
Pervasive translation of Xrn1-sensitive unstable long noncoding RNAs in yeast
- Sara Andjus1,4,
- Ugo Szachnowski2,4,
- Nicolas Vogt2,
- Stamatia Gioftsidi2,
- Isabelle Hatin3,
- David Cornu3,
- Chris Papadopoulos3,
- Anne Lopes3,
- Olivier Namy3,
- Maxime Wery2,5 and
- Antonin Morillon2,5
- 1ncRNA, Epigenetic and Genome Fluidity, Institut Curie, PSL University, Sorbonne Université, CNRS UMR3244, F-75248 Paris Cedex 05, France
- 2ncRNA, Epigenetic and Genome Fluidity, Institut Curie, Sorbonne Université, CNRS UMR3244, F-75248 Paris Cedex 05, France
- 3Université Paris-Saclay, CEA, CNRS, Institute for Integrative Biology of the Cell (I2BC), 91198 Gif-sur-Yvette, France
- Corresponding authors: maxime.wery{at}curie.fr, antonin.morillon{at}curie.fr
-
Handling editor: Ling-Ling Chen
Abstract
Despite being predicted to lack coding potential, cytoplasmic long noncoding (lnc)RNAs can associate with ribosomes. However, the landscape and biological relevance of lncRNA translation remain poorly studied. In yeast, cytoplasmic Xrn1-sensitive unstable transcripts (XUTs) are targeted by nonsense-mediated mRNA decay (NMD), suggesting a translation-dependent degradation process. Here, we report that XUTs are pervasively translated, which impacts their decay. We show that XUTs globally accumulate upon translation elongation inhibition, but not when initial ribosome loading is impaired. Ribo-seq confirmed ribosomes binding to XUTs and identified ribosome-associated 5′-proximal small ORFs. Mechanistically, the NMD-sensitivity of XUTs mainly depends on the 3′-untranslated region length. Finally, we show that the peptide resulting from the translation of an NMD-sensitive XUT reporter exists in NMD-competent cells. Our work highlights the role of translation in the posttranscriptional metabolism of XUTs. We propose that XUT-derived peptides could be exposed to natural selection, while NMD restricts XUT levels.
Keywords
INTRODUCTION
Long noncoding (lnc)RNAs arise from the pervasive transcription of eukaryotic genomes (Jarroux et al. 2017). Although the debate on their functional significance is still ongoing (Ponting and Haerty 2022), some of them are now recognized as important RNA regulators involved in multiple cellular functions (Wery et al. 2011; Kopp and Mendell 2018; Yao et al. 2019; Statello et al. 2021). Consistent with their functional importance, their expression appears to be precisely controlled (Djebali et al. 2012; Lorenzi et al. 2021). Furthermore, the abnormal expression of lncRNAs is associated with human diseases, including cancers (Schmitt and Chang 2016; Renganathan and Felley-Bosco 2017; Saha et al. 2017). However, a full mechanistic description is still required to understand the raison d’être of lncRNAs in cells, as well as the molecular mechanisms regulating their expression.
By definition, lncRNAs are predicted to lack coding potential. However, this assumption has been challenged by several independent observations, showing that cytoplasmic lncRNAs can associate with ribosomes (Ingolia et al. 2011, 2014; Smith et al. 2014; van Heesch et al. 2014; Carlevaro-Fita et al. 2016). In fact, ribosome profiling (Ribo-seq) analyses identified small open reading frames (smORFs) on subsets of lncRNAs (Aspden et al. 2014; Ingolia et al. 2014; Smith et al. 2014; Chen et al. 2020; Chothani et al. 2022), resulting in some cases in the production of functional peptides (Slavoff et al. 2013; Zanet et al. 2015; D'Lima et al. 2017; Matsumoto et al. 2017; van Heesch et al. 2019).
Apart from such examples of functional lncRNA-derived peptides, which are scarce to date, the extent and biological relevance of lncRNA translation remain unclear. An emerging view in the field proposes that lncRNAs could constitute a reservoir of rapidly evolving smORFs which the cell could exploit as a source of potential genetic novelty by producing novel peptides (Ruiz-Orera et al. 2014). If beneficial, lncRNA-derived peptides could be selected, thereby contributing to the emergence of novel protein-coding genes through the evolutionary process known as de novo gene birth (Carvunis et al. 2012; Zhao et al. 2014; McLysaght and Hurst 2016; Schmitz et al. 2018; Van Oss and Carvunis 2019; Blevins et al. 2021; Papadopoulos et al. 2021).
In the budding yeast Saccharomyces cerevisiae, the idea that cytoplasmic lncRNAs can be translated has been suggested by the observation that they are degraded through nonsense-mediated mRNA decay (NMD). NMD is a conserved translation-dependent RNA decay pathway known to target mRNAs bearing premature termination codons (PTCs) in the ORF as well as “normal” mRNAs with a long 3′ untranslated region (UTR) (Muhlrad and Parker 1999; Amrani et al. 2004; Kebaara and Atkin 2009; Celik et al. 2017). Nevertheless, such “aberrant” transcripts represent only one type of NMD substrate (Smith and Baker 2015; Andjus et al. 2021). In fact, 70% of yeast cytoplasmic lncRNAs, known as Xrn1-sensitive unstable transcripts (XUTs) due to their extensive degradation by the cytoplasmic 5′-exonuclease Xrn1 (Van Dijk et al. 2011), were shown to be NMD substrates (Malabat et al. 2015; Wery et al. 2016). However, direct experimental evidence that XUTs are globally translated is still missing. Whether NMD-insensitive lncRNAs (30% of XUTs) are also translated, and if so, what are the molecular features allowing them to escape NMD, remain open questions. The molecular consequences of translation on the posttranscriptional regulation of XUT expression are also far from being understood. Finally, the output and biological relevance of such noncanonical translation events are still largely unknown.
Here, we show that XUTs are pervasively translated. We found that not only NMD-sensitive but also NMD-insensitive XUTs accumulate in wild-type (WT) yeast cells treated with translation elongation inhibitors. In contrast, XUT levels remain unchanged in stress conditions associated with global inhibition of translation initiation. Analysis of 5′ phosphorylated RNA decay intermediates indicates that stalled elongating ribosomes lead to impaired cotranslational degradation of XUTs by blocking Xrn1 in cells treated with translation elongation inhibitors. Ribo-seq experiments confirmed that both NMD-sensitive and -resistant XUTs are bound by ribosomes and identified ribosome-associated smORFs in their 5′-proximal portion. Mechanistic analyses on a candidate XUT demonstrated that its NMD-sensitivity depends on the length of the 3′ UTR downstream from the translated smORF. Finally, we show that the peptide resulting from an NMD-sensitive lncRNA reporter exists in WT cells, suggesting that despite the “cryptic” nature of the transcript, its translation results in a detectable product.
Altogether, our data support a model where translation occupies a central role in the metabolism of cytoplasmic lncRNAs, a rapid binding by ribosomes probably being the default route as they reach the cytoplasm.
RESULTS
Cytoplasmic lncRNAs accumulate upon translation elongation inhibition
The NMD-sensitivity of XUTs suggests that translation determines their decay. We anticipated that inhibiting translation would result in the accumulation of NMD-sensitive XUTs. To explore this idea, we treated exponentially growing WT cells with cycloheximide (CHX), a translation elongation inhibitor which prevents tRNA release and ribosome translocation (Garreau de Loubresse et al. 2014). Samples were collected at different time points after the addition of the drug, then total RNA was extracted and analyzed by northern blot. We observed that the NMD-sensitive XUT1678 and XUT0741 accumulate as soon as 5–10 min after CHX addition (Fig. 1A). This effect is reversible, as XUT1678 and XUT0741 levels decreased after washing the CHX-treated cells and returning them to growth in fresh medium without CHX (Supplemental Fig. S1A). In contrast, the 5′ ITS1 fragment, a well-known physiological target of Xrn1 (Stevens et al. 1991), did not accumulate in CHX-treated WT cells (Fig. 1A), indicating that CHX does not block the activity of Xrn1. Moreover, treating WT cells with anisomycin (ANS), which also inhibits translation elongation but at a different stage than CHX, led to a similar accumulation of XUT1678 and XUT0741 (Supplemental Fig. S1B), reinforcing our hypothesis of a general translation-dependent lncRNA decay process.
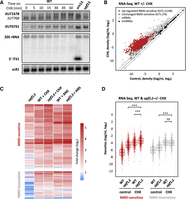
XUTs accumulate upon translation elongation inhibition, independently of NMD. (A) WT (YAM1) cells were grown to mid-log phase in rich (yeast extract–peptone–dextrose, YPD) medium at 30°C. CHX was then added at a final concentration of 100 μg/mL, and samples were collected at different time points. Untreated xrn1Δ (YAM6) and upf1Δ (YAM202) cells, grown under the same conditions, were used as controls. Total RNA was extracted and analyzed by northern blot. XUT1678, XUT0741, the 5′ ITS1 fragment (as well as the 20S pre-rRNA it derives from), and scR1 (loading control) were detected using 32P-labeled AMO1595, AMO1762, AMO496, and AMO1482 oligonucleotides, respectively. Note that probe AMO1595 also detects SUT768, an overlapping shorter stable isoform of XUT1678, which can be detected in WT cells (Wery et al. 2016). (B) Total RNA-seq was performed using total RNA extracted from exponentially growing WT (YAM1) cells (grown as above) treated for 15 min with CHX (100 μg/mL, final concentration) or with an equal volume of DMSO (control). The scatter plot shows the RNA-seq signals (tag densities, log2 scale) for the NMD-sensitive XUTs, mRNAs (light gray dots), and snoRNAs (black dots) in CHX-treated and control WT cells. Up-regulated (CHX/control fold change [FC] >2, P-value < 0.05) and unaffected NMD-sensitive XUTs are represented as red and dark gray dots, respectively (see also Supplemental Table S1). (C) Total RNA-seq was performed in WT (YAM1) and upf1Δ (YAM202) cells, including or not a treatment with CHX (15 min, 100 μg/mL final concentration) or ANS (30 min, 100 μg/mL final concentration). Densities were computed for NMD-sensitive and NMD-insensitive XUTs, using our previously published annotation (Wery et al. 2016). The sensitivity to NMD and/or CHX/ANS of each transcript is shown as a heatmap of the FC (log2 scale) relative to the corresponding control WT cells (treated for the same time with an equal volume of DMSO). Note in the first column that some XUTs (97) previously annotated as NMD-sensitive here show a fold enrichment <2 in the upf1Δ mutant (see Supplemental Table S1), probably reflecting some variability between independent experiments. (D) Same as above. The data are presented as densities (tag/nt, log2 scale) for NMD-sensitive and NMD-insensitive XUTs in control (DMSO) or CHX-treated WT (YAM1) and upf1Δ (YAM202) cells. (***) P-value < 0.001; (ns) not significant upon two-sided Wilcoxon rank-sum test (adjusted for multiple testing with the Benjamini–Hochberg procedure).
These results were extended at the genome-wide level using RNA-seq, showing that 1144 (94%) and 1012 (83%) NMD-sensitive XUTs significantly accumulate (FC > 2, P-value < 0.05) in WT cells treated with CHX (mean FC 7.4) or ANS (mean FC 8.5), respectively (Fig. 1B,C; see also Supplemental Fig. S1C,D; Supplemental Table S1). By comparison, CHX and ANS had a lower effect on cryptic unstable transcripts (mean FC 4.4 and 3.9, respectively; Supplemental Fig. S1E), which are primarily degraded in the nucleus by the exosome (Wyers et al. 2005; Neil et al. 2009; Xu et al. 2009), although some of them can escape nuclear decay and be exported to the cytoplasm (Thompson and Parker 2007).
Notably, 333 (75%) and 254 (57%) NMD-insensitive XUTs also accumulate in CHX- or ANS-treated WT cells, respectively (mean FC 5.0 and 6.2; Fig. 1C,D; see also Supplemental Fig. S1F; Supplemental Table S1), indicating that translation elongation inhibition affects XUTs independently of NMD. To further explore this idea, we performed RNA-seq in CHX-treated upf1Δ cells. This revealed that NMD inactivation and CHX have a synergistic effect on NMD-sensitive XUTs (Fig. 1D), their global levels being significantly higher in CHX-treated upf1Δ cells compared to untreated upf1Δ (P < 2 × 10−26, Wilcoxon rank-sum test) or CHX-treated WT cells (P < 2 × 10−26, Wilcoxon rank-sum test). Similar observations were made with ANS (Supplemental Fig. S1F). Importantly, this synergy between NMD inactivation and CHX- or ANS-induced translation elongation inhibition was only observed for the NMD-sensitive XUTs, but not for the NMD-insensitive ones (Fig. 1D; Supplemental Fig. S1F).
Together, these results show that XUTs globally accumulate in cells treated with translation elongation inhibitors, in a mechanism which is independent of NMD.
LncRNA levels remain globally unchanged upon stress-induced inhibition of translation initiation
At the molecular level, CHX or ANS act by arresting elongating ribosomes on their RNA substrates, a property which is widely exploited in Ribo-seq analyses (Lareau et al. 2014; Wu et al. 2019).
Interestingly, mRNA degradation is known to occur cotranslationally (Hu et al. 2009), and several reports have shown that the physical presence of ribosomes on the mRNA can interfere with its cotranslational degradation by Xrn1 (Pelechano et al. 2015; Serdar et al. 2016). This prompted us to investigate whether the accumulation of XUTs observed upon CHX or ANS treatment reflects a protective effect of the stalled ribosomes, sterically blocking Xrn1 (Fig. 2A). According to this model, XUTs should not accumulate when ribosomes are not loaded on the transcripts, i.e., in conditions where translation initiation is inhibited (Fig. 2A).
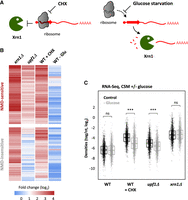
XUT levels remain unaffected upon translation initiation inhibition. (A) Working model showing the presumed effect of CHX-mediated inhibition of translation elongation (left) and of stress-induced inhibition of translation initiation (glucose starvation, right) on Xrn1-dependent degradation of XUTs (red). The red arrow on the XUT represents a smORF. (B) Total RNA-seq was performed in WT (YAM1), xrn1Δ (YAM6), and upf1Δ (YAM202) grown in complete synthetic medium (CSM). WT cells grown in the same conditions and then submitted to a CHX treatment or glucose starvation (−Glu) were also included. Densities (tag/nt) were computed for the 1470 XUTs significantly up-regulated in the xrn1 mutant grown in CSM (see Supplemental Fig. S3A), which were then separated according to their sensitivity to NMD (see Supplemental Fig. S3B). The sensitivity of each of these XUTs to CHX and glucose starvation is presented as a heatmap of the FC (log2 scale). As an indication, the sensitivity of these XUTs to Xrn1 (xrn1Δ/WT) and NMD (upf1Δ/WT) is also presented. (C) Box-plot showing the RNA-seq signals (densities, tag/nt, log2 scale) for the same set of XUTs as in B, in WT (YAM1), upf1Δ (YAM202), and xrn1Δ (YAM6) cells grown in CSM with glucose (control; black) or undergoing glucose starvation for 16 min (−Glucose; gray). An aliquot of WT cells was then treated with CHX for 15 min. (***) P-value < 0.001; (ns) not significant upon two-sided Wilcoxon rank-sum test (adjusted for multiple testing with the Benjamini–Hochberg procedure).
Glucose is the preferred source of carbon of yeast. Previous works have shown that glucose depletion results in a global inhibition of translation at the initiation level, with a rapid loss of polysomes (Ashe et al. 2000; Coller and Parker 2005; Castelli et al. 2011). More recently, the Tollervey lab showed that the stress response induced upon glucose starvation or heat-shock is associated with a displacement of translation initiation factors from mRNAs (Bresson et al. 2020).
To study the impact of translation initiation inhibition on XUTs, we performed RNA-seq using WT cells grown in a glucose-containing medium and then shifted for 16 min into a medium containing glycerol and ethanol (Supplemental Fig. S2A–C). Strikingly, XUT levels remain globally unchanged upon glucose depletion (mean FC 1.0), which is in sharp contrast with the effect of translation elongation inhibition (CHX, mean FC 5.3) observed in glucose-containing medium (Fig. 2B; see also Supplemental Table S1). Similarly, a reanalysis of RNA-seq data obtained in heat-shock conditions (Bresson et al. 2020) showed that this stress does not lead to a global accumulation of XUTs either (mean FC 0.9; see Supplemental Fig. S2D; Supplemental Table S1).
Importantly, we observed that the sensitivity of XUTs to CHX and to NMD significantly decreases upon glucose depletion (Fig. 2C), confirming that translation is inhibited in this condition. In contrast, the sensitivity of XUTs to Xrn1 was unchanged (Fig. 2C), indicating that Xrn1-dependent degradation of XUTs remains fully functional upon glucose depletion.
Altogether, these data show that XUTs do not accumulate in conditions where initial ribosome loading is impaired. This contrasts with the effect of the inhibitors of translation elongation, thereby suggesting that the stabilization of XUTs observed in these conditions is mediated by the stalled elongating ribosomes, sterically protecting them from degradation.
Translational landscape of yeast lncRNAs
A previous Ribo-seq analysis in upf1Δ yeast cells revealed 47 smORFs on 43 lncRNAs, providing a first proof-of-principle that lncRNAs are bound by ribosomes in S. cerevisiae (Smith et al. 2014). To define a more comprehensive translational landscape of yeast lncRNAs, we performed a new Ribo-seq experiment in WT and upf1Δ cells, producing two data sets for each genetic background: one in native conditions (untreated cells), and a second using CHX-treated cells (Fig. 3A).
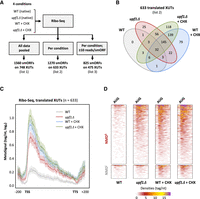
Translational landscape of XUTs. (A) Experimental scheme. Ribo-seq libraries were prepared from biological duplicates of WT and upf1Δ cells grown in native conditions (no CHX) or treated for 15 min with CHX (100 μg/mL final concentration). SmORFs (≥5 codons, starting with an AUG) were detected using the Ribotricer software (Choudhary et al. 2020), pooling all conditions together (list 1) or analyzing them separately (list 2). A third list was produced from list 2 upon application of a signal threshold (≥10 reads/smORF). See lists in Supplemental Table S2. (B) Venn diagram showing the number of XUTs detected as translated by Ribotricer (list 2) in each of the indicated conditions. See also Supplemental Table S2. (C) Metagene of Ribo-seq signals along the 633 translated XUTs (list 2). For each condition, the densities (tag/nt, log2) along the XUTs ±200 nt were piled up, then the average signal was plotted. The shading surrounding each line denotes the 95% confidence interval. (D) Heatmap view of the Ribo-seq signals (densities, tag/nt) from positions −50 to +150 relative to the AUG codon of the smORF showing the highest signal for the 510 NMD-sensitive and 123 NMD-insensitive XUTs detected as translated. A separate heatmap is shown for each condition.
As a first approach, we pooled our Ribo-seq data and searched for smORFs (≥5 codons, starting with an AUG codon) using Ribotricer (Choudhary et al. 2020), which directly assesses the 3-nt periodicity of Ribo-seq data to identify translated ORFs (see Materials and Methods). This led to the identification of 1560 translated smORFs on 748 XUTs (Fig. 3A; list 1 in Supplemental Table S2). We then repeated the same procedure, separating the conditions, which produced a refined list of 1270 smORFs from 633 XUTs, translated in at least one condition (Fig. 3A,B; list 2 in Supplemental Table S2). Applying an additional coverage threshold (≥10 reads/smORF in at least one condition) restricted the list to 825 smORFs for 475 XUTs (Fig. 3A; list 3 in Supplemental Table S2), which corresponds to the most robust candidates within the set of translated smORFs/XUTs, showing the highest levels of translation and being translated in at least one condition. Bearing in mind that lncRNA translation could be transient and occur at low levels, we decided to use the second list of 633 translated XUTs as a compromise for the descriptive analysis below. Figure 3C shows a metagene of the Ribo-seq signals for these XUTs. A similar metagene analysis for the other XUTs (not detected as translated) revealed that the signals are globally lower, suggesting that our analysis captured the XUTs displaying the highest levels of translation (Supplemental Fig. S3A).
Of these 633 XUTs, 510 are NMD-sensitive and 123 are NMD-insensitive (Fig. 3D; see also Supplemental Table S2). Interestingly, 297 of them are detected as translated in the absence of CHX treatment, essentially in the upf1Δ mutant (Fig. 3B). As one would expect, combining NMD inactivation and CHX treatment strongly improves the detection of XUT translation (Fig. 3B). Cumulatively, 411 XUTs were detected as translated in at least two data sets (Fig. 3B).
We note that for half of the XUTs (311/633), Ribotricer detected more than one smORF (Supplemental Fig. S3B). This could reflect the potential of several smORFs on the same XUT to attract the translation machinery and/or the existence of distinct isoforms for the same XUT, possibly encompassing different smORFs. Interestingly, for 75% of the translated XUTs, the smORF showing the highest Ribo-seq signal corresponds to one of the first three smORFs predicted in the XUT sequence (Supplemental Fig. S3C). This is consistent with the observation that ribosomes preferentially bind the 5′-proximal region of the translated XUTs (Fig. 3C). This profile is unlikely to be an artifact due to the CHX treatment, as it is also observed in the absence of the drug (Fig. 3C).
The smORFs detected on XUTs display a median size of 87 nt (Supplemental Fig. S3D), which is in line with the size of noncanonical (n)ORFs recently identified in yeast and the fact that these nORFs are globally shorter than canonical ORFs (Wacholder et al. 2023). On the other hand, these translated smORFs are globally longer than the other ORFs found in XUT sequences, but which are not translated (Supplemental Fig. S3D, P = 5.2 × 10−162, Wilcoxon rank-sum test). They are also longer than the three first ORFs found by chance when randomly shuffling the sequence of XUTs (Supplemental Fig. S3D, P = 6.2 × 10−178, Wilcoxon rank-sum test).
Together, these data show that a substantial fraction of XUTs carry smORFs that are experimentally detected as translated.
Features of translated lncRNAs
The annotation of translated smORFs paved the way for a deeper characterization of the translated XUTs. First, we wanted to strengthen our hypothesis that the stalled elongating ribosomes lead to impaired cotranslational degradation of XUTs by blocking Xrn1 in cells treated with translation elongation inhibitors. To that purpose, we reanalyzed published 5′ phosphorylated RNA-sequencing (5PSeq) data obtained in CHX-treated WT cells (Zhang and Pelechano 2021). This technique allows to follow cotranslational degradation by sequencing 5′ phosphorylated RNA fragments produced as Xrn1 is blocked by the last translating ribosomes (Zhang and Pelechano 2021). As shown in Figure 4A, the translated XUTs display the typical profile of cotranslational degradation, with a peak localized 14 nt upstream of the AUG codon (defined by Ribo-seq), followed by a signal showing a 3-nt periodicity (Pelechano et al. 2015; Zhang and Pelechano 2021).
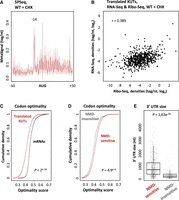
Features of translated XUTs. (A) Metagene of 5PSeq signals along positions −50 to +50 relative to the AUG codon of the smORF showing the highest signal for the translated XUTs. The peak at position −14 corresponds to the 5′ extremity of the footprint of the first ribosome, stalled at the level of the AUG codon. The shading surrounding the main line denotes the 95% confidence interval. Original data were obtained in CHX-treated WT cells by Pelechano's lab (Zhang and Pelechano 2021) and were retrieved from the NCBI Gene Expression Omnibus (accession number: GSE152375). (B) Scatter plot showing the Ribo-seq (TSS to TSS + 50) and RNA-seq signals (tag/nt, log2 scale) for the translated XUTs. The Pearson correlation coefficient (r) is indicated. (C) Average codon optimality score for the 633 translated XUTs (red) versus mRNAs (black), shown as a cumulative frequency plot. For XUTs with several annotated smORFs, we considered the smORF displaying the highest Ribo-seq signal. The indicated P-value was obtained upon the Kolmogorov–Smirnov test. (D) Same as above for the 510 NMD-sensitive (red) and 123 NMD-insensitive (gray) translated XUTs. (E) Box-plot showing the size of the 3′ UTR for the NMD-sensitive and NMD-insensitive translated XUTs. When several translated smORFs have been annotated for the same XUT, we considered the one with the highest Ribo-seq signal. The P-value was obtained upon a two-sided Wilcoxon rank-sum test.
In terms of cellular levels, we found that the translated XUTs are globally more abundant than the other XUTs, not only in CHX-WT cells but also in untreated WT cells (Supplemental Fig. S4A,B), and we observed a positive correlation between ribosome occupancy and XUT abundance (Fig. 4B). This suggests that Ribo-seq might be more prone to capture the translation of the most abundant XUTs.
Then, we analyzed the codon optimality within the translated smORFs of XUTs. Codon optimality affects translation elongation and has been associated with mRNA stability (Presnyak et al. 2015; Hanson and Coller 2018). In fact, unstable mRNAs are enriched in nonoptimal codons, which are supposed to be decoded less efficiently (Presnyak et al. 2015). In addition, “normal” mRNAs (i.e., devoid of PTC) yet targeted by NMD are also enriched in nonoptimal codons (Celik et al. 2017). This prompted us to determine the codon optimality score of the translated XUTs. Globally, we observed that it is significantly lower than for mRNAs (Fig. 4C; P < 2 × 10−16, Kolmogorov–Smirnov test). However, while NMD-sensitivity correlates with a lower codon optimality in the case of mRNAs (Supplemental Fig. S4C; P = 1.1 × 10−16, Kolmogorov–Smirnov test), XUTs display an opposite pattern, the average codon optimality score being significantly higher for the NMD-sensitive XUTs (Fig. 4D; P = 4.9 × 10−4, Kolmogorov–Smirnov test).
Finally, we computed the size of the 3′ UTR of the translated XUTs and observed that it is significantly longer for the NMD-sensitive XUTs than for the NMD-insensitive XUTs (Fig. 4E; median = 733 vs. 236 nt; P = 1.63 × 10−26, Wilcoxon rank-sum test), strongly indicating that the length of the 3′ UTR is a key determinant for XUT degradation by NMD, as for mRNAs. This is also consistent with previous observations supporting the role of the 3′-UTR length in dictating the sensitivity of yeast lncRNAs to NMD (Smith et al. 2014).
In summary, our results are consistent with a model where the accumulation of XUTs in CHX-treated cells is due to the stalled elongating ribosomes, impairing Xrn1-dependent cotranslational RNA degradation. They also reveal that the NMD-sensitive XUT displays a higher codon optimality score and a longer 3′ UTR in comparison to the NMD-insensitive ones.
The long 3′ UTR of XUT0741 is a major determinant of its NMD-sensitivity
In yeast, the length of the 3′ UTR is known to be critical for NMD activation (Muhlrad and Parker 1999; Amrani et al. 2004; Kebaara and Atkin 2009; Smith et al. 2014; Celik et al. 2017). The observation that the 3′ UTR is significantly longer for the NMD-sensitive XUTs compared to NMD-insensitive XUTs suggests that it might also constitute a key determinant of the NMD-sensitivity for XUTs (Fig. 4E). We therefore investigated this hypothesis, using the NMD-sensitive XUT0741 as a model candidate.
XUT0741 belongs to the top list of translated XUTs, with a single 5′-proximal smORF (15 codons), detected by each of our analyses (Supplemental Fig. S5A; Supplemental Table S2). This smORF is followed by a 1.3 kb long 3′ UTR containing multiple stop codons in the same frame (Fig. 5A). To explore the role of the 3′ UTR as a cis element determining its NMD-sensitivity, we designed six mutants of XUT0741 by mutating several of these in-frame stop codons, thereby progressively lengthening the smORF and shortening the 3′ UTR (Fig. 5A; see sequences in Supplemental File S1). These mutant alleles were integrated at the genomic locus in WT and upf1Δ strains. Their expression was then assessed by strand-specific RT-qPCR.
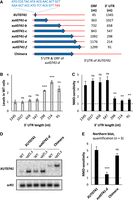
The NMD-sensitivity of XUT0741 depends on its long 3′ UTR. (A) Schematic representation of the native and mutant alleles of XUT0741. The transcript and the coding region are represented as a red line and a blue arrow, respectively. The sequence of the smORF in the native XUT0741 is indicated. The length of the coding region and of the 3′ UTR is shown beside each allele. The chimera construct was obtained by combining the 5′ UTR and coding region of the xut0741-d allele to the long 3′ UTR of the native XUT0741. (B) WT and upf1Δ cells expressing the different alleles of XUT0741 were grown to mid-log phase, at 30°C, in YPD medium. After total RNA extraction, the levels of each transcript were assessed by strand-specific RT-qPCR, and then normalized on scR1. The gray bars correspond to the levels of the different alleles of XUT0741 in WT cells (native XUT set to 1, indicated by the dashed line). (C) NMD-sensitivity of each allele of XUT0741, given by the ratio between the mean levels of each transcript in the upf1 mutant and the WT strain (see Supplemental Fig. S5B). Mean and SD values were calculated from three independent biological replicates. (**) P < 0.01; (***) P < 0.001; (ns) not significant upon t-test. The dashed line indicates a upf1Δ/WT ratio = 1 (i.e., no NMD-sensitivity). (D) WT and upf1Δ cells expressing the native XUT0741, the xut0741-d allele, and the chimera were grown as described above. Total RNA was extracted and analyzed by northern blot. The different alleles of XUT0741 and scR1 (loading control) were detected using 32P-labeled AMO3581 and AMO1482 oligonucleotides, respectively. (E) NMD-sensitivity of each allele, calculated from northern blot signals in WT and upf1Δ cells (see Supplemental Fig. S5D). Mean and SD values were calculated from three independent biological replicates. (***) P < 0.001; (ns) not significant upon t-test. Dashed line: upf1Δ/WT ratio = 1.
Our data show that the abundance of the XUT in WT cells and its NMD-sensitivity remain unchanged in the three first mutants (Fig. 5B,C; Supplemental Fig. S5B,C). However, when the 3′ UTR is shortened to 298 nt in the xut0741-d mutant (which is in the range of 3′-UTR size for NMD-insensitive XUTs; see Fig. 4E), we observed a significant accumulation of the mutated transcript, correlating with a significant decrease of its sensitivity to NMD (Fig. 5B,C; Supplemental Fig. S5B,C). Further shortening the 3′ UTR in mutants –e and –f did not aggravate these effects (Fig. 5B,C). Interestingly, these data are consistent with previous observations indicating that a 3′ UTR longer than 300 nt is critical to determine the sensitivity to NMD of mRNAs (Kebaara and Atkin 2009). Note that the mutations introduced in XUT0741 do not affect the NMD-sensitivity of another XUT (Supplemental Fig. S5C).
Thus, changing the length of the coding region relative to the 3′ UTR not only modifies the abundance of XUT0741 in WT cells, but also its NMD-sensitivity. To discriminate whether the latter depends on the length of the ORF or 3′ UTR, we constructed a chimera combining the extended ORF of the “NMD-resistant” xut0741-d mutant with the long 3′ UTR of the native XUT0741 (Fig. 5A). The fate of this chimera was then analyzed by northern blot. As expected, the corresponding RNA was longer than the native XUT (Fig. 5D). Importantly, the levels of the chimera in WT cells were undistinguishable from the levels of the native XUT0741 (Fig. 5D; Supplemental Fig. S5D), and in contrast to the xut0741-d mutant, the chimera and the native XUT display the same NMD-sensitivity (Fig. 5E). We therefore conclude that the NMD-sensitivity of XUT0741 is determined by its long 3′ UTR, which is consistent with the observation that the 3′ UTR for the translated NMD-sensitive XUTs is significantly longer (Fig. 4E).
The peptide resulting from an NMD-sensitive lncRNA reporter exists in NMD-competent WT cells
All the data described above indicate that translation occupies an important place in the metabolism of cytoplasmic lncRNAs. We asked whether peptides could be produced as these lncRNAs are targeted to NMD (for simplicity, we will systematically use the term “peptide” to refer to the product of the translation of a lncRNA, regardless of its size).
The fact that NMD is triggered as translation terminates makes it possible for the resulting translation product to be released, as previously observed for several PTC-containing mRNA reporters (Maderazo et al. 2000; Kuroha et al. 2009, 2013; Serdar et al. 2020; Chu et al. 2021). We explored whether this could occur with XUTs using the xut0741-b mutant (Fig. 5A), because this mutant displays the same NMD-sensitivity as the native XUT (Fig. 5C) but encodes a larger peptide, easier to detect by western blot. We decided to use this mutant as an artificial NMD-sensitive lncRNA reporter, following the insertion of a C-terminal 3FLAG tag (Fig. 6A; see the sequence in Supplemental File S1). We controlled that the insertion of the 3FLAG tag does not affect the NMD-sensitivity of the transcript (Fig. 6B; Supplemental Fig. S6A). Importantly, despite the very low abundance of the transcript in WT cells, at the protein level we observed a clear band at the expected size by western blotting, demonstrating that the encoded peptide is produced (Fig. 6C, lane 3), its level increasing in the upf1Δ context (Fig. 6C, lane 4).
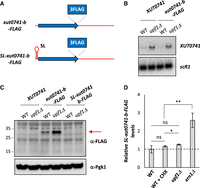
Detection of the peptide derived from an NMD-sensitive XUT reporter. (A) Schematic representation of the tagged xut0741-b alleles, using the same color code as in Figure 5A. (B) WT and upf1Δ cells expressing the native XUT0741 or the xut0741-b allele fused to a C-terminal 3FLAG tag (xut0741-b-FLAG) were grown to mid-log phase, at 30°C, in YPD medium. Total RNA was extracted and analyzed by northern blot. XUT0741 and scR1 were detected as described in Figure 1A. (C) WT and upf1Δ cells expressing the native XUT0741, the xut0741-b-FLAG or the SL-xut0741-b-FLAG alleles were grown as above. Protein extracts (40 μg) were separated by poly-acrylamide gel electrophoresis and then transferred to a nitrocellulose membrane. The size of the protein ladder bands is indicated on the left of the panel. Pgk1 was used as a loading control. (D) WT, upf1Δ and xrn1Δ cells expressing the SL-xut0741-b-FLAG allele were grown as above. For the WT strain, a sample of cells was also treated for 15 min with CHX (100 μg/mL, final concentration). After total RNA extraction, the levels of the SL-xut0741-b-FLAG transcript were assessed by strand-specific RT-qPCR, normalized on scR1 and set as 1 for the untreated WT condition (indicated by the dashed line). Mean and SD values were calculated from three independent biological replicates. (**) P < 0.01; (*) P < 0.05; (ns) not significant upon t-test.
To gain further insight into the relationship between translation and NMD-sensitivity of XUTs, we designed a construct where the translation of our NMD-sensitive lncRNA reporter is blocked in cis by a short stem–loop (SL) element, known to inhibit translation initiation with a minimal effect on RNA decay (Beelman and Parker 1994; Muhlrad et al. 1995). This SL was inserted into our reporter, upstream of the translation start site (Fig. 6A; see sequence in Supplemental File S1). A western blot showed that the production of the peptide is completely lost upon SL insertion, indicating that the transcript is no longer translated in this context (Fig. 6C, lanes 5,6). At the RNA level, this loss of translation correlates with a loss of the sensitivity to CHX and to NMD (Fig. 6D). In contrast, the transcript remained sensitive to Xrn1 (Fig. 6D; Supplemental Fig. S6B).
The experimental detection of the translation product derived from our reporter upon epitope-tagging paves the way toward the characterization of the yeast peptidome using mass spectrometry (MS), bearing in mind that this has been reported to be technically challenging due to the current limitations of peptidomics, especially for lowly expressed microproteins (Wacholder and Carvunis 2023). We failed to detect the peptide derived from the native XUT0741, although we were able to unambiguously detect a synthetic, labeled version of it (Supplemental Fig. S6C). We postulate that the abundance of the native peptide within the total extract is not sufficient enough to be detected by MS.
Besides their low abundance, the amino acid composition of XUT-derived peptides might also impede their detection by MS. In fact, the composition of XUT-encoded peptides differs from the proteins encoded by mRNAs, with an overrepresentation of hydrophobic residues (such as phenylalanine, isoleucine, and leucine), and an underrepresentation of lysine (Supplemental Fig. S6D). We also observed a spectacular depletion of the negatively charged residues (aspartate and glutamate) in the peptides encoded by XUTs (Supplemental Fig. S6D).
Notwithstanding the current lack of robust MS data, our results show that translation of an NMD-sensitive lncRNA reporter gives rise to a peptide in WT cells, while the transcript is being efficiently targeted to NMD. Furthermore, our mechanistic analysis confirms that the CHX- and NMD-sensitivity of XUTs is indicative of an active translation process.
DISCUSSION
Since their discovery, lncRNAs have been considered as transcripts devoid of coding potential, thereby escaping translation. However, accumulating experimental evidence leads us to reevaluate this assumption. In fact, lncRNAs copurify with polysomes in different models, including yeast (Smith et al. 2014) and human cells (van Heesch et al. 2014; Carlevaro-Fita et al. 2016; Douka et al. 2021). In addition, high-throughput sequencing of ribosome-bound fragments using Ribo-seq or related approaches has uncovered smORFs within lncRNAs (Ingolia et al. 2011; Aspden et al. 2014; Ingolia et al. 2014; Smith et al. 2014; Chen et al. 2020; Douka et al. 2021). Finally, several studies reported the identification of peptides resulting from the translation of smORFs carried on lncRNAs (Slavoff et al. 2013; Smith et al. 2014; Zanet et al. 2015; D'Lima et al. 2017; Matsumoto et al. 2017; Chen et al. 2020; Douka et al. 2021).
LncRNA expression in budding and fission yeasts is restricted by the extensive action of nuclear and cytoplasmic RNA decay machineries (Atkinson et al. 2018; Watts et al. 2018; Wery et al. 2018b; Szachnowski et al. 2019), including the 5′-exoribonuclease Xrn1, which degrades a conserved family of cytoplasmic lncRNAs referred to as XUTs (Van Dijk et al. 2011). Strikingly, previous works in S. cerevisiae revealed that most of them are targeted by the translation-dependent NMD pathway, suggesting that XUTs are translated and that translation controls their degradation (Malabat et al. 2015; Wery et al. 2016; de Andres-Pablo et al. 2017).
Here we report several observations supporting this hypothesis. We showed that the majority of XUTs accumulate in WT cells treated with CHX or ANS (Fig. 1), two drugs known to inhibit translation elongation but via different mechanisms (Garreau de Loubresse et al. 2014). Using Ribo-seq, we showed that a substantial fraction of XUTs are actually bound by ribosomes, and we identified ribosome-associated smORFs, which are mainly found in the 5′-proximal region of XUTs (Fig. 3). Mechanistic analyses at the level of a candidate XUT showed that its sensitivity to NMD is determined by the length of the 3′ UTR downstream from the translated smORF (Fig. 5). Finally, we showed that translation of an NMD-sensitive lncRNA reporter results in a peptide which is detected in WT cells, while the transcript is degraded through NMD (Fig. 6).
The fact that NMD-sensitive XUTs accumulate in the presence of translation elongation inhibitors reinforces our model of a translation-dependent decay process. However, the underlying molecular mechanism appears to be more complex than anticipated, as the accumulation of XUTs observed upon CHX/ANS treatment cannot be solely explained by the inability of the cell to trigger NMD when translation is inhibited. First, NMD-insensitive XUTs (which account for 30% of XUTs) also accumulate in the presence of CHX or ANS (Fig. 1C,D; Supplemental Fig. S1F). Second, stress conditions associated with global translation initiation inhibition do not recapitulate the stabilization effect of the translation elongation inhibitors (Fig. 2B,C; Supplemental Fig. S2D). Third, 5PSeq data obtained in CHX-treated cells show that the translated XUTs display the typical profile of cotranslational degradation (Fig. 4A). Together, these observations suggest that the global accumulation of XUTs induced by translation elongation inhibitors is mediated by the elongating ribosomes, stalled on their RNA template and sterically blocking Xrn1 independently of NMD.
Our Ribo-seq analysis allowed us to identify ribosome-associated smORFs for 510 NMD-sensitive XUTs and 123 NMD-insensitive XUTs (Fig. 3D), considerably extending the repertoire of translated lncRNAs in yeast (Smith et al. 2014; Wery et al. 2016). Importantly, our data also indicate that NMD insensitivity does not imply a lack of translation, and that the translational landscape of yeast lncRNAs extends beyond the scope of NMD. This is consistent with the observation that translation elongation inhibition impacts the decay of most NMD-insensitive XUTs (Fig. 1C,D; Supplemental Fig. S1F).
The number of smORFs/XUTs detected as translated depends on the stringency of the approach used to analyze the Ribo-seq signals (Fig. 3A), which is in line with the idea that lncRNA translation is transient and therefore more difficult to detect in comparison to canonical mRNA translation (Wacholder et al. 2023). Besides the low abundance of XUTs even in conditions where they are stabilized (NMD inactivation, CHX treatment), we imagine that the translation of many of their smORFs remains labile, probably reflecting the fact that they are rapidly and continuously evolving. This idea is supported by the idea that the vast majority of nORFs identified in yeast show no conservation (Wacholder et al. 2023). Furthermore, perhaps some constraints associated with canonical mRNA translation could be relaxed in the context of lncRNA translation as a strategy to maximize the potential for genetic novelty, which would be interesting from an evolutionary point of view. But the corollary is therefore the difficulty for us to detect such noncanonical translation events using pipelines that use the marks of canonical translation (e.g., use of an AUG initiator codon, predominance of one phase vs. the two others). The field is therefore in need of dedicated approaches and computational tools to reveal the exhaustive landscape of lncRNA translation.
Together with the observation that the NMD-sensitive XUTs display a longer 3′ UTR compared to the NMD-insensitive ones (Fig. 4E), the mechanistic analysis on the XUT0741 candidate highlights the critical role of the 3′ UTR in determining the NMD-sensitivity of XUTs, as it is also the case for mRNAs (Muhlrad and Parker 1999; Kebaara and Atkin 2009; Celik et al. 2017). However, even in the mutant with the shortest 3′ UTR, the NMD-sensitivity is not fully abolished (Supplemental Fig. S5B). One possible explanation is the existence of an alternative smORF, unaffected in our mutants. Supporting this hypothesis, we observed low Ribo-seq signals upstream of the detected smORF, overlapping the annotated TSS of XUT0741 (Supplemental Fig. S5A). Interestingly, XUT0741 TSS corresponds to the “G” of an “ATG” triplet, followed by 14 codons before the first in-frame stop codon (see sequences in Supplemental File S1). The production of multiple RNA isoforms from the same transcription unit is common in yeast (Pelechano et al. 2013), and we can imagine that any 5′-extended isoforms of XUT0741 would encompass this ATG and therefore carry this alternative smORF. Additional mechanistic analyses combined with RNA isoform profiling would be required to confirm this hypothesis. Nonetheless, the complexity of the yeast transcriptome, with the existence of multiple RNA isoforms displaying different boundaries, might possibly explain the detection of several smORFs per XUT and should be kept in mind when investigating how the position of smORFs relative to its annotated extremities could impact the fate of a XUT.
Independently of the presence of a PTC and of the length of the 3′ UTR, NMD-sensitive yeast mRNAs have been shown to be enriched in nonoptimal codons (Celik et al. 2017). Our data reveal that XUTs display an opposite pattern, the codon optimality score of the NMD-sensitive XUTs being significantly higher in comparison to the NMD-insensitive XUTs (Fig. 4D). More globally, the codon optimality score of XUTs is significantly lower compared to mRNAs (Fig. 4C), which would be consistent with their high instability (Presnyak et al. 2015). Together with the different composition in amino acids of XUT-derived peptides in comparison to proteins (Supplemental Fig. S6D), this also indicates that the primary properties of the smORFs of XUTs are distinct from the canonical ORFs of yeast, possibly reflecting that they are in a very early stage of selection, if only engaged in this process.
One interesting observation made in the context of this work is that the translation product resulting from an NMD-sensitive lncRNA reporter can be detected in a WT context, where NMD is functional. Whether this reporter can still be considered as a lncRNA or whether it becomes an mRNA since its ORF now extends over the arbitrary threshold of 300 nt reflects the debate on the fuzzy boundary between translated lncRNAs and mRNAs. In fact, if the distinction between the two classes was only a matter of ORF size, the MFA1 and MFA2 mRNAs (which encode the mating pheromone a-factor in S. cerevisiae) could be redefined as lncRNAs, since their ORF is 111 nt long (Michaelis and Herskowitz 1988). Besides the semantics, the idea that a peptide resulting from an NMD substrate could exist in the cell is plausible, since NMD is activated as translation terminates at the level of a “normal” stop codon (this is the position of this codon within the transcript which is sensed as “abnormal”). Previous studies using PTC-containing mRNA reporters have shown that the resulting peptides could be detected in yeast and human cells (Maderazo et al. 2000; Kuroha et al. 2009, 2013; Serdar et al. 2020; Chu et al. 2021). However, this has not been explored so far for cryptic lncRNAs. In this context, the observation we made here opens exciting perspectives regarding the possibility that lncRNA translation gives rise to peptides which can exist in the cell, despite the transcripts they originate from are degraded through NMD. Our data pave the way toward the future characterization of the yeast peptidome using MS, searching for native XUT-derived peptides, bearing in mind the actual technical limitations of this approach for the identification of short and lowly expressed peptides (Chothani et al. 2022; Wacholder and Carvunis 2023).
Overall, our data indicate that cytoplasmic lncRNAs are pervasively translated and globally behave like PTC-containing mRNAs, most of them being targeted by NMD. We propose that the resulting peptides could be used as raw material for natural selection.
De novo gene birth has been associated with adaptation to environmental stress (Arendsee et al. 2014). In addition, NMD is known to be repressed under a variety of stress conditions (Mendell et al. 2004; Gardner 2008). It is, therefore, tempting to speculate that despite the fact that the cell has evolved efficient pathways to degrade lncRNAs and control their expression (Smith and Baker 2015), these pathways can be down-regulated under certain conditions (e.g., stress) in order to sample the peptide potential hosted in these lncRNAs.
In conclusion, our work is coherent with the idea that translation plays a major role in the posttranscriptional metabolism of cytoplasmic lncRNAs, and that their definition as “noncoding” is probably not appropriate anymore to describe their actual status. Rather, they might be viewed as noncanonical translated (nct)RNAs oscillating between the “coding” and “noncoding” worlds.
MATERIALS AND METHODS
Yeast strains and media
The strains used in this study are listed in Supplemental Table S3. Mutants were constructed by transformation and were all verified by PCR on genomic DNA (see below).
Yeast cells were grown to mid-log phase (OD600 0.5) at 30°C in YPD medium or CSM, with 2% glucose. In the glucose starvation experiments, glucose was replaced by glycerol and ethanol.
5-Fluoroorotic acid (5-FOA) was used at a final concentration of 1 g/L on solid CSM plates. G418 (Geneticin; Gibco) was used at a final concentration of 100 µg/mL on solid YPD plates. CHX (Sigma) and ANS (Sigma) were used at a final concentration of 100 µg/mL.
Construction of xut0741 mutants
The xut0741-a, -b, -d and -f alleles, flanked by NaeI sites, were produced as synthetic gBlocks DNA fragments (IDT—Integrated DNA Technologies), and then cloned between the KpnI and XbaI sites of the pAM376 backbone vector (Szachnowski et al. 2019), giving the pAM594, pAM596, pAM598, and pAM600 vectors, respectively. The xut0741-c and xut0741-e mutants were constructed by site-directed mutagenesis from xut0741-d and then cloned into the same backbone vector, giving the pAM724 and pAM723 vectors, respectively. The sequence of each allele was verified by Sanger sequencing and is available in Supplemental File S1. The mutant alleles were excised from the pCRII vector using NaeI digestion and transformed into the YAM2831 strain (where the XUT0741/ADH2 locus has been deleted by URA3). After 1 d of growth on a nonselective medium, transformants were replicated on CSM + 5-FOA plates and incubated at 30°C for 4–5 d. The proper integration of the mutant alleles was confirmed by PCR on genomic DNA using oligonucleotides AMO3350-3351. UPF1 was deleted subsequently by transformation with the product of a PCR on YAM202 (upf1Δ::kanMX4) genomic DNA with oligonucleotides AMO2710-2711. The transformants were selected on YPD + G418 plates, and UPF1 deletion was verified by PCR on genomic DNA using oligonucleotides AMO190-2712.
The chimera-encoding plasmid (pAM726) was produced in two steps. First, the 3′ UTR of the native XUT0741 was amplified by PCR on YAM1 genomic DNA using oligonucleotides AMO3471-3382, and then cloned between the KpnI and XbaI sites of a pCRII-TOPO backbone, giving the pAM725 vector. Second, the sequence corresponding to the 5′ UTR and ORF of the xut0741-d mutant was amplified by PCR on YAM2854 genomic DNA using oligonucleotides AMO3379-3497, and then cloned between the KpnI and EcoRI sites of pAM725, giving the pAM726 vector. The sequence of the chimera allele was verified by Sanger sequencing (see Supplemental File S1). Plasmid digestion, transformation in YAM2831 cells, transformants selection and screening, as well as UPF1 deletion, were described above.
Carboxy-terminal 3FLAG tagging of xut0741-b was performed using an “overlap extension PCR” strategy. A first amplicon was produced by PCR on YAM2853 genomic DNA using oligonucleotides AMO3379-3530. A second amplicon was produced by PCR on the same DNA using oligonucleotides AMO3382-3531. After purification on agarose gel, the two amplicons (displaying a 28-bp overlap) were mixed and used as DNA templates for PCR using oligonucleotides AMO3379-3382. The final full PCR product was then digested by KpnI and XbaI and cloned in the same backbone vector as the other xut0741 mutants, giving the pAM728 plasmid (see Supplemental File S1 for insert sequence). All subsequent steps were as above.
The SL (GATCCCGCGGTTCGCCGCGG), previously shown to inhibit mRNA translation (Beelman and Parker 1994), was inserted into the 3FLAG-tagged xut0741-b allele using a similar “overlap extension PCR” strategy, involving the overlapping oligonucleotides AMO3550 (for the 5′ amplicon) and AMO3549 (for the 3′ amplicon), ultimately giving the pAM741 plasmid (insert sequence available in Supplemental File S1). All subsequent steps were as above.
XRN1 was deleted in strains YAM2908 (xut0741-b-3FLAG) and YAM2934 (SL-xut0741-b-3FLAG) by transformation with the product of a PCR on YAM6 (xrn1Δ::kanMX4) genomic DNA with oligonucleotides AMO34-35. The transformants were selected on YPD + G418 plates and screened by PCR on genomic DNA using oligonucleotides AMO3247-1669.
Total RNA extraction
Total RNA was extracted from exponentially growing cells (OD600 0.5) using a standard hot phenol procedure. Extracted RNA was ethanol-precipitated, resuspended in nuclease-free H2O (Ambion), and quantified using a NanoDrop 2000c spectrophotometer and/or a Qubit fluorometer with the Qubit RNA HS Assay kit (Life Technologies).
Northern blot
Ten micrograms of total RNA was separated on denaturing 1.2% agarose gel and then transferred to Hybond-XL nylon membrane (GE Healthcare). 32P-labeled oligonucleotides (listed in Supplemental Table S4) were hybridized overnight at 42°C in ULTRAhyb-Oligo hybridization buffer (Ambion). After hybridization, membranes were washed twice in 2× SSC/0.1% SDS for 15 min at 25°C, and once in 0.1× SSC/0.1% SDS for 15 min at 25°C. Membranes were exposed to Storage Phosphor screens. The signal was detected using a Typhoon Trio PhosphorImager and analyzed with version 10.1 of the ImageQuant TL software (Cytiva).
Strand-specific RT-qPCR
Strand-specific RT-qPCR experiments were performed from three biological replicates, as previously described (Wery et al. 2018a). The oligonucleotides used are listed in Supplemental Table S4.
Total RNA-seq
For each strain/condition, total RNA-seq was performed from two biological replicates. For each sample, 1 µg of total RNA was mixed with 2 µL of diluted ERCC RNA spike-in mix (1:100 dilution in nuclease-free H2O; Invitrogen). Ribosomal (r)RNAs were depleted using the RiboMinus Eukaryote v2 kit (Ambion). Libraries were prepared from the rRNA-depleted RNA using the TruSeq Stranded mRNA Sample Preparation Kit (Illumina) and the IDT for Illumina—TruSeq RNA UD indexes (Illumina). Paired-end sequencing (2 × 50 nt) was performed on a NovaSeq 6000 system (Illumina).
Total-seq data processing and analysis
Reads were trimmed and mapped on the S288C reference genome as previously described (Wery et al. 2023), with the addition of either ERCC RNA spike-in sequences or the Schizosaccharomyces pombe genome (ASM294v2, for the heat-shock data set). All subsequent analyses used uniquely mapped reads. Mapping statistics are presented in Supplemental Table S5.
Gene counts and tag densities were computed as previously described (Wery et al. 2023). The RNA-seq data obtained in the YPD medium (Fig. 1; Supplemental Fig. S1) were first normalized on the ERCC RNA spike-in signal; since snoRNA expression was not affected in the different mutants/conditions analyzed, snoRNA counts were then used for normalization as previously described (Wery et al. 2016, 2018b). The data obtained in CSM ± glucose (Fig. 2; Supplemental Fig. S2) were normalized on the ERCC RNA spike-in signal. For the heat-shock data set (retrieved from the NCBI Gene Expression Omnibus using accession number GSE148166), gene counts were normalized on the S. pombe spike-in RNA (Bresson et al. 2020).
XUTs were defined as up-regulated in a given condition when showing a greater than twofold enrichment in this condition versus the control, with a P-value < 0.05 (adjusted for multiple testing with the Benjamini–Hochberg procedure) upon differential expression analyses using DESeq2 (Love et al. 2014).
Ribo-seq library preparation
Ribo-seq analysis was performed from two biological replicates of YAM1 (WT) and YAM202 (upf1Δ) cells, grown to mid-log phase (OD600 0.5) at 30°C in YPD, then treated or not for 15 min with CHX (100 µg/mL, final concentration). For each sample, 250 mL of cells were harvested by centrifugation at room temperature and directly frozen in liquid nitrogen after supernatant removal.
Cells were lysed in 1× lysis buffer (10 mM Tris-HCl pH 7.4, 100 mM NaCl, 30 mM MgCl2) supplemented by 2× cOmplete Protease Inhibitor Cocktail (Roche), and ribosome-protected fragments (RPFs) were prepared as previously described (Baudin-Baillieu et al. 2016), except that the purification on sucrose cushion was performed before the digestion with RNase I (Ambion, 5 units/UA260). Biotinylated oligonucleotides (IDT—Integrated DNA Technologies) used for ribo-depletion are listed in Supplemental Table S4.
Libraries were then prepared from 10 ng of RPFs using the D-Plex Small RNA-seq kit for Illumina (Diagenode) and the D-Plex Unique Dual Indexes for Illumina—set A (Diagenode). The RPFs were diluted in a final volume of 8 µL before the addition of 2 µL of Dephosphorylation Buffer, 5 µL of Crowding Buffer, and 0.5 µL of Dephosphorylation Reagent. The samples were incubated for 15 min at 37°C. RNA tailing was performed by adding 1.5 µL of Small Tailing Master Mix (1 µL of Small Tailing Buffer + 0.5 µL of Small Tailing Reagent) to the dephosphorylated RNAs, and incubating the samples for 40 min at 37°C. The samples were transferred on ice for 2 min before the addition of 1 µL of the Reverse Transcription Primer (RTPH). The samples were denatured for 10 min at 70°C and then cooled down to 25°C at a 0.5°C/sec rate. A Reverse Transcription Master Mix (RTMM) was prepared by mixing 5 µL of Reverse Transcription Buffer and 1 µL of Reverse Transcription Reagent; 6 µL of this mix was added to the samples, which were then incubated for 15 min at 25°C. After adding 2 µL of Small Template Switch Oligo, the samples were incubated for 120 min at 42°C, then heated for 10 min at 70°C and finally kept at 4°C. For the PCR amplification, 20 µL of D-Plex Primer UDI and 50 µL of PCR Master Mix were added, then the following program was run: initial denaturation at 98°C for 30 sec; 10 cycles including 15 sec at 98°C followed by 1 min at 72°C; final incubation of 10 min at 72°C; hold at 4°C. The libraries were then purified using the Monarch PCR & DNA Cleanup Kit (NEB), using a 5:1 ratio of Binding Buffer:Sample. Purified DNA was eluted in 50 µL of nuclease-free H2O (Ambion). A second cleanup of the libraries was performed using 1 volume of AMPure XP beads (Beckman). Libraries were eluted in 20 µL of nuclease-free H2O (Ambion), and then quantified using the Qubit dsDNA HS assay (Invitrogen). Finally, the size and the molarity of each library were determined using a High Sensitivity D1000 ScreenTape in a 4200 TapeStation (Agilent Technologies).
Single-end sequencing (50 nt) of the libraries was performed on a NovaSeq 6000 system (Illumina).
Detection of translated XUTs/smORFs using Ribotricer
Unique molecular identifiers (UMIs) were extracted using umi_tools (Smith et al. 2017), and then used to discard PCR duplicates. Reads were trimmed using cutadapt v2.10 (Martin 2011), and then mapped using HISAT v2.0.0 (Kim et al. 2019), as above. Reads mapping on rRNA were discarded. Subsequent analyses only used uniquely mapped reads with a size comprised between 25 and 35 nt. Mapping statistics are presented in Supplemental Table S5.
Ribotricer 1.3.1 was used to extract translated ORFs (minimum length of 15 nt) based on S. cerevisiae genome annotation (including XUTs), using ATG as the start codon and a phase-score cutoff of 0.318, as recommended by the authors (Choudhary et al. 2020). The phasing of Ribo-seq data was also analyzed independently (Supplemental Fig. S7), upon prediction of the P-site for different k-mers (25-mers to 35-mers) using RiboWaltz (Lauria et al. 2018). List 1 of translated XUTs was obtained after pooling the bam files from all conditions. List 2 was obtained by analyzing each condition separately, pooling the bam files from the two biological replicates. List 3 was obtained from list 2, upon application of a coverage filter (at least 10 reads per translated smORF).
Protein extraction and western blot
Protein extracts were prepared from exponentially growing cells, using a standard method based on cell lysis with glass beads in “IP” buffer (20 mM HEPES pH 7.5, 100 mM NaCl, 0.5 mM EDTA, 1 mM DTT, 20% glycerol), supplemented with 0.05% NP40, 0.5X cOmplete Protease Inhibitor Cocktail (Roche) and 1 mM AEBSF.
Forty micrograms of total extracts were separated on NuPAGE 4%–12% Bis-Tris gel (Invitrogen) in 1× NuPAGE MOPS SDS running buffer (Invitrogen), and then transferred on a nitrocellulose membrane using iBlot 2 Transfer Stack system (Invitrogen), with program “0.”
The FLAG-tagged peptide and Pgk1 were detected using the anti-FLAG M2 (Sigma F1365; 1:1000 dilution) and anti-Pgk1 22C5D8 (Abcam ab113687; 1:10,000 dilution) monoclonal primary antibodies, revealed using the SuperSignal West Femto Maximum Sensitivity Substrate (Thermo Scientific) and the SuperSignal West Pico Chemiluminescent Substrate (Thermo Scientific), respectively, with a ChemiDoc Imaging System (BioRad). The secondary antibody was the antimouse IgG (whole molecule)–peroxidase antibody produced in rabbit (Sigma A9044; 1:10,000 dilution).
Mass spectrometry
MS was performed starting from crude extracts of upf1Δ (YAM202) cells grown to mid-log phase at 30°C in CSM medium with 0.1% proline as nitrogen source and containing 0.003% SDS, and then treated for 3 h with 50 µM MG-132 proteasome inhibitor (Sigma M7449).
The crude extracts were separated on 4%–12% bis-tris gels (Invitrogen NP0326BOX) in MES buffer (Invitrogen NP0002), then the region of the gel encompassing the small peptides fraction (1–10 kDa) was cut in bands of ∼2 mm and subjected to in-gel trypsin digestion, as previously described (Szabó et al. 2018), before submission to MS analysis. Trypsin-generated peptides were analyzed by nanoLC–MSMS using a nanoElute liquid chromatography system (Bruker) coupled to a timsTOF Pro Mass spectrometer (Bruker). Peptides were loaded on an Aurora analytical column (ION OPTIK, 25 cm × 75 m, C18, 1.6 m) and separated with a gradient of 0%–35% of solvent B for 100 min. Solvent A was 0.1% formic acid and 2% acetonitrile in water, and solvent B was acetonitrile with 0.1% formic acid. MS and MS/MS spectra were recorded from m/z 100 to 1700 with a mobility scan range from 0.6 to 1.4 V s/cm2. MS/MS spectra were acquired with the PASEF (parallel accumulation serial fragmentation) ion mobility-based acquisition mode using a number of PASEF MS/MS scans set as 10. MS and MSMS raw data were processed and converted into mgf files with DataAnalysis software (Bruker). Protein identifications were performed using the Mascot search engine (Matrix Science) against SwissProt and a noncanonical proteins homemade database. Database searches were performed using trypsin cleavage specificity with two possible missed cleavages. Carbamidomethylation of cysteines was set as fixed modification and oxidation of methionines as variable modification. Peptide and fragment tolerances were set at 10 ppm and 0.05 Da, respectively. Only ions with a score higher than the identity threshold and a false-positive discovery rate of <1% (Mascot decoy option) were considered.
As a control, we included a synthetic, labeled AQUA version of the peptide derived from XUT0741 (AQUA Basic Heavy grade, Thermo Scientific). The sequence of the synthetic peptide was MPY(I)TNTAEATMSTV, where (I) corresponds to a stable isotope isoleucine (+7 Da).
DATA DEPOSITION
Raw sequences generated in this work have been deposited to the NCBI Gene Expression Omnibus and can be accessed using accession number GSE203283. Genome browsers for the visualization of processed data are publicly accessible at http://vm-gb.curie.fr/mw4.
SUPPLEMENTAL MATERIAL
Supplemental material is available for this article.
ACKNOWLEDGMENTS
We thank Aaron Wacholder and Anne-Ruxandra Carvunis for sharing unpublished results and fruitful discussions. We are also grateful to all members of our laboratories for discussions and to our colleague Michael Schertzer for his careful reading of this manuscript and English revisions. High-throughput sequencing was performed by the ICGex NGS platform of the Institut Curie. Data management, quality control, and primary analysis were performed by the Bioinformatics platform of the Institut Curie. This work has benefited from the ANR “DNA-life” (ANR-15-CE12-0007) grant and the European Research Council “DARK-616180-ERC-2014” consolidator grant allocated to A.M., and from the ANR “Actimeth” (19-CE12-0004-02) grant allocated to O.N. The ICGex NGS platform of the Institut Curie is supported by grants ANR-10-EQPX-03 (Equipex) and ANR-10-INBS-09-08 (France Génomique Consortium) from the Agence Nationale de la Recherche (“Investissements d'Avenir” program), by the ITMO-Cancer Aviesan (Plan Cancer III) and by the SiRIC-Curie program (SiRIC Grant INCa-DGOS-465 and INCa-DGOSInserm_12554). S.A. has been supported by a PhD fellowship from PSL University and by the Fondation pour la Recherche Médicale (FRM).
Author contributions: M.W., I.H., O.N., and A.M. designed experiments. S.A., N.V., S.G., I.H., D.C., and M.W. performed experiments. U.S., C.P., and A.L. performed bioinformatics analyses. S.A., U.S., I.H., D.C., C.P., A.L., and M.W. analyzed the data. M.W. and A.M. designed the project. M.W. and A.M. supervised the project. A.M. and O.N. acquired funding. M.W. wrote the manuscript, with input from all authors.
Footnotes
-
↵4 Co-first authors.
-
↵5 Co-last authors.
-
Article is online at http://www.rnajournal.org/cgi/doi/10.1261/rna.079903.123.
-
Freely available online through the RNA Open Access option.
- Received November 29, 2023.
- Accepted February 15, 2024.
This article, published in RNA, is available under a Creative Commons License (Attribution-NonCommercial 4.0 International), as described at http://creativecommons.org/licenses/by-nc/4.0/.
REFERENCES
MEET THE FIRST AUTHORS


Meet the First Author(s) is an editorial feature within RNA, in which the first author(s) of research-based papers in each issue have the opportunity to introduce themselves and their work to readers of RNA and the RNA research community. Sara Andjus is the first author of this paper, “Pervasive translation of Xrn1-sensitive unstable long noncoding RNAs in yeast.” Sara is currently working as an R&D researcher in a deep tech startup. She obtained her PhD in October 2022 in Antonin Morillon's lab at Institut Curie under PSL Research University (Paris), working on the biogenesis and decay of long noncoding RNAs in yeast. Ugo Szachnowski was a bioinformatics engineer in Antonin Morillon's lab (Institut Curie) from 2018 to 2021, working on the role of lncRNA in yeast. He started a PhD in 2021 in the same team, working on the heterogeneity of lncRNA expression at the single-cell level in cancer.
What are the major results described in your paper and how do they impact this branch of the field?
Our paper brings to light the intriguing relationship between translation and the fate of unstable long noncoding (lnc)RNAs, challenging conventional notions. Using a simple eukaryotic organism, yeast, we show that RNAs, previously characterized as noncoding, can indeed interact with ribosomes and undergo active translation, consequently affecting how quickly they will be degraded. In fact, we demonstrate that while being bound by elongating ribosomes, lncRNAs are protected from the decay machinery. Using a lncRNA reporter, we show that despite decay machinery targeting the RNA to degradation, its translation product is released, exists in the cell and can be detected. Altogether, in this study, we comprehensively elucidate the functional implications for lncRNAs engaging with the translation machinery in a detailed, robust, and systematic manner. Our findings underscore the significance of translation in shaping the posttranscriptional metabolism of unstable lncRNAs, shedding light on previously unexplored aspects of RNA biology.
What led you to study RNA or this aspect of RNA science?
SA: Studying RNA and its various aspects, including its stability, diverse functions, and the enigmatic world of lncRNAs, despite being technically challenging, presented a captivating challenge and opportunity throughout my PhD journey. RNA molecules have multifaceted capabilities, and we still do not fully understand most of them!
US: Studying lncRNAs was like opening a door on a new world, as this field was not covered during my studies. So many are described and yet the function of most of them is unknown: It is an exciting and challenging field!
If you were able to give one piece of advice to your younger self, what would that be?
SA: It would be to believe more in myself. Also, a small tip—keep the “negative” results somewhere in the drawer—they might reveal to be more useful than you thought.
US: I would say more reading and more thinking. Often, I have been so eager to start running an analysis that I did not take enough time to think in detail of what I was doing, ending in doing it over—really frustrating.
Are there specific individuals or groups who have influenced your philosophy or approach to science?
SA: There are two quotes that followed me throughout my studies. The first one is a quote from Che Guevara: “Be realistic, demand the impossible.” The second one is a citation of Marie Curie: “Nothing in life is to be feared, it is only to be understood. Now is the time to understand more, so that we may fear less.”
US: I really enjoyed Stephen Jay Gould's Punctuated Equilibrium. Science never stops, and all ideas, even the most widely accepted, should be open to debate.
How did you decide to work together as co-first authors?
Having a team that combined expertise in both wet laboratory experimentation and bioinformatics was a must for this project. In fact, all the genome-wide data of this work could not speak by themselves without the bioinformatics analysis behind it. Our collaboration started with the first RNA-seq data set and since then was like a ping pong game. The complementary expertise of the two of us, but also of the other authors of this work, highlights the importance of interdisciplinary collaboration in scientific research, ultimately driving the success of a project—which is also reinforced by chocolate sharing in the afternoons!








