
Suppression of the Escherichia coli rnpA49 conditionally lethal phenotype by different compensatory mutations
- 1Department of Medical Biochemistry and Microbiology
- 2Department of Cell and Molecular Biology, Uppsala University, 751 23 Uppsala, Sweden
- Corresponding author: arianne.babina{at}imbim.uu.se, arianne.babina{at}glasgow.ac.uk
Abstract
RNase P is an essential enzyme found across all domains of life that is responsible for the 5′-end maturation of precursor tRNAs. For decades, numerous studies have sought to elucidate the mechanisms and biochemistry governing RNase P function. However, much remains unknown about the regulation of RNase P expression, the turnover and degradation of the enzyme, and the mechanisms underlying the phenotypes and complementation of specific RNase P mutations, especially in the model bacterium, Escherichia coli. In E. coli, the temperature-sensitive (ts) rnpA49 mutation in the protein subunit of RNase P has arguably been one of the most well-studied mutations for examining the enzyme's activity in vivo. Here, we report for the first time naturally occurring temperature-resistant suppressor mutations of E. coli strains carrying the rnpA49 allele. We find that rnpA49 strains can partially compensate the ts defect via gene amplifications of either RNase P subunit (rnpA49 or rnpB) or by the acquisition of loss-of-function mutations in Lon protease or RNase R. Our results agree with previous plasmid overexpression and gene deletion complementation studies, and importantly suggest the involvement of Lon protease in the degradation and/or regulatory pathway(s) of the mutant protein subunit of RNase P. This work offers novel insights into the behavior and complementation of the rnpA49 allele in vivo and provides direction for follow-up studies regarding RNase P regulation and turnover in E. coli.
Keywords
INTRODUCTION
RNase P is an ancient and essential endoribonuclease found in Bacteria, Archaea, and Eukaryotes that is responsible for the cleavage of 5′-end leader sequences present on premature tRNA (pre-tRNA) transcripts. Since its discovery in the 1970s, numerous studies have characterized different RNase P variants across diverse model organisms (for reviews, see Kazantsev and Pace 2006; Ellis and Brown 2009; Lai et al. 2010; Klemm et al. 2016; Phan et al. 2021; Jarrous and Liu 2023).
In Escherichia coli, as in most other bacteria, RNase P is a heterodimer comprised of a small protein subunit (C5 protein, encoded by rnpA) and an RNA subunit (M1 RNA, encoded by rnpB) (Harris et al. 1998; Altman and Kirsebom 1999). M1 RNA is the catalytic subunit of the enzyme and plays crucial roles in substrate recognition and C5 folding, intracellular solubility, and proteostasis (Guerrier-Takada et al. 1983; Son et al. 2015), as the bacterial C5 protein behaves as an intrinsically unstructured or natively unfolded protein at low ionic strength (Henkels et al. 2001). The C5 protein is important for M1 RNA metabolic stability and has been implicated in electrostatic shielding, substrate binding, product release, and preventing rebinding of the product (Reich et al. 1988; Tallsjö and Kirsebom 1993; Buck et al. 2005; Sun and Harris 2007; Kim and Lee 2009; Kirsebom and Trobro 2009; Lin et al. 2016). In addition to the 5′-end processing of pre-tRNA, bacterial RNase P is essential for the separation of pre-tRNAs from polycistronic operon transcripts (Mohanty et al. 2020) and is involved in the processing of, for example, 4.5S RNA, among other non-tRNA substrates (for a review focusing on E. coli, see Hartmann et al. 2009).
Over the years, many different conditionally lethal mutants and/or inducible complementation systems in the primary bacterial models, E. coli and Bacillus subtilis, have been used to investigate RNase P assembly and activity in vivo. For example, the temperature-sensitive (ts) mutants A49, ts241, and ts709, which exhibit defects in tRNATyrSu3 at higher temperatures, were selected in E. coli. These mutants were found to carry changes in the RNase P protein (C5/rnpA) and RNA (M1 RNA/rnpB) subunits (Schedl and Primakoff 1973; Sakano et al. 1974; Sakamoto et al. 1983; Kirsebom et al. 1988). Conditionally lethal B. subtilis mutants in which rnpA or rnpB expression can be suppressed via inducible promoters have been constructed (Gößringer et al. 2006; Wegscheid et al. 2006; Wegscheid and Hartmann 2007) as well as E. coli strains in which the chromosomal rnpB gene was either deleted (Waugh and Pace 1990) or under control of an arabinose-dependent promoter (Wegscheid and Hartmann 2006). For growth, these mutants typically depend on the expression of the corresponding functional RNase P subunit from plasmids. For this study, we chose to focus on the ts A49 mutation in E. coli, as it was the first RNase P mutant isolated and its mutant phenotype is well-characterized (Schedl and Primakoff 1973; Apirion 1980).
The E. coli ts A49 mutation, also known as the rnpA49 allele, was first isolated from mutagenesis experiments in 1973 and is caused by an A to G transition resulting in the substitution of an arginine to a histidine at position 46 within the C5 protein (C5A49) (Schedl and Primakoff 1973; Apirion 1980; Kirsebom et al. 1988). This mutation reduces the solubility of the C5 protein and is hypothesized to impact the association of the C5 protein and M1 RNA subunits to form the RNase P holoenzyme, resulting in reduced RNase P stability and activity (Baer et al. 1989; Li et al. 2003). Strains containing the rnpA49 allele demonstrate a slight growth defect at permissive temperatures (30°C–33°C) and rapidly accumulate precursor tRNAs and unprocessed polycistronic tRNA transcripts when shifted to the nonpermissive temperature (42°C), eventually leading to arrest of cell growth and death (Schedl and Primakoff 1973; Li et al. 2003; Mohanty and Kushner 2007, 2008; Agrawal et al. 2014).
Previous work has shown that the E. coli rnpA49 ts phenotype can be complemented by plasmid overexpression of either the wild-type or mutant C5 protein (Vioque et al. 1988; Jovanovic et al. 2002), M1 RNA from E. coli (Jain et al. 1982; Motamedi et al. 1984; Baer et al. 1989), or with various C5 protein homologs from other bacterial species (Morse and Schmidt 1992; Pascual and Vioque 1996). Other studies have reported that overexpression of arginine tRNACCG from both E. coli (Kim et al. 1998) and Brevibacterium albidum (Kim et al. 1997) can complement the temperature sensitivity caused by the rnpA49 mutation; however, the mechanism underlying this rescue has not been elucidated. Furthermore, site-directed mutagenesis of the rnpA49 allele has identified a number of amino acid changes that can compensate the ts defect (Jovanovic et al. 2002), and additional hydroxylamine mutagenesis experiments have led to the identification of compensatory mutations in rnpB that can rescue the growth of A49 strains at the nonpermissive temperature (Morse and Schmidt 1993). Lastly, recent work has shown that the deletion of poly(A) polymerase I (PAP I, encoded by pcnB) can also partially rescue the ts defect caused by the rnpA49 allele (Mohanty et al. 2020).
Although these studies provide invaluable insights into RNase P assembly, structure, and function in vivo and demonstrate that there are many ways to rescue the E. coli rnpA49 ts phenotype, they relied on the use of artificial approaches to examine complementation, such as plasmid overexpression, gene deletions, and mutagenesis. Subsequently, there remains a gap in the literature regarding how E. coli can naturally compensate for and suppress the deleterious effects brought about by the rnpA49 mutation at nonpermissive temperatures. In this work, we isolated and characterized naturally occurring second-site suppressor mutations of the ts phenotype in an E. coli strain harboring the rnpA49 allele. Consistent with past plasmid overexpression studies, we found that E. coli can compensate for a defective RNase P by increasing the copy number of either rnpA49 or rnpB via large genome amplifications. We also identified RNase R loss-of-function mutations, as well as numerous mutations impacting Lon protease expression and activity. These latter findings present novel links between the mutant RNase P, RNase R, and Lon protease, setting the stage for further investigations into RNase P regulation, degradation, and turnover in E. coli.
RESULTS AND DISCUSSION
Isolation of suppressor mutants that enable the growth of E. coli A49 at the nonpermissive temperature
Suppressor mutants of an E. coli strain carrying the ts rnpA49 allele were isolated from standard fluctuation assays performed at the nonpermissive temperature. Briefly, ∼108 cells from 40 independent E. coli A49 cultures grown at permissive temperature were plated on LB-agar and incubated for 48 h at the nonpermissive temperature (42°C). From these experiments, the mutation rate for E. coli A49 ts revertants generated by spontaneous mutation was determined to be ∼10−10 to 10−9 per cell per generation using the bz-rates web tool (Gillet-Markowska et al. 2015).
After incubation, a single colony from each plate was randomly selected, restreaked, and retested for growth at 42°C, and then locally sequenced to confirm the retention of the rnpA49 allele. Seventeen isolates carrying no additional mutations in the rnpA49 locus were selected for additional Sanger and/or whole-genome sequencing to identify potential second-site suppressor mutations and for further characterization (Fig. 1A; Table 1; Supplemental Table S1). These suppressor isolates were subsequently classified as one of three mutation types, which are discussed in detail below: (i) gene amplifications of rnpA49 or rnpB, (ii) mutations in rnr, and (iii) mutations in lon. To assess the extent of complementation, the relative growth rate of each suppressor strain was determined from growth assays in liquid medium at the permissive temperature (30°C) and at a sublethal temperature (40°C). We chose to perform growth assays at 40°C to enable comparison with the original A49 parental strain, as it was the maximum temperature at which our A49 strain reproducibly grew in liquid medium while still demonstrating a significant growth defect (Fig. 1B; Table 1).
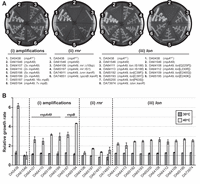
Suppressor mutations rescue the growth of Escherichia coli A49 at the nonpermissive temperature. (A) Growth of the E. coli A49 suppressor strains carrying (i) amplifications of rnpA49 or rnpB or loss-of-function mutations in (ii) rnr or (iii) lon on LB-agar after 24 h incubation at the nonpermissive temperature (42°C). DA5438 is E. coli MG1655 carrying the wild-type rnpA allele; DA61546 is the E. coli A49 parental strain carrying the ts rnpA49 allele. Additional control strains containing kanR deletion cassettes from the KEIO collection in the parental DA61546 A49 background are included for comparison. (B) Relative exponential phase growth rates of the E. coli A49 suppressor strains and relevant control strains as compared to the DA61546 A49 parental strain (set to 1) at the corresponding temperature. Growth measurements were performed in LB medium at both 30°C (permissive temperature) and 40°C (sublethal temperature). The values reported represent the mean of at least three independent biological replicates (with two technical replicates each); error bars represent the standard deviation of the mean.
Summary of E. coli A49 suppressor strains isolated from this study
Complementation via gene amplifications of rnpA49 or rnpB
Of the 17 suppressor strains selected for further sequencing and study, four strains (DA64107, DA64110, DA65198, and DA65199) were found to have amplifications of the genomic region containing the rnpA49 allele, encoding the mutant C5A49 RNase P protein subunit, and two strains (DA65187 and DA65194) were found to contain amplifications of the genomic region containing the rnpB gene, encoding the M1 RNA subunit of RNase P (Table 1; for a schematic explaining gene amplifications, see Supplemental Fig. S1). The copy number of the amplified units varied among the different isolates, ranging from ∼2×–5× for the units containing rnpA49 and 7×–16× for the units containing rnpB, and the size of the amplified chromosomal regions ranged from 29 to 192.5 kbp for the units containing rnpA49 and 17.2–34.5 kbp for the units containing rnpB (Supplemental Table S2). Apart from a synonymous mutation in lsrA (predicted to encode the ATP-binding component of an AI-2 ABC transporter) that was found in one suppressor strain with an rnpA49 amplification (DA64110) and a nonsynonymous mutation in one instance of insA (one of the IS1 elements on the chromosome) in a suppressor strain with an rnpB amplification (DA65187), no additional mutations were identified (Table 1; Supplemental Table S1). As is commonly observed for genomic amplifications in bacteria, the boundaries of the majority of the amplified chromosomal units were flanked by regions homologous to transposases or insertion sequences, specifically IS1 and, in one instance, IS4 (DA64107). However, no repeat sequences or regions of homology were identified within the breakpoints of the amplified units in one rnpA49 amplification strain (DA64110), suggesting an alternate recombination mechanism not involving homologous segments (Supplemental Table S2; Tlsty et al. 1984; Reams and Roth 2015; Nicoloff et al. 2019).
In the growth assays in liquid medium, the suppressor strains containing amplifications of either rnpA49 or rnpB demonstrated a slight 1.2-fold increase in growth rate relative to the original A49 parental strain at the permissive temperature. At the sublethal temperature, the strains carrying rnpA49 amplifications exhibited an ∼2.5-fold to threefold increase in relative growth rate, whereas the two strains with rnpB amplifications demonstrated a three- to fourfold increase in relative growth rate at 40°C (vs. a sixfold increase in relative growth rate for an E. coli strain with the wild-type rnpA allele at 40°C, DA5438), indicating partial complementation of the ts phenotype (Fig. 1B).
The correlation between gene copy number and protein abundance was confirmed via tandem mass tag (TMT)-based proteomics of strain DA65198 (Mosier et al. 2015; Paulo et al. 2016; Zecha et al. 2019; Li et al. 2020). Strain DA65198 carries an estimated 5× amplification of the genomic region containing the rnpA49 allele and correspondingly exhibited a four- to fivefold increase in the abundance of the mutant C5A49 protein relative to the original A49 parental strain (DA61546) (Fig. 2A). These findings are in agreement with past plasmid complementation studies and confirm that increasing expression and/or gene copy number of either rnpA49 or rnpB can rescue the ts defect of E. coli A49. As the rnpA49 mutation is hypothesized to reduce the efficiency with which C5A49 assembles with the M1 RNA, overexpression of either subunit of the mutant RNase P heterodimer likely shifts the equilibrium of assembly toward formation of the RNase P holoenzyme. This is also consistent with previous work demonstrating that wild-type rnpA overexpression increases the steady-state levels of the RNase P holoenzyme (Wegscheid and Hartmann 2006; Gößringer et al. 2023).
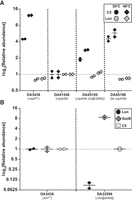
Select suppressor mutations increase C5A49 protein abundance. (A) Relative abundance of either the wild-type or mutant C5 protein and Lon protease during early-to-mid exponential phase growth at the permissive (30°C) or sublethal temperature (40°C), as compared to the original A49 parental strain (DA61546, set to 1). Strains DA61546, DA65195, and DA65198 carry the rnpA49 mutant allele (encoding C5A49). (B) Relative abundances of the wild-type C5 protein and Lon protease in lon+ (DA5438) and lon− (DA22599) E. coli MG1655 strains during early-to-mid exponential phase growth at 30°C, as compared to the E. coli MG1655 wild-type control strain (DA5438, set to 1). Strain DA22599 contains a partial deletion of the lon coding region in an E. coli MG1655 genetic background such that the overlapping coding region for the small RNA SraA and the overlapping promoter for the downstream hupB gene remain intact (Nicoloff and Andersson 2013). GadE, a known Lon substrate, is included as a control to demonstrate the loss of Lon activity in DA22599 (Heuveling et al. 2008). For both A and B, total proteome analysis was performed via TMT-based relative quantification using two biological replicates for each strain (at each temperature for A). Briefly, peptides isolated from each sample were labeled with unique isobaric TMT tags, pooled, and analyzed using mass spectrometry. Peptides were identified by matching the resulting ion peaks with those reported in fragment databases and relative peptide abundance was quantified by comparing TMT reporter ion intensities across all samples. Horizontal bars represent the mean between the two biological replicates; vertical I-bars represent the range. Some data points overlap.
Interestingly, there is no clear linear relationship between complementation efficiency and gene copy number or level of amplification. It is well-established that genome amplifications inherently carry a substantial fitness cost (Nicoloff et al. 2019; Pereira et al. 2021); thus, there are likely trade-offs between the size of the amplified genomic regions, the degree of overexpression from the genes encoded within the amplified units, and overall cell fitness. For example, the strain carrying the 7× rnpB amplification (DA65194) exhibited a greater increase in relative growth rate and subsequent extent of complementation in comparison to the strain carrying the 16× rnpB amplification (DA65187) (Fig. 1B). The higher copy number coupled with the larger size of the amplified unit (34.5 kbp for DA65187 vs. 17.2 kbp for DA65194) likely render suppressor strain DA65187 slightly less fit than DA65194 (Supplemental Table S2).
The strains carrying rnpB amplifications demonstrated the greatest increase in gene copy number relative to those carrying amplifications of rnpA49 (maximum 16× for rnpB vs. 5× for rnpA49). As previous work has shown that high-level overexpression of C5 protein is toxic to E. coli (Jovanovic et al. 2002), perhaps there is an upper limit to which the rnpA49 allele can be amplified before the toxicity of C5A49 overexpression outweighs complementation of the ts phenotype at the nonpermissive or sublethal temperature. Furthermore, the sizes of the amplified chromosomal units in the two rnpB amplification strains were on average smaller than those found in the strains containing the rnpA49 amplifications (Supplemental Table S2). Therefore, the smaller size of the amplified units may also contribute to the average higher increase in gene copy number observed with the rnpB amplification strains. It would be interesting to examine the extent to which subsequent evolution experiments would improve strain growth at the nonpermissive temperature via further amplification, and/or whether the amplifications would eventually be reduced or lost upon accumulation of additional suppressor mutations.
Complementation via loss-of-function mutations in rnr
Two suppressor strains were found to have loss-of-function mutations in rnr, encoding RNase R, a 3′–5′ exoribonuclease that is involved in rRNA and tmRNA maturation and turnover, as well as the degradation of polyadenylated RNAs (Andrade et al. 2009; Condon et al. 2021). One suppressor strain (DA64106) contained a 10 bp deletion in the sequence encoding the N terminus of RNase R, resulting in a +1 frameshift after codon 59. The other strain (DA65197) contained an IS1 insertion in rnr, as well as an IS186 insertion in trmA, which encodes a tRNA methyltransferase that also acts as a tRNA chaperone facilitating tRNA folding (Table 1; Supplemental Table S3; Supplemental Fig. S2).
Of all the suppressor mutants isolated, the two strains carrying rnr mutations exhibited the weakest complementation of the ts phenotype, with smaller colonies on plates incubated at the nonpermissive temperature and slower growth rates in liquid culture (Fig. 1). Both rnr suppressor strains grew comparable to the A49 parental strain at 30°C and exhibited only a modest 1.5-fold increase in growth rate relative to the parental A49 strain at 40°C, the sublethal temperature assayed.
To further confirm that rnr loss-of-function mutations can complement the ts phenotype, we generated and assayed the growth of an A49 strain containing the Δrnr::kanR deletion cassette from the KEIO collection (DA74911) (Baba et al. 2006). Although this deletion strain grew slower than the original A49 parental strain at the permissive temperature, it grew similarly to the two rnr suppressor strains recovered from our screen at the sublethal temperature (Fig. 1B; Table 1). The growth defect of the Δrnr::kanR control strain at 30°C may be due to fitness costs associated with a full deletion of rnr and the subsequent disruption of the genetic context surrounding the rnr open reading frame (ORF), constitutive expression of the kanR cassette, and/or disruption to other pathways regulated by RNase R. Additionally, previous studies have shown that rnr mutants exhibit growth defects at both standard (i.e., 37°C) and lower temperatures (Cairrão et al. 2003).
It is possible that the rnr loss-of-function mutations act via the same complementation mechanism/pathway as the pcnB (PAP I) deletion described by Mohanty et al. (2020). Because RNase P function is compromised with the rnpA49 allele, 5′-unprocessed pre-tRNA transcripts accumulate and become substrates for 3′-end polyadenylation by PAP I, which destabilizes the available pre-tRNA pool and exacerbates the ts growth defect. Deleting PAP I facilitates the accessibility of pre-tRNA 3′-ends for further processing, and studies have shown that the majority of pre-tRNAs that retain their 5′-leaders (i.e., because of a defective RNase P) but have mature 3′-ends are sufficient substrates for aminoacylation (Mohanty et al. 2020). Thus, the absence of pcnB ultimately improves the processing and/or aminoacylation of pre-tRNAs to promote survival at the nonpermissive temperature.
Alternatively, it is also possible that RNase R is involved in the turnover and degradation of M1 RNA, as the preprocessed primary M1 RNA transcript is subject to PAP I-mediated poly(A)-dependent degradation (Kim et al. 2005). Thus, the inactivation of rnr may directly improve the assembly and stability of the mutant RNase P holoenzyme at the nonpermissive temperature. To investigate whether inactivation of rnr improves mutant RNase P stability and/or abundance, we quantified M1 RNA transcript and C5A49 protein levels in rnr mutant suppressor strain DA65197 (rnr::IS1) during early-to-mid exponential phase growth at both the permissive (30°C) and sublethal temperature (40°C) using RT-qPCR and mass spectrometry-based proteomics, respectively. Although we observed a very slight increase (1.3-fold) in C5A49 protein relative abundance at 40°C (Supplemental Fig. S3), we measured a modest, but not statistically significant decrease in M1 RNA transcript levels in DA65197 at the sublethal temperature relative to the DA61546 A49 parental strain (Supplemental Fig. S4), suggesting that RNase R may not be directly involved in M1 RNA stability and/or degradation. However, follow-up experiments outside the scope of this study are necessary to confirm this postulation. Although RNase E is involved in M1 RNA maturation (Lundberg and Altman 1995; Ko et al. 2008), and studies have shown that the mature M1 RNA is metabolically stable and that association with the C5 protein is required for this stability (Jain et al. 1982; Kim and Lee 2009), the full suite of enzymes involved in M1 RNA turnover in E. coli is still unknown.
Lastly, connections between the RNase R and PAP I-mediated poly(A)-dependent degradation pathways and the roles disruptions to these pathways play in suppressing the rnpA49 ts phenotype are further supported by the fact that our A49 parental strain containing the ΔpcnB::kanR deletion cassette from the KEIO collection (DA74851) behaves similarly to both the naturally occurring rnr suppressor mutants isolated from our screen and the constructed A49 Δrnr::kanR control strain at the sublethal temperature (Fig. 1). Like the Δrnr::kanR strain, the ΔpcnB::kanR strain also exhibits a slight growth defect at the permissive temperature relative to the parental A49 strain. Again, this defect may be due to costs associated with kanR expression, potential disruption of the regulatory sequences surrounding or overlapping the fully deleted pcnB ORF, and/or disruption of other regulatory pathways involving PAP I.
Complementation via promoter disruption and nonsynonymous mutations in lon
The majority of mutations recovered from our fluctuation assays affected lon, encoding the ATP-dependent AAA+ protease, Lon. In bacteria, Lon is a global regulator that coordinates a number of important processes such as stress response, DNA replication and repair, and virulence via the degradation of misfolded proteins, rapid turnover of key regulatory proteins, and, in some instances, acting as a protein-folding chaperone (Tsilibaris et al. 2006; Van Melderen and Aertsen 2009; Gur 2013). In E. coli, Lon subunits equilibrate between hexamers and dodecamers under physiological conditions (Wohlever et al. 2013). Two strains were found to have an IS186 insertion in the lon promoter region and seven strains were found to contain SNPs resulting in nonsynonymous mutations in the lon protein-coding region. Six of the nine lon mutant suppressor strains were mucoid when grown at 30°C (DA64109, DA64111, DA64112, DA64113, DA65193, and DA65195), a characteristic phenotype of E. coli lon mutants (Table 1; Supplemental Fig. S5; Bush and Markovitz 1973).
Previous studies have shown that the lon promoter is a hotspot for IS186 insertion and that IS186-mediated lon promoter disruption is a fairly frequent mutation that inhibits lon expression under specific selective conditions, such as in the presence of certain antibiotics (Supplemental Table S3; SaiSree et al. 2001; Nicoloff et al. 2007; Nicoloff and Andersson 2013). The lon::IS186 suppressor strains demonstrated an ∼1.3-fold increase in relative growth rate at 30°C, and a twofold increase in relative growth rate at the sublethal 40°C when compared to the original A49 parental strain (Fig. 1B).
Similarly, the suppressor strains carrying the nonsynonymous changes in the lon protein-coding sequence also exhibited a 1.3-fold increase in relative growth rate at the permissive temperature. However, these strains complemented the ts defect slightly better than the lon::IS186 suppressor strains, with an average 2.6-fold increase in relative growth rate at the sublethal temperature. Of the seven lon protein-coding mutants isolated, four strains contained substitutions at or near residue E240 (Table 1). The E240K mutation has previously been shown to favor Lon's dodecamer conformation and affects substrate recognition and degradation activity. Furthermore, the region surrounding residue 240 has been implicated in substrate recognition and activation of Lon's protease and ATPase activity (Ebel et al. 1999; Cheng et al. 2012; Wohlever et al. 2013). This finding suggests that the suppressor mutants carrying single nonsynonymous mutations within this region (DA64108: Q225P, DA64112: L230R, DA64109: E240G, and DA65195: E240K) likely express a Lon protein with partial or complete loss of function. It is less clear how the suppressor mutations located closer to the N terminus (DA65192: C39F, DA65193: C39Y, and DA65200: P63S) might affect Lon expression and/or function. Although nonsynonymous mutations can directly impact protein function and assembly (as described above), mutations near the 5′-end of a protein-coding gene can also have significant effects on mRNA stability, as well as transcription and/or translation initiation and efficiency (Meyer 2017). Nevertheless, these three suppressor strains behave similar to the isolated lon suppressor strains that carry mutations known to alter Lon expression (lon::IS186 promoter disruptions) and function (nonsynonymous mutations surrounding residue 240), and the suppressor strain with the C39Y mutation (DA65193) is mucoid at 30°C, indicating that nonsynonymous mutations close to the N terminus can indeed impact Lon expression and/or activity. Lastly, a control A49 strain containing the Δlon::kanR deletion cassette from the KEIO collection (DA73974) was constructed and assayed, confirming that loss of Lon function can partially complement the ts defect caused by the rnpA49 allele (Fig. 1).
To our knowledge, the wild-type C5 protein subunit of RNase P has not yet been identified as a substrate for Lon protease (Heuveling et al. 2008), and the observation that the abundance of the wild-type C5 protein is similar in lon+ (DA5438) and lon− (DA22599) E. coli MG1655 strains supports this (Fig. 2B). However, it is possible that the mutant C5A49 protein is degraded by Lon. Thus, lon loss-of-function mutations would stabilize C5A49 levels and the overall mutant RNase P holoenzyme to partially suppress the rnpA49 temperature sensitivity.
To investigate how Lon protease loss-of-function mutations impact the relative abundance of both the wild-type and mutant C5 protein, we performed total proteome analysis on select E. coli strains during early-to-mid exponential phase growth at both the permissive (30°C) and sublethal temperatures (40°C) (Fig. 2A). Although relative abundances of the mutant Lon[E240K] protein and M1 RNA transcript in suppressor strain DA65195 remained similar to those of both the A49 parental strain (DA61546) and the MG1655 wild-type strain (DA5438) (Fig. 2A; Supplemental Fig. S4), suppressor strain DA65195 demonstrated an approximately two- to threefold increase in the relative abundance of the mutant C5A49 protein subunit as compared to the original ts A49 parental strain (Fig. 2A). This increase in C5A49 protein abundance coincides with the almost threefold increase in the suppressor strain's relative growth rate at the nonpermissive temperature (Fig. 1) and indicates that the mutant C5A49 protein is potentially a substrate of Lon protease.
This observation is consistent with findings from a similar study in which a mutant RNA polymerase σ subunit was found to be a direct substrate for Lon proteolysis and lon loss-of-function mutations suppressed the ts phenotype caused by the mutant rpoD800 allele (Grossman et al. 1983). Moreover, recent work suggests that Lon-mediated proteolysis may serve as a quality control measure to prevent the accumulation of nonfunctional RNA-binding proteins (RBPs). In particular, RNA-binding deficient mutants of ProQ and Hfq, the two major RBPs facilitating sRNA activity in bacteria, have been found to be targets of Lon degradation in Salmonella (El Mouali et al. 2021). Although naturally occurring lon loss-of-function mutations were not characterized in this study, the ProQ and Hfq mutants demonstrated increased stability and/or abundance in Δlon genetic backgrounds. Unlike the A49 suppressor strains carrying lon mutations, the mutant phenotypes of the ProQ- and Hfq-deficient strains could not be rescued by increasing the abundance of the mutant proteins, as the mutations directly impacted protein function rather than protein complex or holoenzyme assembly. Regardless, the present study potentially adds the E. coli C5 protein to the list of bacterial RBPs regulated by Lon-mediated quality control mechanisms.
Previous work demonstrating that M1 RNA acts as a chaperone to facilitate C5 protein folding, solubility, and stability further supports our proposed Lon-mediated degradation model (Son et al. 2015). When the C5 protein is misfolded or mutated (as is the case with the rnpA49 mutant allele), Son et al. found that weakened interactions with M1 RNA are sufficient to prevent aggregation of otherwise insoluble mutant C5 proteins and facilitate their proteolytic degradation. Investigations of this hypothesis in a lon-deficient strain of E. coli showed that mutant C5 proteins aggregated in the presence of M1 RNA. It is possible that this increased aggregation also indicates increased mutant C5 protein abundance because of the lon deletion, reinforcing our findings that suggest mutant C5 proteins are potentially targets for Lon-dependent proteolysis.
Because of its role as a chaperone and global regulator, it is also possible that Lon indirectly impacts C5A49 protein stability via the regulation of other proteases and/or pathways. Nevertheless, our understanding of RNase P metabolism in E. coli remains incomplete, and additional studies are required to further elucidate the pathways and mechanisms involved in regulating E. coli RNase P levels.
Concluding remarks
In this study, we report for the first time naturally occurring second-site compensatory mutations of the rnpA49 ts phenotype in E. coli. Although a relatively small screen, similar mutations were recovered multiple independent times across the 17 suppressor strains isolated, suggesting that these are frequent targets for compensatory evolution (Fig. 3). The compensatory mechanisms underlying the genomic amplifications of the regions containing the genes encoding either subunit of the mutant RNase P complex are well-established from past plasmid overexpression studies, and there is precedent for the role of RNase R loss-of-function mutations in the literature. However, follow-up studies are necessary to determine the exact mechanism behind the rnr suppressor mutations. Additionally, we found that E. coli frequently suppresses the ts defect caused by the rnpA49 allele via mutations that likely inactivate or alter the activity or expression of Lon protease and that select suppressor strains with lon mutations have increased C5A49 protein abundance. This finding suggests that the mutant C5A49 protein is potentially a substrate for Lon degradation and/or Lon protease is indirectly involved in the regulation of C5A49 protein levels. Previous work indicates that Lon protease plays a role in bacterial RBP quality control by specifically degrading mutant and/or nonfunctional RBPs and not their wild-type counterparts. Again, additional investigations are needed to understand Lon's role in RNase P metabolism in greater detail.
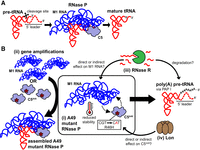
Schematic summary of the E. coli A49 second-site suppressor mutations isolated from this study. (A) Among other roles, RNase P is responsible for the 5′-end maturation of tRNAs by cleaving the 5′ leader sequences found on pre-tRNA transcripts. In E. coli strains carrying the wild-type rnpA allele, the C5 protein and M1 RNA subunits readily assemble to form the functional holoenzyme. (B) (i) The rnpA49 mutant allele encodes a ts mutation that compromises the assembly and stability of the RNase P holoenzyme at nonpermissive temperatures. This leads to an accumulation of pre-tRNAs with unprocessed 5′-ends, which are often polyadenylated by PAP I and targeted for degradation. (ii) The rnpA49 ts phenotype can be partially suppressed by increasing the gene copy number of either subunit of the mutant RNase P complex (rnpA49 or rnpB) via large genome amplifications, which shifts the equilibrium of assembly to favor the formation of the mutant RNase P holoenzyme. (iii) Rescue of the rnpA49 ts phenotype can also occur via the acquisition of loss-of-function mutations in RNase R. RNase R targets polyadenylated RNAs for degradation, thus inactivation of RNase R likely stabilizes the unprocessed, polyadenylated pre-tRNAs for aminoacylation and/or may also contribute to increased M1 RNA stability and improved assembly of the mutant RNase P holoenzyme. (iv) Lastly, rnpA49 suppressor mutations inactivating Lon protease increase C5A49 protein abundance to promote mutant RNase P holoenzyme assembly and stability.
Although the isolated strains grow comparable to wild-type E. coli (DA5438) at the permissive temperature, the suppressor mutations only partially complement the ts phenotype at the sublethal and nonpermissive temperatures, and growth rates are not restored to wild-type levels at the higher temperatures. This can potentially be attributed to the inherent costs of the suppressor mutations themselves. As mentioned previously, large genome amplifications can have substantial impacts on cell fitness (Nicoloff et al. 2019; Pereira et al. 2021), high-level C5 protein overexpression is known to be toxic to E. coli (Jovanovic et al. 2002), and the disruption of the various regulatory functions of RNase R and Lon likely have off-target effects on overall fitness. Furthermore, only single relevant suppressor mutations were identified in each strain. Therefore, it is possible that additional mutations could arise with subsequent evolution experiments to more fully complement the ts defect and/or compensate for the fitness costs brought about by the original suppressor mutations.
Apart from the overexpression of either RNase P subunit via genome amplifications, the other second-site suppressor mutations recovered were loss-of-function mutations in other enzymes, suggesting that there is no promiscuous and/or moonlighting activity from alternative E. coli RNases or other proteins that can compensate for the mutant RNase P. To determine if the overexpression of other E. coli ORFs can in fact rescue the rnpA49 ts defect, we introduced a plasmid library containing pooled clones from the ASKA collection (representing 4000 cloned ORFs from E. coli) into our E. coli A49 strain and screened for ORFs that could enable strain growth at the nonpermissive temperature (42°C) (Kitagawa et al. 2006). The gene encoding the wild-type C5 protein, rnpA, was the only “positive hit” recovered from this screen, further supporting our observation that complementation is primarily driven by overexpression of RNase P or loss-of-function mutations in other genes, rather than via gain-of-function mutations and/or increased expression of other potentially promiscuous enzymes (Supplemental Fig. S6).
It is worth noting that ts mutant strains may have phenotypes that are not directly linked to the function, regulation, and metabolism of the wild-type enzyme, as functional complementation studies need to be performed under temperature stress conditions, which can globally effect gene expression and exacerbate the mutant phenotype (Li et al. 2003). For example, the proposed role of yeast RNase P in the maturation of box C/D small nucleolar RNAs was initially identified in a yeast ts strain (Coughlin et al. 2008); however, this phenotype was not observed in a yeast RNase P RNA deletion strain complemented with an eukaryotic protein-only form of RNase P (PRORP) under conditions of normal temperature growth (Weber et al. 2014). This may also be the case with our observation that Lon protease is involved in regulating C5A49 protein abundance but has no effect on wild-type C5 protein levels. Nevertheless, recent work suggests that Lon protease might act on mutant RBPs as a quality control measure in bacteria; therefore, targeting mutant C5 protein variants for Lon-dependent degradation might be an innate control measure that is also found in wild-type E. coli genetic backgrounds (El Mouali et al. 2021). In addition, past studies with the ts A49 strain have revealed previously unknown RNase P substrates and functional connections between RNase P and different metabolic pathways (Li et al. 2003). Thus, despite the aforementioned caveats, investigations using ts strains can provide invaluable insights that can inform routes for further investigation in wild-type genetic settings and/or under more standard or relevant growth conditions.
For decades, the molecular, biochemical, and structural elements governing RNase P function have been the subject of hundreds of studies across different organisms. Nevertheless, the regulation of RNase P expression, its turnover and degradation, and the mechanisms underlying certain RNase P mutations are still not fully understood, especially in E. coli. Although informative, previous efforts to examine the suppression of the E. coli rnpA49 ts phenotype have been limited to “artificial” plasmid overexpression, gene deletion, and mutagenesis approaches. Our work complements these past studies and describes new mechanisms by which E. coli A49 could naturally compensate for the RNase P assembly and fitness defects caused by the mutant rnpA49 allele. All of the suppressor mutations isolated from our study appear to favor the formation and stability of the mutant RNase P holoenzyme (rnpA49 and rnpB amplifications, lon mutations) or the stability of the enzyme's pre-tRNA substrates (rnr mutations) to ultimately improve cell survival at the nonpermissive temperature. Our findings suggest novel links between E. coli RNase P and the regulatory pathways involving RNase R and Lon protease and implicate the mutant C5A49 RNase P protein subunit as a potential target for Lon proteolysis. This work provides direction for future investigations into RNase P regulation and metabolism in E. coli, especially in more wild-type genetic contexts and conditions.
MATERIALS AND METHODS
Bacterial strains and growth conditions
The ts rnpA49 allele was transferred from the original mutagenized E. coli A49 strain [E. coli Genetic Stock Center Strain 6465; Strain Designation N2020; F-, lacZ8(Am), trpA36, glyA34, argA52, rpsL999(strR), rnpA49(ts), ilvG866(Act)] (Apirion 1980) to an E. coli MG1655 background (F-, λ-, rph-1) using P1 transduction (Thomason et al. 2007). The presence of the ts mutation of interest was confirmed via lack of growth at the nonpermissive temperature (42°C) and local and whole-genome sequencing (MG1655 A49 strain designated as DA61546). For all experiments, strains were grown in lysogeny broth (LB Miller; 10 g/L NaCl, 10 g/L tryptone, 5 g/L yeast extract; Sigma-Aldrich) or plated on LB supplemented with 1.5% (w/v) agar (LB-agar; Millipore). All strains generated in this study were cryo-preserved at −80°C in LB supplemented with 10% DMSO. A49 strains containing additional gene deletions were constructed by P1 transduction of the kanR cassette from the corresponding KEIO collection strain and selection on LB-agar supplemented with 50 μg/mL kanamycin at 30°C (Baba et al. 2006; Thomason et al. 2007). See Supplemental Table S1 for the list of strains used, isolated, and/or constructed for this study.
Fluctuation assays
Twenty cultures of E. coli A49 strain DA61546 inoculated from independent colonies were grown overnight in 1 mL LB at 30°C with shaking at 190 rpm. To confirm the amounts of cells to be plated (see below), serial dilutions of each overnight culture were plated on LB-agar and incubated overnight at 30°C, and colonies were counted. For the fluctuation assay, ∼108 cells (50 µL) from each overnight culture were plated on LB-agar prewarmed to 42°C, and then incubated at 42°C (nonpermissive temperature) for a total of 48 h. Plates were monitored and colonies were counted after 16, 20, and 48 h incubation. After 48 h, a single colony was randomly chosen from each plate with growth, reisolated on LB-agar plates incubated at 30°C, retested for growth at 42°C, and saved for future studies. Mutation rate was determined using the bz-rates webtool, which implements the Ma–Sandri–Sakar maximum likelihood estimator (Hamon and Ycart 2012; Gillet-Markowska et al. 2015). This experiment was performed independently twice, for a total of 40 independent colonies from A49 strain DA61546 being assayed.
Whole-genome sequencing
Before whole-genome sequencing, the rnpA49 and lon loci of select ts revertant strains isolated from the fluctuation assays were PCR-amplified and Sanger sequenced (Eurofins) to confirm the presence of the rnpA49 and wild-type lon alleles (see Supplemental Table S4 for the list of oligos used for PCR and local sequencing). These strains were then grown for whole-genome sequencing in 1 mL LB medium overnight at 30°C with shaking at 190 rpm. Genomic DNA was extracted from 0.5 mL of each culture using an Epicentre MasterPure Complete DNA and RNA Purification Kit according to the manufacturer's protocol. DNA was resuspended in nuclease-free water (Sigma), concentrations were determined using a Nanodrop 1000 (Thermo Scientific) and Qubit 2.0 fluorometer (DNA Broad-Range kit, Invitrogen), and sequencing was performed in-house using an Illumina MiSeq platform with a Nextera XT DNA library preparation kit. Sequences were mapped to the E. coli MG1655 reference genome (GenBank U00096.3) and mutations were identified using CLC Genomic Workbench software (QIAGEN). The extent of DNA amplification was estimated by dividing the average sequence coverage of the amplifications by the average sequence coverage of the rest of the chromosome in CLC Genomic Workbench (QIAGEN) as described previously (Nicoloff et al. 2019). All raw sequence reads for the whole-genome sequencing from this study are deposited in the Sequence Read Archive (SRA) at the National Center for Biotechnology Information (NCBI) under BioProject Accession PRJNA1032291.
Growth assays
Growth curves were obtained using a Bioscreen C instrument (Oy Growth Curves AB, Ltd.). Overnight cultures (1 mL LB) of select strains were grown in triplicate from independent colonies at 30°C with shaking at 190 rpm and used to inoculate 1 mL fresh LB cultures (1 µL, 1:1000 dilution). From this dilution, two 300 µL aliquots were transferred to Bioscreen C honeycomb plates to serve as technical replicates. Plates were incubated in the Bioscreen C for 24 h (30°C assays, permissive temperature) or 4 days (40°C assays, sublethal temperature) with shaking, and the OD600 was measured every 4 min. The background OD600 measurements from wells containing blank, uninoculated medium were subtracted from the OD600 measurements. Growth rates were calculated from the linear slope of ln(OD600) during the exponential phase [ln(OD600) = −3.5 to −2.5], and the relative growth rate was calculated by dividing the growth rate of each suppressor or control strain by that of the parental A49 strain (DA61546) in the corresponding condition. The reported values represent the mean of at least three biological replicates, with two technical replicates each; the reported error is the standard deviation of the mean.
Proteomics sample preparation
Cultures (5 mL LB) started from two or three independent colonies from select suppressor and control strains were grown overnight at 30°C with shaking (190 rpm). These were used to inoculate two 500 mL flasks containing 60 mL LB to a starting OD600 of ∼0.06. For each strain/biological replicate, one flask was incubated at 30°C with shaking (190 rpm) and one flask was incubated at 40°C (water bath) with shaking (175 rpm), until an OD600 ∼0.2–0.25 was reached. Flasks were then cooled on ice for 10–20 min, cells were pelleted at 4500 rpm, 15 min at 4°C, washed twice with 1 mL phosphate-buffered saline (PBS; 8 g/L NaCl, 0.2 g/L KCl, 1.44 g/L Na2HPO4, and 0.24 g/L KH2PO4), and then stored at −80°C until proteomics experiments were performed.
The bacterial samples were homogenized by bead-beating using a FastPrep-24 instrument (MP Biomedicals) in SDS buffer, and protein concentration was determined using Pierce BCA Protein Assay Kit (Thermo Scientific) on a SpectraMax iD3 (Molecular Devices). Proteins were reduced in 10 mM dithiothreitol at 56°C for 30 min, then alkylated with 20 mM chloroacetamide at room temperature for 10 min. Protein samples were added to washed hydrophobic and hydrophilic Sera-Mag SpeedBeads (Carboxylate-Modified, Cytiva) in a bead-to-protein ratio of 10:1. The SP3 workflow was adapted from the protein and peptide cleanup for mass spectrometry protocol provided by the manufacturer. In short, proteins were precipitated on the beads by acetonitrile, washed with ethanol, and dried at room temperature. For digestion, 50 mM TEAB and LysC+trypsin (Promega) were added in a protein-to-enzyme ratio of 100:1, and incubation took place overnight at 37°C while shaking. An additional portion of trypsin (Thermo Fisher Scientific) was added and digested for 3 h. The peptide concentration was determined, and aliquots of 30 µg peptides were labeled in 100 mM TEAB using TMTpro 18-plex isobaric mass tagging reagents (Thermo Fisher Scientific) according to the manufacturer's instructions. Samples were pooled into one TMT-set each, and peptides were purified using a HiPPR detergent removal kit and Pierce peptide desalting spin columns (both Thermo Fisher Scientific), according to the manufacturer's instructions. The TMT-sets were fractionated by basic reversed-phase chromatography using a Dionex Ultimate 3000 UPLC system (Thermo Fisher Scientific). Peptide separations were performed using a reversed-phase XBridge BEH C18 column (3.5 μm, 3.0 × 250 mm, Waters Corporation) and a stepped gradient from 3% to 54% solvent B over 65 min followed by an increase to 80% solvent B at a flow of 200 µL/min. Solvent A was 25 mM ammonia and solvent B was 84% acetonitrile. The 96 primary fractions were combined into 15 final fractions, which were evaporated and reconstituted in 3% acetonitrile and 0.1% trifluoroacetic acid for LC-MS3 analysis.
LC-MS3 analysis
The above fractions were analyzed on an Orbitrap Eclipse Tribrid mass spectrometer equipped with the FAIMS Pro ion mobility system and interfaced with an Easy-nLC1200 liquid chromatography system (both Thermo Fisher Scientific). Peptides were trapped on an Acclaim Pepmap 100 C18 trap column (100 μm × 2 cm, particle size 5 μm, Thermo Fisher Scientific) and separated on an in-house packed analytical column (39 cm × 75 μm, particle size 3 μm, Reprosil-Pur C18, Dr. Maisch) using a stepped gradient from 4% to 80% acetonitrile in 0.2% formic acid over 75 min at a flow of 300 nL/min. FAIMS Pro was alternating between the compensation voltages (CVs) of −50 and −70, and the same data-dependent settings were used at both CVs. The precursor ion mass spectra were acquired at a resolution of 120,000 and an m/z range of 375–1500. Using a cycle time of 1.5 sec, the most abundant precursors with charges 2–7 were isolated with an m/z window of 0.7 and fragmented by collision-induced dissociation (CID) at 30%. Fragment spectra were recorded in the ion trap at a Rapid-Scan rate. The 10 most abundant MS2 fragment ions were isolated using multinotch isolation for further MS3 fractionation. MS3 fractionation was performed using higher-energy collision dissociation (HCD) at 55%, and the MS3 spectra were recorded in the Orbitrap at 50,000 resolution and an m/z range of 100–500.
Proteomic data analysis
Identification and relative quantification were performed using Proteome Discoverer (Thermo Fisher Scientific). The data were matched against the E. coli SwissProt database (5385 entries, May 2023). Database matching was performed using SEQUEST as a search engine with a precursor tolerance of 5 ppm and a fragment ion tolerance of 0.6 Da. Tryptic peptides were accepted with one missed cleavage; methionine oxidation was set as a variable modification and cysteine carbamidomethylation, TMTpro on lysine, and peptide N termini were set as fixed modifications. Percolator was used for PSM validation with a strict FDR threshold of 1%. For quantification, TMT reporter ions were identified in the MS3 HCD spectra with 3 mmu mass tolerance, and the TMT reporter intensity values for each sample were normalized on the total peptide amount. The SPS Mass Match threshold was set to 65%, and a SEQUEST XCorr threshold score of 2 was chosen. Only unique peptides were used for relative quantification, and proteins were required to pass a protein FDR of 5%. The mass spectrometry proteomics data have been deposited to the ProteomeXchange Consortium via the PRIDE partner repository with the data set identifier PXD047498 (Perez-Riverol et al. 2019). For the experiments using three biological replicates, the relative abundance of select proteins from select A49 suppressor strains was compared to that of the DA61546 A49 parental strain at the corresponding temperature using a two-tailed Mann–Whitney test in GraphPad Prism. Values were considered significantly different if P < 0.05.
RT-qPCR
As above, cultures (5 mL LB) started from three independent colonies from select suppressor and control strains were grown overnight at 30°C with shaking (190 rpm). These were used to inoculate two 500 mL flasks containing 60 mL LB to a starting OD600 of ∼0.06. For each strain/biological replicate, one flask was incubated at 30°C with shaking (190 rpm) and one flask was incubated at 40°C (water bath) with shaking (175 rpm), until an OD600 of ∼0.2–0.25 was reached. Flasks were then cooled on ice for 10–20 min, and 0.5 mL aliquots of each culture were combined with 1 mL RNAprotect Reagent (QIAGEN), incubated on ice for 5–10 min, and pelleted for 2 min at full speed and 4°C. Total RNA was extracted from the cell pellets using the RNeasy Mini Kit (QIAGEN) according to the manufacturer's protocol, and genomic DNA was removed from the RNA samples (∼4 μg) using the TURBO DNA-free Kit (Invitrogen). RNA integrity was confirmed via agarose gel electrophoresis, RNA concentration was determined using a NanoDrop 1000 and Qubit fluorometer (RNA Broad-Range assay kit, Invitrogen), and cDNA was synthesized using ∼500 ng of each DNase-treated RNA sample and the High Capacity cDNA Reverse Transcription Kit (Applied Biosystems). M1 RNA (rnpB) transcript levels (Loveland et al. 2014) were measured via quantitative PCR (qPCR) using the resulting cDNA as template, PerfeCTa SYBR Green FastMix (Quantabio), and an Illumina Eco Real-Time PCR system. Primers targeting housekeeping genes hcaT and cysG were used as internal normalization controls (see Supplemental Table S4 for oligos used; Zhou et al. 2011). Reactions lacking reverse transcriptase were also used as template for qPCR to confirm the effective removal of genomic DNA. Data for each strain at each temperature are represented by three independent biological replicates, and qPCR was performed with three technical replicates for each biological replicate. To determine statistical significance, M1 RNA transcript levels from select A49 suppressor strains were compared to those of the DA61546 A49 parental strain at the corresponding temperature using a two-tailed Mann–Whitney test in GraphPad Prism. Values were considered significantly different if P < 0.05.
ASKA library screen
A plasmid library (representing 4000 cloned ORFs from E. coli) from ∼460,000 pooled clones from the ASKA collection (Kitagawa et al. 2006) was isolated using an EZNA Plasmid DNA Mini Kit I (Omega Bio-Tek). Electrocompetent E. coli A49 strain DA61546 cells were prepared as described previously, but with incubation at 30°C (Knopp et al. 2019), and ∼300 ng of the ASKA plasmid library or the empty pCA24N ASKA plasmid were transformed into 40 µL cells via electroporation. Cells were immediately recovered in 1 mL LB at 30°C with shaking at 190 rpm for 1 h. After incubation, a dilution series for each transformation was plated on LB-agar supplemented with 20 μg/mL chloramphenicol, which was then incubated overnight at 30°C, and the colonies were counted to determine the transformation efficiency and subsequently estimate the diversity of the library covered (∼106 total transformants). The remaining transformant recovery cultures were plated on LB-agar supplemented with 20 μg/mL chloramphenicol and 1 mM IPTG and allowed to incubate for ∼30 h at 42°C. Any colonies that appeared were restreaked on LB-agar supplemented with 20 μg/mL chloramphenicol and 1 mM IPTG and incubated overnight at 42°C to confirm the rescue of the ts phenotype. Plasmids were isolated from these “positive hits” using the EZNA Plasmid DNA Mini Kit I (Omega Bio-Tek), sequenced using Sanger sequencing (Eurofins) to identify the cloned ORF recovered (Supplemental Table S4), retransformed into DA61546, and retested for growth at the nonpermissive temperature as described above.
SUPPLEMENTAL MATERIAL
Supplemental material is available for this article.
ACKNOWLEDGMENTS
This work was supported by grants from the Wallenberg Foundation (grant no. 2018.0168 to D.I.A.) and the Swedish Research Council (grant no. 2019-02091 to D.I.A.). The funders had no role in study design, data collection and analysis, decision to publish, or preparation of the manuscript. The quantitative proteomics analysis was performed by Evelin Berger at the Proteomics Core Facility, Sahlgrenska Academy, Gothenburg University, with financial support from SciLifeLab and BioMS. The authors thank Omar Mahmud Warsi and Hervé Nicoloff for their assistance with whole-genome sequencing and analysis, Roderich Römhild for help with growth curve analysis, and Nikolaos Fatsis-Kavalopoulos for help with data visualization and statistics. The thermometer image in Figure 3 is by vectorspoint from https://www.flaticon.com.
Footnotes
-
Handling editor: Eric Westhof
-
Article is online at http://www.rnajournal.org/cgi/doi/10.1261/rna.079909.123.
-
Freely available online through the RNA Open Access option.
- Received December 5, 2023.
- Accepted April 12, 2024.
This article, published in RNA, is available under a Creative Commons License (Attribution-NonCommercial 4.0 International), as described at http://creativecommons.org/licenses/by-nc/4.0/.
REFERENCES
MEET THE FIRST AUTHOR

Meet the First Author(s) is an editorial feature within RNA, in which the first author(s) of research-based papers in each issue have the opportunity to introduce themselves and their work to readers of RNA and the RNA research community. Arianne M. Babina is the first author of this paper, “Suppression of the Escherichia coli rnpA49 conditionally lethal phenotype by different compensatory mutations.” Arianne is a new Lecturer in Bacteriology in the School of Infection and Immunity at the University of Glasgow and an Affiliate Researcher in the Department of Medical Biochemistry and Microbiology at Uppsala University. Her research currently focuses on how bacterial genetic regulators (i.e., small proteins and noncoding RNAs) and subsequent regulatory networks arise de novo and evolve over time in response to environmental changes.
What are the major results described in your paper and how do they impact this branch of the field?
Since the 1970s, complementation of the temperature-sensitive phenotype caused by the rnpA49 mutant allele in E. coli has been an invaluable tool for investigating the biochemical and biophysical mechanisms underlying RNase P assembly and function. Although informative, previous studies often relied on the use of artificial approaches to examine complementation, including plasmid overexpression, gene deletion, and mutagenesis. As a result, there has been a knowledge gap regarding how E. coli naturally suppresses the fitness defects resulting from the rnpA49 mutation. In this study, we isolated and characterized naturally occurring second-site suppressor mutations of the temperature-sensitive phenotype in an E. coli strain carrying the rnpA49 allele. Briefly, we found that E. coli can partially compensate for the rnpA49 mutation via large genome amplifications of the regions encoding either subunit of the mutant RNase P complex (rnpA49 or rnpB) or via loss-of-function mutations in RNase R or Lon protease. These findings agree with past plasmid overexpression studies and importantly suggest novel links between the A49 mutant RNase P and the regulatory pathways involving Lon protease and RNase R, a crucial step toward elucidating the many still unknown players and pathways involved in E. coli RNase P regulation. It is my hope this work paves the way for follow-up studies in the field, particularly those involving RNase P regulation, turnover, and degradation.
What led you to study RNA or this aspect of RNA science?
I was serendipitously introduced to the RNA field during an undergraduate research opportunity, after which I became fascinated by the many diverse and highly specialized biological roles of RNA, which go far beyond that of simply encoding proteins. Since then, I have been involved in a number of projects aimed at discovering and characterizing different RNAs and their interactions with various proteins and small molecules in a range of model organisms, including plants, archaea, and both Gram-negative and Gram-positive bacteria. The remarkable plasticity of RNA has further expanded my interests to include evolution and fitness, particularly within bacterial model systems. Wherever my science takes me, I always somehow manage to bring it back to RNA!
During the course of these experiments, were there any surprising results or particular difficulties that altered your thinking and subsequent focus?
The entire project was a bit of a surprise, actually. Despite E. coli A49's use in numerous studies over the course of many decades, it was surprising (and exciting!) that none of the rnpA49 suppressor mutations we found were previously reported in the literature. Similarly, during a conversation with coauthor Leif Kirsebom about RNase P mutants, he mentioned that he occasionally came across a “slimy” looking E. coli A49 colony. It was really rewarding to eventually link that anecdotal observation to the mucoid Lon mutants that we now describe in this study.
Are there specific individuals or groups who have influenced your philosophy or approach to science?
I am forever grateful to Dieter Söll for giving me my first research opportunity as an undergraduate student and for introducing me to the field of “RNAs in bacteria.” After a particularly frustrating week of experiments, he once told me “Science doesn't always work the way you want it to.” Those reassuring words are still something I carry with me; I often think of them during a tough day in the laboratory, and I often find myself repeating them to colleagues and mentees in similar situations. It has become a mantra that both comforts and motivates me to continue moving forward in my research. Sure, it can be frustrating when something does not go as anticipated, but it is also the promise that things will not always work that encourages me to learn more, delve deeper, and become a better scientist.








