-2a) cells obtained from ATCC (CCL-131) were grown in Opti-MEM Medium plus DMEM, high glucose with L-glutamine, with 5% FBS and penicillin/streptomycin at 37°C and 5% CO2. Cells were grown in biological triplicates.
For gene-specific knockdown, cells were transfected with 10 nM pools of gene-specific siRNA (siGENOME, Dharmacon) using RNAiMax, according to manufacturer recommendations (Life Technologies). Cells were harvested 72 h after transfection for RNA-seq analysis. RNA was extracted using TRI Reagent and ZYMO Direct-zol RNA Miniprep kits.
RNA-seq library preparation and analysis
Libraries were prepared using NEBNext Ultra II poly(A)-enriched kits for Illumina sequencing, followed by quality control measurements via Qubit and Agilent TapeStation. A total of 150 bp paired-end reads were generated on an Illumina HiSeq 2000 short-read sequencer, and reads were mapped to the mouse genome (GRCm38) with STAR (Dobin et al. 2013) (version 2.5.3a). Each condition was sequenced in biological triplicate, with an average of 41.5 million paired-end reads per replicate, and an average of 74.1% uniquely mapped reads per replicate. All sequencing reads and data are deposited at NCBI Gene Expression Omnibus (GEO) database (accession GSE267257). Previously published prp-40 mutant RNA-seq data from C. elegans are available at the NCBI SRA (accession PRJNA684142).
Gene-specific counts were tabulated for each sample using HTSeq (Anders et al. 2015) (version 0.9.1), and statistically significant differentially expressed transcripts were identified with DESeq2 (Love et al. 2014) (version 1.36.0). Differential alternative splicing analysis was carried out using the junction usage model (Wang and Rio 2018) (JUM 2.0.2). For visualization of aligned reads, bam files were generated by samtools and subsequently, sashimi plots were generated by the Integrative Genomics Viewer (IGV).
SUPPLEMENTAL MATERIAL
Supplemental material is available for this article.
ACKNOWLEDGMENTS
This work was supported by the National Institute of Neurological Disorders and Stroke of the National Institutes of Health (R01NS111055).
Footnotes
-
Handling editor: Benjamin Blencowe
-
Article is online at http://www.rnajournal.org/cgi/doi/10.1261/rna.080142.124.
- Received June 18, 2024.
- Accepted September 29, 2024.
This article is distributed exclusively by the RNA Society for the first 12 months after the full-issue publication date (see http://rnajournal.cshlp.org/site/misc/terms.xhtml). After 12 months, it is available under a Creative Commons License (Attribution-NonCommercial 4.0 International), as described at http://creativecommons.org/licenses/by-nc/4.0/.
REFERENCES
MEET THE FIRST AUTHOR

Meet the First Author(s) is an editorial feature within RNA, in which the first author(s) of research-based papers in each issue have the opportunity to introduce themselves and their work to readers of RNA and the RNA research community. Bikash Choudhary is the first author of this paper, “Conserved role for spliceosomal component PRPF40A in microexon splicing.” Bikash is an Assistant Project Scientist in the Department of Biochemistry, University of California, Riverside. The lab goal is to understand the regulation of alternative splicing of transcripts in a cell/tissue-specific manner.
What are the major results described in your paper and how do they impact this branch of the field?
This article indicates the role of PRPF-40A in regulation of alternative splicing of microexons, which is slightly distinct from SRRM4-mediated regulation. This opens a completely new field for exploring the role of the spliceosomal complex in alternative splicing of microexons.
What led you to study RNA or this aspect of RNA science?
This is an interesting question. It was a completely serendipitous finding that in a forward genetic screen conducted on the genetic model C. elegans for identifying the regulators of alternative splicing in the nervous system, we identified prp-40 as one of the candidates (Choudhary et al. 2021). When we performed RNA-seq analysis for alternative splicing, we found that inclusion of alternative microexons was drastically affected in these animals. This led us to investigate the mammalian homolog PRPF40A, which has a role in the mammalian nervous system. And that is what this article is about.
What are some of the landmark moments that provoked your interest in science or your development as a scientist?
I grew up in a countryside which was surrounded by dense vegetation. I was fascinated about how plants make their food (when I learned in middle school about photosynthesis). This led me to follow a career in agricultural sciences. In graduate school, I was fascinated about neurons. And since then I was involved in understanding the physiological basis of neurons. Finally, in my present group, I am interested in how the splicing of neuronal transcripts is regulated. Moreover, how splicing is different in different classes of neurons.
Are there specific individuals or groups who have influenced your philosophy or approach to science?
Yes, I am very much influenced by my former mentor Professor Eva Mandelkow and Professor Eckhard Mandelkow. I feel that my short tenure in this lab completely changed my way of thinking and understanding of research. They made me an independent thinker. From my present supervisor, Professor Adam Norris, I learned the concept of “patience” in science.
What are your subsequent near- or long-term career plans?
I would like to be an independent researcher and train the young minds of future scientists.








