
Challenges to mapping and defining m6A function in viral RNA
- 1Department of Integrative Immunobiology, Duke University School of Medicine, Durham, North Carolina 27710, USA
- 2Department of Medicine, Duke University School of Medicine, Durham, North Carolina 27710, USA
- Corresponding author: stacy.horner{at}duke.edu
Abstract
Viral RNA molecules contain multiple layers of regulatory information. This includes features beyond the primary sequence, such as RNA structures and RNA modifications, including N6-methyladenosine (m6A). Many recent studies have identified the presence and location of m6A in viral RNA and have found diverse regulatory roles for this modification during viral infection. However, to date, viral m6A mapping strategies have limitations that prevent a complete understanding of the function of m6A on individual viral RNA molecules. While m6A sites have been profiled on bulk RNA from many viruses, the resulting m6A maps of viral RNAs described to date present a composite picture of m6A across viral RNA molecules in the infected cell. Thus, for most viruses, it is unknown if unique viral m6A profiles exist throughout infection, nor if they regulate specific viral life cycle stages. Here, we describe several challenges to defining the function of m6A in viral RNA molecules and provide a framework for future studies to help in the understanding of how m6A regulates viral infection.
VIRAL AND CELLULAR RNA MOLECULES ARE REGULATED BY m6A
Both viral and cellular RNA molecules contain chemical modifications that regulate their function and provide regulatory complexity beyond the primary or secondary RNA sequence. While there are over 170 chemical modifications that can be present on RNA molecules, likely in combinatorial profiles (Cappannini et al. 2024), here we focus on N6-methyladenosine (m6A), the most abundant internal mRNA modification. This modification is present in 20%–40% of all cellular mRNAs, which have a median methylation level of 40% (Dominissini et al. 2012; Meyer et al. 2012; Liu et al. 2023). While m6A can be found across an mRNA molecule, it is typically enriched in the 3′ UTR and near the stop codon. The role of m6A on an mRNA molecule is to regulate many aspects of mRNA function and metabolism, including RNA stability and mRNA translation (Flamand et al. 2023). These and other functions of m6A on a particular RNA molecule are controlled by RNA-binding proteins that detect the modification (Zaccara et al. 2019; Flamand et al. 2023). The major RNA-binding proteins that recognize m6A contain YTH domains, including the YTHDF family members, which alter mRNA stability or translation. Additionally, YTHDC1 and YTHDC2 can regulate mRNA splicing and nuclear export (Xiao et al. 2016; Roundtree et al. 2017), or mRNA translation and degradation (Hsu et al. 2017; Wojtas et al. 2017), respectively. In addition to these well-described m6A-binding proteins, numerous other RNA-binding proteins can sense the presence or absence of m6A, often due to changes in RNA secondary structure, to control various RNA functions (Zaccara et al. 2019).
Several mechanisms regulate how m6A is added to mRNA. For cellular mRNA, a large m6A-methyltransferase complex methylates nascent mRNA in the nucleus as it is being transcribed by RNA polymerase II (Garcias Morales and Reyes 2021; He and He 2021). Key members of this complex include the catalytic core proteins METTL3 and METTL14, which play a role in RNA substrate recognition. In addition, WTAP aids in proper localization and RNA targeting of the complex. Another distinct m6A-methyltransferase catalytic enzyme, METTL16, acts independently of METTL3 and METTL14. It modifies a small number of RNAs with specialized sequence and structural features (Pendleton et al. 2017; Warda et al. 2017). Importantly, m6A is reversible, and it can be removed from mRNA by the demethylases ALKBH5 and FTO (Flamand et al. 2023). The mechanisms by which viral or cellular RNA molecules are targeted for methylation by METTL3/METTL14 are governed by RNA intrinsic and extrinsic features (He and He 2021; Aufgebauer et al. 2024). RNA intrinsic features that regulate methylation include sequence motifs, such as the DRACH motif (D = A,G,U; R = A,G; H = A,C,U) and RNA secondary structure, while RNA extrinsic features would include trans-acting factors, such as RNA-binding proteins. For example, the exon junction complex can physically occlude the m6A-methyltransferase complex (Yang et al. 2022; He et al. 2023; Luo et al. 2023; Uzonyi et al. 2023), while transcription factors and specific histone modifications in chromatin can target the m6A-methyltransferase complex (Patil et al. 2016; Barbieri et al. 2017; Bertero et al. 2018; Huang et al. 2019; Wu et al. 2020). Indeed, we know that m6A can undergo regulated changes in response to stress as external stimuli, such as heat shock, hypoxia, or viral infection, can result in increased methylation of specific transcripts (Meyer et al. 2015; Zhou et al. 2015; Gokhale et al. 2020; Liu et al. 2023).
Similar to host mRNA, the presence of m6A on viral RNA molecules, the mRNAs derived from RNA or DNA viruses, or viral RNA genomes or antigenomes, enables tunable regulation of viral RNA function. The methylation of viral mRNA molecules transcribed in the nucleus is likely to have a similar role in regulating gene expression as cellular mRNA methylation, such as promoting RNA degradation or mRNA translation. Indeed, this has been demonstrated for the DNA virus Kaposi's sarcoma-associated herpesvirus (KSHV), where the interaction of YTHDF2 with the m6A-marked mRNA encoding ORF50 (RTA) negatively regulates its RNA stability, inhibiting viral replication (Hesser et al. 2018; Tan et al. 2018). In contrast, viral RNA genomes from RNA viruses that replicate in the cytoplasm typically do not enter the nucleus nor are they spliced. Thus, the methylation of viral genomic RNA may have different biological consequences. Overall, though, the molecular mechanisms that control how m6A functions on any given viral RNA molecule mirror those that act on cellular mRNA; both are regulated by differential m6A interactions with viral RNA-binding proteins (Girardi et al. 2021). These RNA-binding proteins include the antiviral RNA sensor RIG-I, whose interaction with the RNA from several viruses is prevented by m6A (Kim et al. 2020; Lu et al. 2020, 2021; Li et al. 2021; Qiu et al. 2021; Xue et al. 2021). m6A can also promote novel protein interactions with viral RNA. For example, in KSHV, the Tudor domain-containing protein SND1 interacts with m6A on ORF50 (RTA) to stabilize the viral transcript and promote lytic replication (Baquero-Perez et al. 2019). For the positive-strand RNA virus, hepatitis C virus (HCV), the presence or absence of m6A in the RNA of the E1 gene controls interactions between the cellular YTHDF binding proteins or the viral core protein to regulate infection (Gokhale et al. 2016). Thus, m6A on viral RNA can regulate a diverse set of viral or cellular RNA-binding protein interactions to control infection.
The ultimate role of m6A on infection by any virus depends on the biological consequence of methylation of that specific m6A site within the viral RNA. However, we know that viral RNA molecules can contain multiple m6A-containing regions and that m6A in these regions can have seemingly heterogeneous effects on infection, as seen in several viruses, including for SARS-CoV-2, human immunodeficiency virus-1 (HIV-1), and HCV (Gokhale et al. 2016; Riquelme-Barrios et al. 2018; Kim et al. 2020; Kim and Siddiqui 2021; Li et al. 2021; Liu et al. 2021; Zhang et al. 2021a). For example, m6A in the E1 gene of HCV RNA limits infection, while others found that m6A in the internal ribosome entry site (IRES) or NS5B gene promotes infection (Gokhale et al. 2016; Kim et al. 2020; Kim and Siddiqui 2021). One explanation for these seemingly opposing functions of m6A on HCV is that they do not occur simultaneously on all HCV RNA molecules, but rather that individual HCV molecules with specific m6A profiles make up a heterogeneous population of modified HCV RNA (Fig. 1A). In fact, we know that for HCV RNA, the packaged viral RNA and intracellular viral RNA have different levels of methylation (Gokhale et al. 2016). This suggests that there is likely more than one function of m6A on the viral RNA during infection. However, our current experimental studies on viral m6A RNA cannot uncouple these differential functions. Typically, experimental studies use a combination of an m6A mapping strategy that identifies the presence of m6A in viral RNA regions, along with the depletion of m6A-machinery proteins. However, to define how m6A on viral RNA regulates infection, we need to be able to generate single-nucleotide resolution m6A maps of full-length viral RNA molecules specific to viral life cycle stages and then selectively inactivate or deposit m6A at specific sites on the viral RNA during the viral life cycle stage of interest.
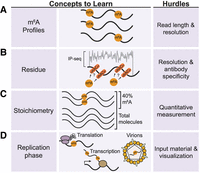
Challenges to m6A mapping of viral RNA. (A) Identification of viral m6A profiles requires the ability to map m6A residues and long-read sequencing to identify isoforms of modified viral RNA molecules. (B) Single-nucleotide mapping of viral m6A is needed for increased resolution that is difficult to obtain with immunoprecipitation (IP)-based m6A mapping strategies. (C) Quantifying the stoichiometry of m6A in a pool of expressed transcripts requires a fully quantitative method of measurement not possible using IP-based m6A mapping methods. (D) Measuring m6A in populations of viral RNA in different phases of viral replication is hampered by the limitation of enriching enough RNA to meet the input requirements of many m6A mapping methods.
METHODS TO MAP m6A ON VIRAL RNA AND THEIR LIMITATIONS
While m6A maps have been generated for many viruses (for review, see Baquero-Perez et al. 2021), limitations in experimental strategies for profiling m6A on RNA molecules have impacted our ability to understand how m6A, or any combination of internal RNA modifications, on viral RNA regulates infection. This is because most approaches to profile m6A only define the relative abundance and distribution of m6A across all viral RNA molecules in the infected cell. While a comprehensive review of m6A mapping methods is outside the scope of this article (Li et al. 2017; Hartstock and Rentmeister 2019; Owens et al. 2021; Zhang et al. 2024), here, we highlight several methods that have potential use for offering new insights into how m6A acts on viral RNA. These methods include antibody-dependent mapping strategies, which have been used extensively to map m6A in viruses, and antibody-independent methods, which have not been widely used by the virology community to date but have the potential to add enormous versatility to the toolbox of methods to study m6A in viruses.
The most widely used methods to map m6A typically use an m6A-specific antibody to enrich m6A-containing nucleotides during an RNA immunoprecipitation and then perform high-throughput sequencing of the immunoprecipitated and input RNA fractions (referred to as meRIP-seq or m6A-seq) (Dominissini et al. 2012; Meyer et al. 2012). In these antibody-dependent mapping methods, bulk mRNA is typically fragmented into ∼100 to 200 nt fragments before immunoprecipitation. Then, sequencing of the immunoprecipitated and input RNA reveals short RNA molecules enriched for m6A, referred to as m6A peaks. Within these peaks, the presence of a DRACH motif has been used to suggest where the m6A is located; however, we do know that non-DRACH motifs can also contain m6A (Liu et al. 2023; Miyake et al. 2023; Zhang et al. 2023). While meRIP-seq and other antibody-related m6A mapping methods are relatively straightforward for most researchers, they have several major limitations (McIntyre et al. 2020). First, they do not provide a single-base resolution map of m6A sites, and thus, it is impossible to know which motifs, DRACH or others, contain m6A (Fig. 1B). In fact, a recently developed antibody-independent m6A mapping method (discussed below) did not detect m6A sites in 30% of meRIP peaks, and only 45% of the quantifiably detected m6A sites fall within a meRIP peak (Liu et al. 2023). The reasons underlying this major discrepancy are unclear. Still, these findings suggest that while meRIP-seq can determine the methylation status of a particular mRNA molecule, determining the methylated site requires validation using an alternative method. A second major limitation of the antibody-based m6A mapping methods is that they are not quantitative, often providing little information on m6A stoichiometry (Fig. 1C). That is, it is impossible to know how many copies of an RNA molecule are modified. These methods are subject to false positives and negatives, as m6A antibodies have some non-m6A-related RNA preferences related to sequence and structure (Liu et al. 2018; McIntyre et al. 2020; Zhang et al. 2021b). Another limitation of meRIP-seq is that it requires large amounts of input RNA from millions of cells and requires a fragmentation step before the immunoprecipitation. Thus, meRIP-seq cannot be performed on low input levels of RNA, such as from virally infected patient samples, nor can it identify the methylation profiles of single viral RNA molecules that may have different roles during viral infection (Fig. 1D). While meRIP-seq has been a powerful tool for defining the biological roles of m6A, including during viral infection, to gain a comprehensive understanding of how m6A regulates viral infection, we need alternative approaches for mapping this modification.
One strategy to overcome the shortcomings of antibody-dependent sequencing methods is to use antibody-independent sequencing methods. Currently, several methods have been developed that either engineer cells to “mark” RNA that contains m6A or treat RNA harvested from cells, chemically or enzymatically, such that m6A residues can be identified (Cerneckis et al. 2024). In the category of engineering cells to mark m6A, mapping strategies that hold great promise to map m6A in viral RNA include RNA molecular recorders such as DART (deamination adjacent to RNA modification targets) or YTH-TRIBE (targets of RNA-binding proteins identified by editing) approaches (Fig. 2A; McMahon et al. 2016; Meyer 2019; Flamand et al. 2022). In these methods, the m6A RNA-binding domain of the YTHDF proteins is linked to the catalytic domain of a deaminase enzyme, either APOBEC1 for DART, or ADAR for TRIBE. These fusion proteins can indirectly mark YTH-bound m6A sites by generating unique mutational signatures, either C-to-U for DART or A-to-I for ADAR, which standard cDNA-based RNA sequencing methods can easily detect. The advantages of these methods over m6A antibody-dependent mapping approaches include their ability to detect m6A in low-abundance RNA and single cells, their potential to provide relative quantifications of m6A abundance, and their adaptability for m6A measurements of extracted viral RNA in vitro. Importantly, when combined with platforms that can sequence long-reads, for example, Nanopore or PacBio, they can reveal methylation signatures specific to individual RNA isoforms (Meyer 2019; Brannan et al. 2021; Flamand et al. 2022; Lin et al. 2023). There are some limitations to using these methods. First, they are indirect readouts of m6A, as they measure YTH domain-RNA binding, which does not always completely overlap with m6A (Wang et al. 2014, 2015). Second, they do not directly mark the m6A site, and so in the absence of a single-nucleotide reference m6A map of the RNA of interest, the actual methylated adenosine likely needs to be experimentally determined. In addition, the APOBEC or ADAR enzymes that generate the mutational signatures have RNA structure and sequence dependence, as well as potential off-target effects (Rosenberg et al. 2011; McMahon et al. 2016; Abruzzi et al. 2023), some of which can be overcome by evolved RNA molecular recorders (Lin et al. 2023). Finally, it is possible that expression of the DART or YTH-TRIBE constructs could indirectly inhibit infection by making C-to-U or A-to-I edits in viral RNA or inhibit infection by other activities (Xu et al. 2020). If this is the case, in vitro DART could be performed on extracted viral RNA (Meyer 2019). In summary, with careful design and optimization, molecular recording techniques hold potential for studying m6A in viral RNA.
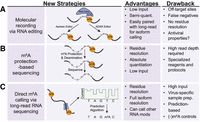
Recent strategies in m6A mapping and quantification that could be applied to viral infection studies. (A) “Molecular recording” techniques in which m6A readers are fused to RNA editing enzymes can address issues with input requirements, quantitative measurement, and isoform (when paired with cDNA long-read sequencing). (B) Protection-based sequencing techniques selective for m6A can directly map m6A at single-nucleotide resolution with absolute quantification. (C) Measuring m6A using long-read direct RNA sequencing can identify individual m6A RNA profiles across a population of transcripts at high resolution.
The antibody-independent m6A mapping techniques that use chemical or enzymatic methods to induce a mutation at an m6A site also have the potential for generating robust viral m6A RNA profiles (Zhang et al. 2024). For example, recent approaches have adopted a “protection-based” strategy whereby m6A is selectively protected from global deamination and thus can be measured by the presence of a “persistent A” signature following sequencing (Fig. 2B; Liu et al. 2023; Shao et al. 2023; Xiao et al. 2023). Importantly, these methods generate quantitative and single-nucleotide resolution maps of m6A, as they can measure the ratio of intact m6A relative to deaminated A. While some of these methods use evolved enzymes to perform the selective deamination that may not be widely available, one uses common chemical reagents to perform glyoxal and nitrite-mediated deamination of unmethylated adenosines (GLORI) (Liu et al. 2023). As these techniques have only recently been published, it remains to be seen how easily they are adaptable to common use; however, they hold great potential as methods that can be used for quantitative, single-nucleotide resolution m6A mapping of viral RNA.
The methods described for mapping m6A so far primarily rely on short-read sequencing data that cannot determine distant (>250 nt) m6A signatures on individual viral RNA molecules. One method that could revolutionize our understanding of how unique m6A signatures regulate the function of single viral RNA molecules is direct RNA sequencing, coupled with machine learning to readout modification, for example by using Nanopore (Liu et al. 2019; Abebe et al. 2022). In this method, polyadenylated RNA or target RNAs are loaded onto a flow cell as a cDNA–RNA hybrid after first-strand cDNA synthesis, and the native RNA strand is threaded through a protein pore that measures changes in ionic current as it passes through the application-specific integration circuit on the membrane. These changes are ultimately converted to base calls, based on probability scores, and RNA modifications are inferred using supervised learning-based tools to define these base-call error rate signatures (Fig. 2C). Theoretically, a spectrum of RNA modifications and their co-incidence could be mapped at site-specific resolution on a single viral RNA molecule. Indeed, the m6A profile of adenoviral RNA, which has a complex transcriptome with overlapping spliced mRNAs, was defined by using direct RNA sequencing (Price et al. 2020). Despite its great potential, direct RNA sequencing does have several limitations. First, the computational methods for accurately calling m6A by direct RNA sequencing are still being fully developed and rely on using bioinformatics to infer probabilities of m6A-marked bases (Zhong et al. 2023). As such, direct RNA sequencing requires large data sets and input material to bolster the computational power. Second, direct RNA sequencing is most powerful at predicting m6A sites when the experimental sample is compared to an m6A-depleted control sample, because its base-calling accuracy is not 100% (Liu et al. 2019; Price et al. 2020). In viruses, which often replicate as a quasispecies population, the m6A-depleted control sample is essential for accounting for this diversity of sequences that could be present (Domingo and Perales 2019). For positive-strand RNA viruses, in vitro transcribed viral RNA can serve as the m6A-depleted control sample. For other viral infections, m6A-depleted control samples could be generated by infecting METTL3 KO cells, which are hard to generate, or by rapidly degrading METTL3 and METTL14 by using auxin-inducible degrons or recently developed proteolysis-targeting chimera molecules (PROTACs) (Price et al. 2020; Chelmicki et al. 2021; Poh et al. 2022; Du et al. 2024; Marayati et al. 2024). Alternatively, RNA can be demethylated post-harvest by treatment with recombinant FTO protein (Xiao et al. 2018). Other limitations include the need for high input levels of RNA, the challenge of getting reads that cover the entire viral genome, and the need to enrich for viral RNA genomes that may have different RNA features than cellular RNA (Jain et al. 2022). For example, for HCV, which encodes a nonpolyadenylated RNA molecule of ∼9600 nt (Lindenbach and Rice 2005), these limitations mean that experimental variables such as sequencing depth and custom capture probes must be optimized. Overall, despite these limitations, direct RNA sequencing for mapping m6A in single viral RNA molecules will likely be of great use for defining the unique roles of m6A during viral infection.
METHODS TO TEST THE FUNCTION OF VIRAL m6A AND THEIR LIMITATIONS
Ultimately, the generation of high resolution of maps of m6A on viral RNA is a means to testing the direct function of m6A on viral infection. In early viral m6A studies, experiments to define the function of m6A typically deplete or overexpress the m6A machinery or use enzymatic inhibitors (Yankova et al. 2021). However, we know that any virologic outcomes that result from manipulation of the m6A machinery are undoubtedly a combination of altering the methylation profile of viral RNA and host mRNA molecules that encode factors that can regulate viral infection. For example, expression of the transcripts encoding interferon-stimulated genes and even interferon-β itself are regulated by m6A (Rubio et al. 2018; Winkler et al. 2019; McFadden et al. 2021). Beyond this, viral host factors can also be modified by or indirectly control m6A (Gokhale et al. 2020; Denolly et al. 2023). Thus, while experiments that manipulate the expression of m6A machinery can reveal if m6A plays any role in viral replication, resulting phenotypes cannot be used to conclude if m6A on viral RNA itself controls infection. Thus, to define the function of m6A in regulating a specific virus, one needs to uncouple changes in host methylation from those that occur on viral RNA, as we have done previously for HCV (Sacco et al. 2022). In addition, it is also important to remember that any phenotypic differences that arise from the manipulation of viral RNA m6A levels could vary between cell lines depending on the pool of m6A-selective RNA-binding proteins they express (Hesser et al. 2018).
To overcome the challenges of uncoupling host and viral m6A regulation, m6A must be directly altered on the viral RNA. To do this, one first needs to confirm that the viral RNA contains m6A and, in tandem, identify the location of m6A in the viral RNA. Then, one would measure the impacts on viral replication after synonymously mutating or inactivating a putative m6A site. Mutational strategies to inactivate a single m6A in viral RNA can be challenging because any nucleotide changes must be synonymous and the viral RNA structure must be preserved, ideally defined experimentally (Boerneke et al. 2019). Nonmutation-based strategies for selective m6A deposition and/or removal on viral RNAs would be ideal, and CRISPR–Cas-based strategies to target m6A-methylase and demethylase enzymes to specific viral RNA sequences hold great potential (Sun et al. 2022). In either strategy, as m6A can be deposited in clusters in an RNA molecule, it is unclear if one would need to inactivate all m6A sites in a region or gene to have a biological function (Tegowski et al. 2022; Liu et al. 2023). It may be important to analyze viral genetic conservation in nucleotide composition when deciding which m6A site to inactivate within a viral RNA. For both mutation- and nonmutation-based strategies for selective m6A removal, validation of changes in methylation should be confirmed. These validation methods could include meRIP-qPCR-based strategies with primers to amplify the region of interest, perhaps in the presence or absence of METTL3 inhibitors (Sacco et al. 2022), SCARLET (site-specific cleavage and radioactive-labeling followed by ligation-assisted extraction, and thin-layer chromatography) (Liu et al. 2013), the related, but easier technique called SCARPET (Mirza et al. 2024), or SELECT (single-base elongation- and ligation-based qPCR amplification method) (Xiao et al. 2018; Castellanos-Rubio et al. 2019). In summary, any attempts to alter m6A on a viral RNA require independent validation.
FRAMEWORK FOR IMPROVED METHODS TO DEFINE THE FUNCTION OF m6A IN VIRAL RNA MOLECULES
To define how m6A on viral RNA regulates infection, we need to be able first to generate single-nucleotide resolution m6A maps of full-length viral RNA molecules specific to viral life cycle stages, and then second, selectively inactivate or deposit m6A on the viral RNA during the viral life cycle of interest. None of the currently described m6A mapping techniques individually provide the resolution required to generate high-resolution m6A maps of viral RNA molecules. However, we propose integrating the data from multiple mapping techniques to improve our understanding of m6A on viral RNA. For example, chemical m6A mapping methods, such as GLORI, could provide a high-resolution, quantitative map of m6A on bulk viral RNA. This map could serve as a reference standard for all future experiments. Then, direct RNA sequencing, using m6A-deficient control samples, could be used to define single RNA molecule m6A maps, complete with phasing. While not experimentally described yet, the coupling of chemical labeling of m6A with long-read sequencing could aid in the resolution of m6A on a single viral RNA molecule, even removing some of the bioinformatic challenges to calling m6A using RNA sequencing (Cerneckis et al. 2024).
Generating viral life cycle–specific m6A maps is another challenge we have not overcome yet. This is because purifying viral RNA from a specific life cycle stage is laborious and requires a large amount of starting material. While it is straightforward in general to isolate the virion-associated viral RNA from the intracellular viral RNA, in the case of RNA viruses, isolating other life cycle stages, or even enriching for the antigenome, is not as straightforward, as in many cases the factors that directly regulate distinct life cycle stages are unknown, or straightforward methods for purifying these viral RNA molecules from cells do not yet exist. To this latter point, methods that can report on m6A in virally infected cells could be useful. The use of RNA molecular recorders such as DART or YTH-TRIBE to map m6A holds promise in this respect, especially when combined with viral mutants or depletion of host factors to trap the viral infection in specific life cycle stages (Meyer 2019; Flamand et al. 2022). Importantly, the DART or YTH-TRIBE can edit viral RNA in cells, and thus, less RNA input is required as there is no immunoprecipitation step or need for high-depth data to compute m6A probabilities. In addition, because A-to-I or C-to-U editing can easily be identified in sequencing data, these methods can be paired with cDNA long-read sequencing methods, allowing for quantitative profiling of m6A across an entire single RNA (Meyer 2019; Brannan et al. 2021). These indirect m6A mapping methods may not exactly mark the methylated adenosine; however, using the gold-standard reference maps generated by high-resolution techniques such as GLORI, one could undertake mutational studies to track each edit to methylated adenosine. Thus, we propose that a combination of m6A mapping techniques should be used to define viral RNA m6A profiles as the first step for defining how m6A on viral RNA can control viral life cycles.
Ultimately, the generation of single-nucleotide, full-length viral RNA genome, and life cycle–specific m6A maps of viral RNA molecules is the first step to elucidating novel roles for m6A in viral infection. Once we know this information, strategies that use selective m6A deposition and/or removal on viral RNAs, for example by using CRISPR–Cas-based strategies described above (Sun et al. 2022), will be an important next step. This information will be essential to define how specific viral m6A profiles are enriched during each viral life cycle stage to control infection, and how these m6A-modified viral RNAs themselves are regulated during infection. Ultimately, defining how distinct single viral RNA molecule m6A profiles or combinatorial modification profiles regulate viral infection will reveal new therapeutic targets for viral infection.
ACKNOWLEDGMENTS
We thank current and past members of the Horner lab for valuable feedback and discussion. Research on m6A in the Horner lab has been supported by the Burroughs Wellcome Fund and National Institutes of Health grant R01AI125416, with M.G.T. supported by ACS PF-21-117-01-RMC.
Footnotes
-
Article is online at http://www.rnajournal.org/cgi/doi/10.1261/rna.079959.124.
-
Freely available online through the RNA Open Access option.
This article, published in RNA, is available under a Creative Commons License (Attribution-NonCommercial 4.0 International), as described at http://creativecommons.org/licenses/by-nc/4.0/.








