
Understanding the redundant functions of the m6A-binding YTHDF proteins
- 1Department of Systems Biology, Columbia University Irving Medical Center, New York, New York 10032, USA
- 2Department of Pharmacology, Weill Cornell Medicine, Cornell University, New York, New York 10065, USA
- Corresponding authors: srj2003{at}med.cornell.edu, sz3145{at}cumc.columbia.edu
Abstract
N6-methyladenosine (m6A) is the most prevalent modified nucleotide in mRNA, and it has important functions in mRNA regulation. However, our understanding of the specific functions of m6A along with its cytosolic readers, the YTHDF proteins, has changed substantially in recent years. The original view was that different m6A sites within an mRNA could have different functions depending on which YTHDF paralog was bound to it, with bound YTHDF1 inducing translation, while bound YTHDF2 induced mRNA degradation. As a result, each YTHDF was proposed to have unique physiologic roles that arise from their unique binding properties and regulatory effects on mRNA. More recent data have called much of this into question, showing that all m6A sites bind all YTHDF proteins with equal ability, with a single primary function of all three YTHDF proteins to mediate mRNA degradation. Here, we describe the diverse technical concerns that led to the original model being questioned and the newer data that overturned this model and led to the new understanding of m6A and YTHDF function. We also discuss how any remaining questions about the functions of the YTHDF proteins can be readily resolved.
Keywords
INTRODUCTION
m6A is the most prevalent internal modified nucleotide in mRNA. m6A was initially detected in 1974 as part of studies that focused on understanding the nucleotide composition of mRNA (Desrosiers et al. 1974; Perry and Kelley 1974). The m6A biosynthetic enzyme METTL3 was subsequently purified and cloned by Bokar and Rottman in 1997 (Bokar et al. 1994, 1997), which enabled studies of the importance of m6A. These studies, which included the inactivation of the yeast homolog IME4, showed that m6A is important for sporulation (Clancy et al. 2002). Subsequently, in 2008 Fray and colleagues showed that loss of MTA, the METTL3 homolog in plants, led to highly selective defects in seed development (Zhong et al. 2008). This study proposed that m6A was likely to have highly selective effects on specific mRNAs, and thus helped prompt the development of transcriptome-wide m6A mapping methods. These mapping methods developed by us (Meyer et al. 2012) and the Rechavi group (Dominissini et al. 2012) revealed the key principles of m6A distribution in the transcriptome, including the selective enrichment in specific mRNAs, the presence of m6A in specific consensus sequences, and the enrichment of m6A in the coding sequence and 3′ UTR. Subsequent studies of METTL3 depletion by Hanna (Geula et al. 2015) and by Chang (Batista et al. 2014) in mouse embryonic stem cells demonstrated the conserved role of m6A in developmental processes that were originally found in plants.
In addition to these seminal studies, another breakthrough was the discovery of YTH domain proteins YTHDF2 and YTHDF3 as m6A-binding proteins by the Rechavi group (Dominissini et al. 2012). YTH domains had previously been identified as RNA-binding domains (Stoilov et al. 2002; Zhang et al. 2010), but the link to m6A was first established by Rechavi (Dominissini et al. 2012) and then further confirmed by others (Wang et al. 2014). Specifically, the YTHDF family is composed of three highly similar paralogs: YTHDF1, YTHDF2, and YTHDF3. The immediate question was how these YTH domain proteins mediate the effects of m6A in mRNA.
Notably, the function of m6A was identified by J. Darnell in seminal studies in 1978 (Sommer et al. 1978). In this study, using metabolic radionucleotide labeling of m6A, Darnell and colleges showed that mRNAs containing m6A were rapidly depleted in cells compared to mRNAs that lacked m6A. This work demonstrated that m6A promotes mRNA degradation. Thus, a major question was whether the YTHDF proteins mediate this and/or other functions.
Soon after the discovery of the YTH proteins as m6A readers, a series of studies from the He lab presented a model for how to understand the function of YTHDF proteins (Wang et al. 2014, 2015; Tirumuru et al. 2016; Shi et al. 2017). These studies had three major conclusions: First, they demonstrated that the YTHDF proteins bind to sometimes overlapping but often distinct m6A sites in the transcriptome. Thus, some m6A sites are bound by YTHDF1, some by YTHDF2, and others by YTHDF3. Second, these studies identified distinct functions and roles for the YTHDF proteins. YTHDF1 was found to regulate translation, YTHDF2 was proposed to regulate mRNA stability, while YTHDF3 was proposed to have a novel function as an mRNA-transferring protein, interacting with m6A-mRNAs as they exit the nucleus, and then facilitating their transfer to either YTHDF1 or YTHDF2 (Shi et al. 2017). Third, these studies proposed that YTHDF1, YTHDF2, and YTHDF3 have different physiologic roles that are due to their different RNA-binding sites and their different effects on their bound mRNAs.
These major studies provided the framework for understanding the function of YTHDF proteins, and indeed, the overall function of m6A. These studies showed that each individual m6A site needed to be studied in detail to determine whether YTHDF1, YTHDF2, or YTHDF3 was bound to the m6A site. Based on the specific YTHDF protein bound to the m6A site, the function of that m6A site could be understood. For example, if the m6A site binds YTHDF1, and not YTHDF2, its primary function would be translation enhancement.
However, a series of new studies from our lab (Zaccara and Jaffrey 2020), as well as another study from the Hanna lab (Lasman et al. 2020), as well as several follow-up studies by other groups (Kontur et al. 2020; Li et al. 2020; Arribas-Hernández et al. 2021; Niu et al. 2022), have led to a very different model for the function of m6A and their YTHDF readers. We found that all m6A sites have equal ability to bind YTHDF1, YTHDF2, and YTHDF3. We examined mRNAs that were previously described as uniquely bound to YTHDF1 or YTHDF2 and showed clear evidence that these mRNAs instead bind all three paralogs. Additionally, we found that YTHDF1 has no major effect on translation, thus contradicting this major concept related to m6A biology. Instead, we found that YTHDF1, along with YTHDF2 and YTHDF3, promote m6A-mRNA degradation.
As described in more detail below, we found that one of the original data sets that was used to support a translational role for YTHDF1 had a technical problem, possibly YTHDF1 overexpression, rather than the intended YTHDF1 knockdown. We also found technical problems and data analysis issues that were used to generate the original conclusion that YTHDF proteins bind different m6A sites. Additionally, the idea that YTHDF proteins have different functions and binding properties seemed counterintuitive since we and others found that the YTHDF proteins exhibit colocalization in cells, show similar protein-binding partners, and have nearly identical amino acid sequences and structures. Based on this, the natural expectation would be that each of these three proteins would have highly similar functions rather than highly divergent functions. These concerns stimulated our reinvestigation of the function of the YTHDF proteins.
The new studies led to a fundamentally different model about m6A and its function. We showed that m6A has one primary function in the cytosol: to promote degradation. Additionally, we showed that m6A sites generally have the same, rather than different functions. Thus, there is no need to determine the function of each m6A site in an mRNA. m6A is therefore primarily a degradation signal and maintains mRNAs at low levels in cells. This work led to a shift in thinking from m6A as an unpredictable modification that could impact translation or stability, to the new understanding that m6A is a mediator of mRNA degradation.
In this Perspective, we discuss the evidence in favor and against the initial model of m6A and YTHDF function, and the newer data that provided a new model for YTHDF function. We also discuss divergent opinions that may remain about m6A and ways that the binding properties and function of YTHDF proteins can be experimentally reconciled.
YTHDF PROTEINS—EARLY EVIDENCE THAT THEY BIND DIFFERENT m6A SITES
The binding sites of YTHDF proteins in the transcriptome were established using RNA immunoprecipitation and sequencing (RIP-seq) and CLIP-seq (cross-linking and immunoprecipitation sequencing). The CLIP method is widely used to study RNA-binding proteins and relies on an in-cell UV cross-linking step so that RNA-binding proteins are cross-linked to their bound RNAs (Ule et al. 2003). Then, cells are lysed and any RNA-binding protein of interest is immunoprecipitated along with any bound RNA. Nonspecifically bound RNA is removed by denaturing gel electrophoresis followed by transfer to a protein-binding nitrocellulose membrane, which inefficiently binds RNA unless it is cross-linked to the RNA-binding protein. The cross-linked RNA is recovered from the membrane and subjected to deep sequencing. The sequencing reads are aligned to the transcriptome and form read-pileups, which can be used to identify putative binding sites for the RNA-binding protein of interest.
Shortly after the identification of YTHDF proteins as m6A readers, the He group performed CLIP experiments to establish the binding sites of each of the YTHDF proteins (Wang et al. 2014, 2015; Tirumuru et al. 2016; Shi et al. 2017). RIP-seq and CLIP-seq experiments were performed in HeLa cells and resulted in the same conclusion, that is, that YTHDF1, YTHDF2, and YTHDF3 exhibited partially overlapping, but generally distinct binding patterns throughout the transcriptome (Fig. 1A). Many mRNAs only bind one of the YTHDF paralogs, some mRNAs bind two, and some mRNAs bind all three YTHDF proteins (Wang et al. 2015; Shi et al. 2017).
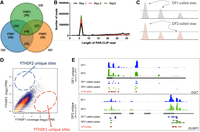
YTHDF proteins show similar binding to all m6A sites. (A) Shown is a Venn diagram depicting the highly selective nature of YTHDF paralog binding to different transcripts. This image, using data taken from Shi et al. (2017), shows the previous view that some transcripts are uniquely bound by YTHDF1, YTHDF2, or YTHDF3, while others are bound to two or three of the paralogs. This model implies that the function of each m6A site will depend on the YTHDF protein that binds to it. (B) Shown is a quantification of read length size from the PAR-CLIP data that identified YTHDF-binding sites in the HIV genome (Tirumuru et al. 2016). The PAR-CLIP data were comprised of extremely short reads, which cannot be reliably mapped to the transcriptome, and which explained why this study initially reported highly different binding patterns of the YTHDF proteins. (C) Schematic representation of the problem with using cutoff thresholds to call peaks. Shown are two examples of CLIP data sets in which three peaks are clearly seen. When a threshold is applied, some peaks fall below the threshold while others are above the threshold. However, qualitatively the binding properties appear essentially identical. If the threshold is lowered slightly, different peaks are called resulting in a different conclusion, that is, that the patterns of binding in the two data sets are identical. This highlights how threshold cutoffs can give the misleading conclusion that specific m6A sites are only bound by YTHDF1 (the middle site), and certain sites are only bound by YTHDF (right site). (D) Reexamination of YTHDF1 and YTHDF2 binding properties using a correlation analysis. In this approach, every m6A site in the transcriptome was analyzed, and the read counts from a YTHDF1 CLIP data set and a YTHDF2 CLIP data set were used to determine the position on the scatterplot. As can be seen, a strongly linear correlation is seen, demonstrating that every m6A site exhibits highly correlated levels of YTHDF1 and YTHDF2 binding. The dashed circle shows the region in which m6A sites would be located if they selectively bound YTHDF2 or YTHDF1. These sites were not found, demonstrating that there are no YTHDF1-unique or YTHDF2-unique sites. (E) Shown is CLIP data from HEK293 cells for YTHDF1, YTHDF2, and YTHDF3 on two transcripts proposed to be a YTHDF1-unique (top) and YTHDF2-unique transcript (bottom). As can be seen, the overall pattern of binding is essentially the same. Shown in red is the miCLIP data set which shows the location of m6A peaks, with sites indicated as red dots (taken from Zaccara and Jaffrey 2020).
Another study by the He group provided separate evidence that YTHDF proteins can bind different m6A sites. In this study, CLIP experiments were performed in HeLa cells that were infected with the human immunodeficiency virus in order to map binding sites of each YTHDF protein in the HIV RNA genome (Tirumuru et al. 2016). In this study, the precise locations of YTHDF1, YTHDF2, and YTHDF3 binding in the HIV genome were determined and clearly showed different binding sites (Tirumuru et al. 2016). This study further highlighted the stark differences in the binding sites of the YTHDF proteins in RNAs.
HOW CAN NEARLY IDENTICAL YTH DOMAINS BIND DIFFERENT m6A SITES?
In our view, the idea that YTHDF proteins would bind different m6A sites was counterintuitive based on our understanding of m6A and the high sequence identity of the YTHDF proteins. Each m6A site might bind a different reader if m6A sites were found in different sequence contexts. However, our mapping studies (Linder et al. 2015) had demonstrated that m6A has a single sequence motif, consistent with earlier biochemical work (Harper et al. 1990). Thus, it is not clear how any m6A site might be recognized as different from any other m6A site.
Another problem with this model is the similarity in the YTH domains of YTHDF1, YTHDF2, and YTHDF3. The three YTH domains are nearly identical in amino acid sequence, and have complete amino acid identity on the surface of the YTH domain that faces the RNA. If the YTH domains have different binding preferences, then they should have different sequences and structures in their YTH domains that could mediate these differences. Indeed, a more recent comparative analysis of the three YTH domains using molecular dynamic simulations further shows that the YTH domains are essentially identical in structure and are expected to have the same binding specificity (Li et al. 2020).
Conceivably the YTHDF proteins could bind different m6A sites due to the presence of a second nearby motif which might confer YTHDF paralog specificity. However, the crystal structures of the YTH domain show a single RNA-binding domain, which is the m6A-binding domain (Luo and Tong 2014; Li et al. 2020). No other RNA-binding surface was evident. Thus, an alternative model in which binding specificity is mediated by m6A along with more distal sequences appeared unlikely.
A PARTIAL EXPLANATION—CLIP DATA SETS COMPRISED OF 5 NT-LONG READS
We therefore asked how it would be possible for YTHDF1, 2, and 3 to bind to different m6A sites. When we started examining this question, another group published a report that identified a technical flaw in the CLIP-seq analysis of YTHDF binding to the HIV genome. Cullen and colleagues reanalyzed the CLIP data and showed that the reads were typically 5 nt in length, with a range of 5–7 nt in length, rather than the traditional >20 nt-long reads (Fig. 1B; Tirumuru et al. 2016). An RNA fragment that is 5–7 nt in length cannot be used to map the binding site of an RNA-binding protein since it would align to many places in the HIV genome. Longer fragments are required in CLIP experiments since they would align to a single location in the genome. For unclear reasons, short reads were used.
After this problem was identified, a correction was subsequently published with a properly generated CLIP data set (Tirumuru et al. 2016). Although not discussed in the correction, the new data sets showed the high-confidence YTHDF1, YTHDF2, and YTHDF3 binding patterns were now essentially identical. Thus, at least in the case of the YTHDF proteins and their binding to HIV, the published data no longer supported the idea that m6A sites bind different YTHDF proteins.
THE PROBLEM WITH “PEAKS”
We next wanted to address the other CLIP studies which examined YTHDF binding throughout the transcriptome and showed different binding properties of the different YTHDF proteins (Wang et al. 2014, 2015; Shi et al. 2017). We noticed that these studies relied on a peak-calling approach to identify binding sites for YTHDF proteins. In CLIP experiments, multiple reads that overlap a specific region of a gene comprise a peak. Despite the low background in CLIP experiments, nonspecific reads are present, which therefore requires “peak calling” algorithms. These algorithms call a peak based on peak height relative to other regions in the transcript.
However, as in any NGS-based method, some peaks are true positives (i.e., actual binding sites) and some are false positives. Therefore, a statistical cutoff threshold is used to decide which peaks will be used for further analysis. If the goal is to only have true positives, then the threshold will be high, that is, a more stringent P-value is applied, even if other true binding sites are lost. In this way, false positives are minimized. If the goal is to capture as many binding sites as possible, even if some false sites are included, then the cutoff is set to a low position. Thus, the threshold is tuned based on the needs of the subsequent analysis. Importantly, calling peaks using a cutoff is not the appropriate method for identifying all the binding sites of an RNA-binding protein—this is not the purpose of using a cutoff threshold. Instead, cutoffs are designed to miss true binding sites (when the threshold is high) or to capture as many true binding sites at the cost of some false binding sites (when the cutoff is low).
This can be easily seen in Figure 1C in which a true-binding site peak for YTHDF1 is just below a threshold while a peak for YTHDF2 at the same site in the mRNA is just above the threshold and therefore called as a YTHDF2-binding site (Fig. 1C). Since the cutoff threshold is arbitrary, a slightly lower threshold would have called both peaks. Thus, using cutoffs creates an incorrect impression that an mRNA binds one YTHDF protein and not the other.
In the analysis of YTHDF1, YTHDF2, and YTHDF3-binding sites, thresholds were used. The peaks that passed the threshold for YTHDF1, YTHDF2, and YTHDF3 were interpreted as binding sites for each YTHDF protein and used to create a Venn diagram to demonstrate that the mRNAs can bind one, two, or all three YTHDF proteins (Wang et al. 2014, 2015; Shi et al. 2017) (Fig. 1A).
In some experiments, PAR-CLIP (photoactivatable ribonucleoside CLIP) (Hafner et al. 2010) was used, a variant of CLIP. PAR-CLIP relies on the incorporation of 4-thiouridine (4SU), a photoactivatable nucleoside, into nascent RNA transcripts. The labeled RNAs are excited in living cells with UVB light (>310 nm) to create cross-links between RNA and interacting RNA-binding proteins. During reverse transcription, the cross-links cause the introduction of site-specific mutations. Thus, at the analysis step, RNA-binding protein-interacting sites can be recognized because of a signature mutation (T → C mutation). The ability to call T → C mutations is influenced by sequencing coverage and background sporadic transitions. Thus, similar to cutoffs used for peak calling, PAR-CLIP uses an arbitrary threshold value to call the minimum number of T → C mutations in order to call a site with the desired false positive and false negative rate.
IF NOT PEAKS, HOW SHOULD CLIP DATA SETS BE COMPARED?
So how should the binding sites of YTHDF1, YTHDF2, and YTHDF3 be compared? This is a long-standing question for many types of NGS data sets. An early study asked whether peak comparisons should be made when comparing ChIP data sets (Yang et al. 2014). To test this, the authors called peaks using cutoff thresholds in biological replicates. Since the samples are biological replicates, the binding sites should be identical. However, even the biological replicates had many different called peaks. Therefore, instead of using peaks that were called using cutoffs, the authors instead compared read numbers at predicted binding sites. In this case, the read numbers in each replicate showed high correlation at each site. Based on these and other studies, the commonly used method to determine if data sets show similar or different binding, involves performing a correlation of read numbers at predicted binding sites when two data sets are meant to be compared (Shao et al. 2012). Overall, peak-calling methods are likely to produce meaningless differences even in replicates.
Since we found that the CLIP data sets were compared using thresholds, rather than by comparing read abundances, we reexamined the published CLIP data sets, including the He data sets and our own. We examined YTHDF binding at each m6A site in the transcriptome, based on our earlier single-nucleotide resolution map of m6A (Linder et al. 2015). We measured peak heights for YTHDF1, YTHDF2, and YTHDF3 at each m6A and performed pairwise comparisons (Zaccara and Jaffrey 2020). If any m6A site exhibits selective binding to either YTHDF1, YTHDF2, or YTHDF3, we should see read coverage for only those YTHDF proteins. However, we found an essentially perfect correlation between peak heights when comparing each read coverage at each m6A site in the transcriptome for all three YTHDF proteins (Fig. 1D). Thus, this relatively straightforward analysis showed that no m6A site has a preference for any of the YTHDF proteins. Importantly, we analyzed the PAR-CLIP data sets from the He group, demonstrating that their data showed no difference between YTHDF1 and YTHDF2 binding patterns when analyzed in this manner.
Thus, our analysis contradicted previous studies that had concluded that there would be many m6A sites that only bound YTHDF1 or YTHDF2. To further test whether some mRNAs show specific binding to YTHDF1 or YTHDF2, we visualized the binding using the CLIP data plotted as genome browser tracks. Importantly, we used the PAR-CLIP data from the studies that claimed that binding was specific for specific mRNAs. When we tried to reanalyze the YTHDF3 PAR-CLIP data set, we noticed that it was unusual for several reasons. First, ∼44% of all reads mapped to a single site in a single mitochondrial transcript, MT-RNR2. These reads were identical, consistent with a PCR duplication event, and were not found in the YTHDF1 or YTHDF2 PAR-CLIP data sets. Second, of the remaining reads, only a small percentage of these were mappable. For this reason, the YTHDF3 PAR-CLIP data set lacked sufficient read depth to detect endogenous YTHDF3-binding sites in the transcriptome as efficiently as the other PAR-CLIP data sets. Thus, when searching for specific binding sites of YTHDF1 or YTHDF2 while using a low-coverage YTHDF3 data set, peak-calling and mutation-calling pipelines are even more problematic.
Regardless, when using simple visual inspection, it is clear that the peak patterns are identical for all three YTHDF proteins (Fig. 1E). This powerful approach provides compelling visual proof that mRNAs claimed to be specific for a single YTHDF protein are clearly binding all three YTHDF proteins. Thus, even the published data that was used to argue for YTHDF1, YTHDF2, and YTHDF3-unique binding sites clearly show that there are no m6A sites that selectively bind YTHDF1, YTHDF2, or YTHDF3.
Despite the diverse data that we and others (Lasman et al. 2020) have presented using new CLIP data and data from the He lab to show that m6A sites are not selective for specific YTHDF proteins, He and colleagues continue to refer to “YTHDF1-unique,” YTHDF2-unique mRNAs, etc., and use these pools of mRNAs to study the function of YTHDF proteins (Zou et al. 2023a,b). In their report, they do not address the evidence that has been used to show that YTHDF proteins show identical binding. In our view, identifying the correct targets of the YTHDF proteins is important for understanding their function. For this reason, studies of YTHDF “unique” mRNAs could produce misleading results. The methods for calling YTHDF paralog-unique proteins rely on near-threshold stochastic peak calling which occurs most commonly for low-abundance mRNAs. These low-abundance mRNAs are likely to be different from other RNAs, for example, they may have very high instability, or may suffer from a lower coverage after sequencing.
Our data suggest that the function of YTHDF proteins should be made by studying the effect of YTHDF paralog depletion on all m6A-mRNAs. It is expected that the effect of any YTHDF protein will depend on the number of m6A sites—since the more m6A sites in an mRNA, the more YTHDF protein that will be bound. An even better method is to classify mRNAs based on the m6A levels per transcript based on the m6A stoichiometry at each site (e.g., see Cruciani et al. 2023; Liu et al. 2023; Xiao et al. 2023). Thus, a dose-response should be seen with increasing m6A levels. For example, we showed that the YTHDF-dependent destabilization effect of m6A is correlated with the number of m6A sites (Zaccara and Jaffrey 2020). In our view, this is a more valid set of mRNAs to study in YTHDF depletion experiments, and allows comparison between mRNAs with varying levels of bound YTHDF proteins.
HOW CAN YTHDF1-UNIQUE OR YTHDF2-UNIQUE SITES BE DEMONSTRATED?
Despite the high correlation of YTHDF paralog binding at all m6A sites across multiple independent CLIP sites, we recognize that some may still question whether there are m6A sites that bind only one YTHDF protein. Here, we suggest some relatively simple experiments that can be performed to unambiguously demonstrate whether an m6A site is selective for one or all three YTHDF paralogs. First, we propose binding assays. For any mRNA proposed to bind selectively to one YTHDF protein, the affinity should be measured for each YTH domain for each YTHDF protein. This can be achieved by simply synthesizing a region of the target mRNA with A or m6A, and measuring affinity with microscale thermophoresis or quantitative electrophoretic mobility shift assays. The binding affinity should be notably higher for one of the YTHDF paralogs compared to the others. Second, we suggest that the CLIP browser tracks be presented at m6A sites that are thought to uniquely bind one YTHDF protein and not others, with care to normalize for library size. These data would clearly demonstrate that an m6A site selectively binds one of the YTHDF proteins and not others. These experiments must include control m6A sites that are known to bind all three YTHDF proteins. These types of experiments will show that specific m6A sites are selective rather than nonselective, as our data has shown.
Additionally, in order to claim that YTHDF proteins show different patterns of binding in the transcriptome, it will also be very important to establish a mechanism for how this can occur. Based on current in vitro binding studies, the YTHDF proteins appear to bind m6A regardless of the sequence context (Miller et al. 2023). Molecular dynamics simulations using structures of the YTHDF YTH domains also suggest that the YTH domains should have essentially identical binding properties (Li et al. 2020). The fundamental question that needs to be answered is: What would prevent one of the YTHDF proteins from binding an m6A site when all three proteins bind m6A and would have equal access to any m6A site since the YTHDF proteins are colocalized together in the cytosol? Conceivably, the YTHDF proteins could localize to different complexes in the cell. However, current data show that the YTHDF proteins colocalize and coassociate with each other in complexes in the cell (Youn et al. 2018). Since the idea that YTHDF1, YTHDF2, and YTHDF3 can have different binding properties is not compatible with a wealth of biochemical and structural data, it will be important to present a clear molecular explanation for how unique binding can be achieved. In our view, as of now, there is currently no plausible scientific mechanism for selective binding given the nearly identical sequence identity, structure, cellular localization, and binding properties of the YTHDF proteins.
WOULD YTHDF PROTEINS BE EXPECTED TO HAVE DIFFERENT OR SIMILAR FUNCTIONS?
The central question in the m6A field is the function of m6A. Since the YTHDF proteins appear to be the major readers of m6A, at least in the cytosol, the function of the YTHDF proteins can reveal the function of m6A. Prior to our study, the prevailing view developed by the He group was that m6A sites had different functions, with some m6A sites regulating translation and some m6A sites regulating stability, based on whether the m6A site bound YTHDF1 or YTHDF2 (Wang et al. 2014, 2015). YTHDF3 was proposed to bind m6A-mRNAs emerging from the nucleus and then transfer them to YTHDF1 or YTHDF2 (Shi et al. 2017). Each YTHDF protein was thus proposed to have a completely unrelated function.
As will be described below, several publications from the He group further developed this concept, linking the unique binding with the unique functions of YTHDF1 in the brain (Shi et al. 2018) and YTHDF2 in zebrafish (Zhao et al. 2017). These studies all supported the idea that different YTHDF proteins have different functions.
However, the prevailing model seemed inconsistent with the remarkably similar amino acid sequence and domain structure. The YTHDF proteins each have a large low-complexity domain region comprising the majority of the protein, and a C-terminal YTH domain. Perhaps more notable is their lack of different domains to mediate their different functions. YTHDF1 would be expected to have certain domains for interacting with translational machinery, and YTHDF3 would have different domains involved in mRNA transfer functions. However, all YTHDF proteins have a similar low-complexity domain.
Conceivably the YTHDF proteins may have highly different functions due to slight differences in their low-complexity domains. We previously compared the low-complexity domains of YTHDF1, YTHDF2, and YTHDF3 (Ries et al. 2019; Zaccara and Jaffrey 2020). Different alignment algorithms can be used to compare the sequence identity of these domains. In these analyses, ∼50% of residues within the low-complexity domain are identical. The low-complexity domains share even higher levels of sequence similarity. There are many different classes of low-complexity domains, each characterized by very distinct sequence compositions. These different sequence compositions (e.g., arginine–serine-rich vs. proline–glutamine-rich) have different functions by forming different types of cellular structures (Wang et al. 2018). However, the low-complexity domains of the YTHDF proteins fall in the same sequence class (Patil et al. 2018).
Even though the low-complexity domains of YTHDF proteins are all proline–glutamine–asparagine-rich domains (Patil et al. 2018), we wanted to know if there were still differences between these domains. To test this, we scanned the length of each low-complexity domain to examine the physiochemical properties in different regions, and whether these patterns were seen in all three YTHDF low-complexity domains. In this approach, we measured regions of net charge and hydrophobicity. This analysis showed regions of charge and hydrophobicity were enriched in specific areas of the low-complexity domains and were positioned at the exact same location along the length of all three paralogs (Fig. 2A; Zaccara and Jaffrey 2020). Since the low-complexity domains of the YTHDF proteins are similar, they would be expected to assemble into similar intracellular condensates (Ditlev et al. 2018). Therefore, we found it problematic that the YTHDF proteins would have completely different functions yet exhibit highly similar low-complexity domains and YTH domains.
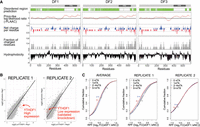
Technical problems with YTHDF1 knockdown accounts for the putative translation-promoting effect of YTHDF1. (A) The YTHDF paralogs exhibit similar disordered regions, prion-like regions, net charge, and hydrophobicity along the length of each protein. The disordered region score (green) was calculated using D2P2. This algorithm utilizes multiple different disordered protein prediction tools to generate scores for disordered regions (Oates et al. 2013). PLAAC (Lancaster et al. 2014) was used to calculate the prion-like likelihood ratio. A sliding window of 10 amino acids was used to calculate net charge per residue, probability of charged residues, and hydrophobicity. The plots were generated by CIDER (Holehouse et al. 2017) and ProtScale (ExPASy). All physiochemical parameters are similar along the length of all YTHDF paralogs, consistent with the idea that the low-complexity domains have similar properties and functions. (Image taken from Zaccara and Jaffrey 2020.) (B) We downloaded the data sets listing the ribosome-protected fragment number for each gene in each YTHDF1 replicate from Wang et al. (2015). We also downloaded the data for the matched control siRNA. In the case of Replicate 2, YTHDF1 expression is very low, consistent with knockdown. However, in Replicate 1, the expression of YTHDF1 is much higher than the control expression, which is inconsistent with a knockdown. Instead, this is more consistent with an overexpression or some other type of artifact. Because of this problem with Replicate 1, it was analyzed separately below. (C) Reexamination of the effect of YTHDF1 knockdown for each replicate separately. Because we could not confirm that Replicate 1 was a knockdown, we analyzed Replicate 1 and Replicate 2 separately. In the case of Replicate 1, we could see a clear decrease in translation, as measured by the number of ribosome-protected fragments for each gene which contained high levels of m6A. However, this replicate does not appear to be a knockdown because it has high levels of YTHDF1. If this is an overexpression, the reduced translation may represent YTHDF1 aggregates which trap m6A-containing mRNA. In the case of Replicate 2, which is a clear knockdown as seen above, there is no effect on the number of ribosome-protected fragments for highly methylated RNAs compared to nonmethylated RNAs. Thus, the original data set does not support the idea that YTHDF1 promotes the translation of m6A-containing mRNA based on Replicate 2, which is the only valid datable replicate from this study.
He and colleagues have argued that the subtle differences between the low-complexity domains could result in distinct functional roles, that is, translation for YTHDF1 and mRNA degradation for YTHDF2 (Zou et al. 2023a). They found that bacterially purified YTHDF1 formed precipitates during prolonged storage that were different from YTHDF2, which they argued supports the idea that the low-complexity domains could have different cellular functions (Zou et al. 2023a). Although we think the physiologic relevance of these precipitation observations is difficult to know, the experiments described below would more directly address YTHDF1 and YTHDF2 function.
WHY WAS THE LINK BETWEEN YTHDF1 AND TRANSLATION QUESTIONED?
In the original studies of the YTHDF proteins, their function was determined by knockdown of each YTHDF transcript and measuring mRNA stability and translation (Wang et al. 2014, 2015; Shi et al. 2017). This lead to the major discovery that the function of YTHDF1 is to promote the translation of m6A-mRNAs (Wang et al. 2015). The authors observed a reduction in translation upon YTHDF1 knockdown using ribosome profiling, the standard assay for measuring translation rates (Ingolia et al. 2011). This method involves using nucleases to digest mRNAs that are being translated. The regions of mRNA covered by a ribosome are protected from digestion, leading to a ribosome-protected fragment. The number of ribosome-protected fragments that map to a specific gene reflects the number of ribosomes that are translating an mRNA, and thus reflect the translation rate of an mRNA.
A problem arose when we reanalyzed the published YTHDF1 knockdown ribosome profiling data sets that reported the measured number of ribosome-protected fragments for each gene (Wang et al. 2015). The experiment involved two control siRNA and two YTHDF1-specific siRNA data sets. When we re-analyzed this data, we observed a very concerning problem with the deposited data sets. In any knockdown data set, the gene that is being knocked down should be lowly expressed, and at the very least, below the expression seen in control knockdown cells. Thus, in this case, there should be few or no ribosome-protected fragments derived from YTHDF1 mRNA in the knockdown cells. As expected, in one of the YTHDF1 knockdown replicates, there were almost no ribosome-protected fragments (plots are based on GSE63591_C-Y1-Ribosome_profiling.xlsx within GEO GSE63591 in Wang et al. 2015, Fig. 2B). To our great surprise, the other YTHDF1 knockdown replicate showed extremely high levels of YTHDF1-derived ribosome-protected fragments (Fig. 2B). The expression was far above levels of YTHDF1 in control siRNA cells. It is difficult to explain the cause of this increased YTHDF1 expression. It could reflect some unknown technical problem. This high level of expression is consistent with YTHDF1 overexpression rather than knockdown. Although we do not know the cause of this high YTHDF1 expression, it is clear that results from this data set should not be used to understand the effects of YTHDF1 knockdown.
Because only one of the two knockdown data sets showed YTHDF1 knockdown, we analyzed each of these ribosome profiling data sets separately. The published data used an average of the two putative knockdown data sets to show that YTHDF1 depletion caused target mRNAs to be less translated (Wang et al. 2015). However, when we analyzed these replicates separately, we observed a very different result. In the case of the ribosome profiling data set where YTHDF1 knockdown was validated, we found no change in the number of ribosome-protected fragments in methylated RNAs compared to nonmethylated RNAs (Fig. 2C). However, in the sample which exhibited high levels of YTHDF1-derived fragments, the methylated mRNAs showed reduced ribosome-protected fragments compared to nonmethylated RNAs. Thus, the only the data set that supported the idea that YTHDF1 depletion lowers translation was the data set where YTHDF1 was elevated. In contrast, the validated YTHDF1 knockdown data set showed no effect on translation. We therefore came to two conclusions: First, the published data sets do not appear to reflect two knockdown replicates, and second, the one sample that was clearly a YTHDF1 knockdown does not show any YTHDF1-dependent effects on translation.
In summary, our re-analysis explains why the original study reported reduced translation in the YTHDF1 knockdown datasets. Since the average of the effects from each replicate (i.e., no effect in one replicate and translational repression in the other) is translational repression, the interpretation was that knockdown of YTHDF1 leads to translational repression. Overall, our reanalysis shows that the central core finding of the YTHDF1 study (Wang et al. 2015) comes from the knockdown data set which does not appear to be a knockdown (Fig. 2B).
RIBOSOME PROFILING SHOWS THAT YTHDF1 DOES NOT REGULATE m6A-mRNA TRANSLATION
Our finding that one of the YTHDF1 knockdown data sets was not consistent with knockdown, but instead possibly a YTHDF1 overexpression, drove us to reanalyze the role of YTHDF1 on the fate of m6A-mRNAs. To do this, we repeated the ribosome profiling experiments in the same HeLa cell type used in the original study (Zaccara and Jaffrey 2020). We also knocked down YTHDF1, YTHDF2, and YTHDF3 separately, and all three YTHDF proteins together. Knockdown was confirmed based on loss of ribosome-protected fragments and gene expression. In each case, we found no effect on the translation of methylated RNA as compared to unmethylated RNAs. When all three YTHDF proteins were knocked down, we observed a slight increase, rather than a decrease, in the number of ribosome-protected fragments. Although this was a small effect, it raises the possibility that the YTHDF proteins may actually repress the translation of mRNA, possibly by recruiting them to granules as has been seen in the context of stress (Ries et al. 2019, 2023).
Hanna and colleagues also attempted to detect a role for YTHDF1 in translation (Lasman et al. 2020). In their ribosome profiling experiments in mouse embryonic stem cells, they found slightly elevated translation rates for Ythdf1 unique, Ythdf2 unique, Ythdf3 unique, and all m6A-modified mRNAs in their control cells, but this was not affected by knockout of Ythdf1 or any Ythdf gene, or by triple knockout. They thus concluded that Ythdf1 does not affect translation.
Meyer and colleagues also addressed this question using their innovative YTHDF-Apobec fusion DART-seq technology, which induces C → U edits adjacent to m6A in mRNAs bound to either YTHDF1, YTHDF2, or YTHDF3 (Flamand et al. 2022). They found that actively translated m6A-mRNAs in polysomes had no preferential association with YTHDF1.
Taken together, data from different laboratories using different approaches fail to show evidence of YTHDF1 as a translational regulator of m6A-mRNAs.
YTHDF1 MEDIATES m6A-mRNA DEGRADATION
Our search for a function for YTHDF1 turned toward RNA degradation since (as described below), we found that all YTHDF paralogs interacted with mRNA degradation proteins. Our experiments demonstrated that YTHDF1, as well as the other YTHDF proteins, mediate mRNA degradation (Zaccara and Jaffrey 2020). YTHDF2 depletion led to a clear increase in mRNA stability, and this effect was further enhanced when combined with either YTHDF1 or YTHDF3 depletion, and further enhanced when all three proteins were depleted. Importantly, we examined how depletion of YTHDF proteins affected mRNAs with no m6A, as well as mRNAs with increasing numbers of m6A sites. The nonmethylated mRNAs were not stabilized by YTHDF depletion, demonstrating that depletion of all three YTHDF proteins did not lead to nonspecific mRNA stabilization. In contrast, stabilization was correlated with the number of m6A sites. Additionally, it should be noted we examined every pairwise combination of YTHDF1, YTHDF2, and YTHDF3 knockdown. Each of these showed that loss of two proteins was not as effective as the knockdown of all three, indicating that all three YTHDF proteins contribute to mRNA degradation (Zaccara and Jaffrey 2020).
It should be noted that YTHDF2 was previously shown to mediate mRNA degradation (Wang et al. 2014) consistent with seminal studies that first showed that m6A is associated with mRNA instability (Sommer et al. 1978). Our data confirmed this finding, but showed that YTHDF1 and YTHDF3 also mediate mRNA degradation (Fig. 3).

Model for YTHDF function in regulating m6A. (A) The original model of the YTHDF function was that each YTHDF paralog had a different effect on mRNA. (B) In the revised model, each of the YTHDF proteins functions cooperatively so that the cumulative amount of YTHDF protein determines the magnitude of m6A-mRNA degradation.
Recent studies by He and colleagues have now confirmed that depletion of all three YTHDF proteins exhibits a synergistic effect to stabilize mRNA (Zou et al. 2023a). However, they argue that this effect is not strictly m6A dependent. In their hands, triple knockdown of all three YTHDF proteins leads to increased cellular P-body formation and global stabilization of most mRNAs, regardless of their methylation status. In contrast, in our data, we did not observe any prominent change in P-bodies or stabilization of nonmethylated mRNAs. They suggested that we may not have seen this effect due to a lack of spike-ins to normalize expression. However, they missed that we also used spike-ins; thus, the absence of spike-ins normalization cannot explain the discrepancy between the two studies. It may be possible that the global effects observed by He and colleagues (Zou et al. 2023a) could reflect the general cell toxicity seen with prolonged YTHDF depletion. In our studies, we measured the effects of mRNA stabilization induced by YTHDF depletion shortly after knockdown. Regardless, our experiments demonstrate that m6A-mRNAs are selectively stabilized by YTHDF1, YTHDF2, and YTHDF3 depletion, while nonmethylated mRNAs are only minimally stabilized.
Overall, in our studies, we found that the maximal effect on m6A-mRNA stabilization was seen when all three YTHDF proteins were knocked down. This is compatible with a model where the total amount of YTHDF protein is the critical factor in determining mRNA degradation rates. We observed a partial effect when only YTHDF2 was knocked down and minimal effects when only YTHDF1 or YTHDF3 was knocked down. These data are consistent with the relatively high abundance of YTHDF2 in most tissues (Patil et al. 2018). As a result, knocking down YTHDF2 might be expected to have the most prominent effect. However, the other YTHDF proteins contribute since their knockdown further enhances mRNA stability. Notably, Hanna and colleagues showed that different YTHDF proteins might be more abundant in some cell types which may make those cell types more dependent on certain YTHDF paralogs (Lasman et al. 2020).
Despite the data presented by us and by others (Lasman et al. 2020), He and colleagues have continued to argue that YTHDF1 controls translation (Zou et al. 2023a,b). He and colleagues have suggested that errors in our analysis may have caused us to miss the translation-regulatory role of YTHDF1. However, several of their arguments reflect misunderstanding of our methods. First, it was erroneously argued that our protocol for the isolation of the translating mRNAs had a pretreatment step with cycloheximide, which can adversely affect ribosome profiling data (Zou et al. 2023a). However, no pretreatment with cycloheximide was used in our study. As described in the methods section of our study, snap freezing using liquid nitrogen was used to stop the runoff of ribosomes. Thus, if YTHDF1 had a role on active translation, our data set should reveal it. One major difference in our analysis is the threshold decided to call ribosome-associated mRNAs. The process of ribosome purification assumes that 28–30 nt-long fragments represent ribosome-occupied regions. However, during the purification, indirect detection of complexes that occupy a region similar in size may occur. To remove this background noise, we decided to apply all quality control steps previously described to be essential for the correct analysis of the translating mRNAs, including the removal of reads within the untranslated region of the mRNAs and the correction for the P-site position of the ribosomal read (McGlincy and Ingolia 2017).
He and colleagues have questioned how mRNAs were selected for analysis in our ribosome profiling analysis. We selected only mRNAs with more than zero counts in the control condition, and asked how that pool is affected by YTHDF1 silencing. These mRNAs should show reduced ribosome-protected fragments if YTHDF1 is promoting their translation. According to He and colleagues, interrogating functions of YTHDF1 only based on actively translated RNA may lead to the incorporation of indirect effects in the analysis (Zou et al. 2023a). However, if YTHDF1 promotes translation, mRNAs would need to have some detectable translation in order for the level to drop upon YTHDF1 silencing. Thus, our analysis has the ability to detect the removal of mRNAs from the active translation pool upon YTHDF1 silencing.
Despite the questions about our methods, He and colleagues have not addressed the key question, that is, why their YTHDF1 knockdown data set appears to be an overexpression, and why this data set should be used. The He group has not replicated their ribosome profiling data set in HeLa cells after YTHDF1 knockdown. Because ribosome profiling is the main method for implicating any protein, including YTHDF1, as a translational regulator, the core issue is the validity of the published data sets.
He and colleagues reexamined our YTHDF1 knockdown ribosome profiling data sets and found a small reduction in the translation of mRNAs that they defined as “YTHDF1-unique” mRNAs. Based on this, they argued that YTHDF1 regulates translation. However, they found the same effect with YTHDF2 depletion, which was not addressed in their report (Zou et al. 2023a). Thus, no selective effect of YTHDF1 on translation was seen. The very small effects that were seen for both YTHDF1 and YTHDF2 knockdown could simply be the result of small variation in background read counts on lowly translated genes, even in normal conditions.
Thus, we have yet to see any ribosome profiling data which supports the idea that YTHDF1 is a selective regulator of translation compared to YTHDF2.
It should be noted that the He group has examined how YTHDF1 depletion leads to reduced translation of specific mRNAs in other contexts. These effects were analyzed on single genes rather than transcriptome-wide ribosome profiling analysis. For example, in the context of hippocampal neurons, potassium stimulation induced noticeable increases in the synthesis of proteins involved in long-term potentiation (Camk2b, Grin2a) in control neurons but much less in Ythdf1 knockout neurons (Shi et al. 2018). Similarly, a reporter RNA was shown to exhibit higher translation following potassium-mediated neuronal depolarization, and this effect was reduced in YTHDF1-depleted neurons. In our view, without global ribosome profiling data, these are likely to reflect the indirect effects of YTHDF1 depletion. These indirect effects could include stabilization of specific mRNAs that encode regulators of synaptic translation or potassium depolarization-induced signaling.
In a recent study, He and colleagues studied glycosylation of YTHDF proteins and included ribosome profiling data (Chen et al. 2023). In this study, the roles of YTHDF1 and YTHDF3 were studied together to determine their effect on translation. However, this ribosome profiling experiment used an unusual YTHDF1/YTHDF3 overexpression coupled with YTHDF1/YTHDF3 knockdown in the same cells (Chen et al. 2023). The key experiment that is needed is a simple YTHDF1 knockdown side-by-side with a YTHDF2 knockdown to show that m6A-mRNAs exhibit selective reduction in translation only with YTHDF1 depletion. However, to date, no ribosome profiling data set has been published, which shows that YTHDF1, and not YTHDF2, affects the translation of m6A-containing mRNAs. Thus, the idea that YTHDF1 is a promoter of translation still lacks support. In our view, Replicate 2 from Wang et al. 2015 clearly and unambiguously demonstrates that there is no effect of YTHDF1 on translation of methylated mRNAs. Any claim that YTHDF1 regulates translation needs to address this data, as well as other ribosome profiling datasets showing no effects of YTHDF1 on translation. Nevertheless, the controversy can be resolved by simply presenting YTHDF1 and YTHDF2 knockdown ribosome profiling data side-by-side and demonstrating selective effects on m6A-mRNA translation upon depletion of YTHDF1.
INTERACTOME ANALYSIS SUPPORTS A COMMON mRNA DEGRADATION FUNCTION FOR YTHDF PROTEINS
Although our major concern was that the ribosome profiling data set was not a YTHDF1 knockdown, there were other major problems with the concept that YTHDF1, YTHDF2, and YTHDF3 have different functions. In principle, if they have different functions, they should bind different effector proteins. These different proteins would be linked to those unique functions. We therefore mined a BioID data set in which Gingras and colleagues performed a BioID analysis on 139 RNA-binding proteins (Youn et al. 2018). In this experiment, each protein was tagged on the N terminus, C terminus, or both, with TurboID, and nearby proteins were biotinylated. In the published analysis, which we reanalyzed to enable side-by-side comparisons of YTHDF1, YTHDF2, and YTHDF3, we found a remarkable similarity in the binding partners, rather than highly distinct binding partners. We anticipated that YTHDF1 would be associated primarily with translational machinery, whereas YTHDF2 would be associated with degradation machinery. However, we found a low probability association of translation machinery proteins for all three YTHDF paralogs while we found a high significant probability of interaction with the RNA degradation machinery. This supports the idea that all YTHDF proteins participate in RNA degradation.
He and colleagues (Zou et al. 2023a) have argued that a reanalysis of the interactome data shows that YTHDF1 exhibits preferential binding to translation machinery compared to YTHDF2. Their approach used an interesting principal component analysis to examine global patterns of binding from the Gingras study (Youn et al. 2018). This analysis showed a subtle increase in the association of YTHDF1 with translation-associated proteins (Zou et al. 2023a). However, most of these interactions were based on interactions that were designated low confidence by Gingeras and colleagues due to poor peptide coverage in mass spectrometry. For this reason, we think the most reliable way to measure the interactors of YTHDF proteins is to use the conventional approach of looking at their high-confidence interactors.
He and colleagues (Zou et al. 2023a) have raised a concern that we compared YTHDF interactomes in which one YTHDF protein was tagged on the C terminus, while the other YTHDF protein had interactomes generated with versions of the protein tagged either at the N terminus or C terminus (Zou et al. 2023a). However, a recent study reported comprehensive interactomes of similarly tagged YTHDF proteins (Moch et al. 2023). The results confirm our earlier finding that there is a general association of all YTHDF proteins with the RNA degradation machinery. Additionally, in vitro binding studies of recombinant YTHDF proteins have shown that all three YTHDF proteins interact with the CCR4/NOT RNA degradation proteins (Du et al. 2016). Thus, diverse interactome studies from multiple independent groups support the idea that all YTHDF proteins interact with RNA degradation pathway proteins and have a similar low probability of interaction with proteins implicated in translation.
The idea that YTHDF1 promotes translation also seemed somewhat problematic because m6A, and thus the recruited YTHDF1 proteins, are primarily found near stop codons (Dominissini et al. 2012; Meyer et al. 2012). However, translation regulators nearly always bind to the 5′ UTR to promote ribosome entry in front of the start codon (Pestova et al. 2001). A common mechanism involves the recruitment of eIF3, a ribosome-recruiting complex, to 5′ UTRs. Indeed, YTHDF1 was proposed to bind eIF3 (Wang et al. 2015). However, recruitment of YTHDF1 to stop codons appears to be the wrong place to recruit eIF3, and thus ribosomes, to enhance translation. It should be noted that poly(A)-binding protein (PABP) binds to the poly(A) tail of mRNAs and interacts with cap-binding proteins to increase their affinity for the cap (Pestova et al. 2001). In this way, mRNAs with poly(A) tails exhibit enhanced translation. Thus, it remains possible that YTHDF1 could have some sort of similar long-distance interaction. However, the original study did not demonstrate how YTHDF1 could control translation from the stop-codon region.
Lastly, regulators of translation initiation are typically validated using in vitro translation assays in which purified initiation factors are incubated with mRNA, and ribosome entry into mRNA can be measured using various footprinting methods or by measuring the translation of reporter RNAs (Pestova et al. 2001). This sort of data was not presented, making it unclear if YTHDF1 is indeed a direct inducer of translation initiation. Notably, in some of the published experiments reporters were used in which YTHDF1 or YTHDF2 was overexpressed. Overexpressing any protein with a low-complexity domain is problematic since they can form aggregates in the cell. We showed that this was the case with YTHDF proteins, which form variable levels of aggregates in different transfected cells (Zaccara and Jaffrey 2020). Thus, YTHDF overexpression should be avoided due to possible effects caused by the sequestration of mRNAs in the aggregates.
In summary, the key issue is the ribosome profiling. Additionally, other aspects of the model of YTHDF1 as a translational regulator, including YTHDF1 interaction partners and its binding location in mRNA, cannot be readily reconciled with a role in translation.
YTHDF PARALOGS DO NOT APPEAR TO HAVE UNIQUE PHYSIOLOGIC FUNCTIONS
An independent line of experiments has also suggested that each YTHDF protein has different physiologic roles and functions. The core idea is that each YTHDF protein will have unique physiological roles due to their different effects on mRNA stability or translation and their ability to regulate different mRNAs. These studies, also by the He lab and colleagues, helped establish the idea that YTHDF1 has specific physiological functions that are distinct from YTHDF2. One of these was a study of YTHDF1 depletion in the hippocampus, which was associated with systematic defects in synaptic plasticity in the hippocampus (Shi et al. 2018). Although YTHDF2 was not tested, this study provided a strong example of how YTHDF1 could mediate unique effects, presumably by promoting the translation of specific mRNAs that uniquely bind YTHDF1.
However, more recent studies were unable to replicate these findings. Using Ythdf1- and Ythdf2-specific knockout mice, Ji and colleagues demonstrated that YTHDF1 depletion has no effect on synaptic plasticity in the hippocampus (Zhuang et al. 2023). Instead, this group found the opposite result—YTHDF2 was the major driver of synaptic plasticity in the hippocampus. A way to explain this incongruency is that the mice in one laboratory had high levels of YTHDF1 and the other had high levels of YTHDF2. The key point is that the specific YTHDF protein does not matter. Instead, the critical factor is the amount of YTHDF proteins, not the specific YTHDF protein.
A similar example is a major study in zebrafish by He and colleagues which argued that YTHDF2 had a specific function in catalyzing maternal mRNA degradation in the zygote (Zhao et al. 2017). However, when another group attempted to replicate this, they found that YTHDF2 was not the critical driver of maternal mRNA degradation (Kontur et al. 2020). Instead, they found that all the YTHDF proteins contributed to mRNA degradation. Again, this highlights that no specific YTHDF protein has a unique effect. Instead, the effects of YTHDF proteins will be driven by the cumulative action of YTHDF proteins, with phenotypes being evident upon depletion of the most abundant YTHDF protein in that cell type.
These attempts to replicate the original studies, and the subsequent failures, have reshaped our thinking of YTHDF proteins along the lines that we presented earlier, which is that the YTHDF proteins likely perform their functions together, with the major role to mediate mRNA degradation.
ARE YTHDF PROTEINS FULLY REDUNDANT?
An important question that came from our studies was whether the YTHDF proteins are fully redundant with each other. Our data clearly show that each YTHDF paralog contributes to m6A-mRNA degradation. However, it is possible that one of the paralogs is more effective than another. For example, in some cells, one of the YTHDF paralogs may be inactivated by a post-translational modification, which could make that paralog less able to mediate degradation than another paralog. One such mechanism might be glycosylation. Recent studies have highlighted that YTHDF1, YTHDF2, and YTHDF3 are glycosylated, which affects their binding partners, although the levels of glycosylation for each paralog appear to differ based on cell type (Chen et al. 2023; Li et al. 2023). The ability of the YTHDF proteins to be differentially regulated can allow one paralog to be more effective at mediating mRNA degradation in some cellular contexts, thus fine-tuning YTHDF-dependent m6A-mRNA degradation.
In addition, it should be noted that the different YTHDF paralogs are not equally expressed and can have very different expression levels in tissues (Lasman et al. 2020). Therefore, it is not surprising that knockdown of one of the paralogs in some cell types might have minimal effects, since the other paralogs can fully compensate and mediate m6A-mediated mRNA degradation. However, this does not mean that the most abundant YTHDF paralog in the cell is the most important. As mentioned above, glycosylation or other post-translational modifications may inactivate one of the paralogs, thus making the others more important, even if they are not the most abundant. It should be noted that these forms of regulation do not negate the idea that all three of the proteins promote mRNA degradation, and the “active” forms of each YTHDF paralog work together to cumulatively mediate the overall magnitude of the m6A degradation response.
An interesting experiment will be to generate mice in which YTHDF1 is swapped with YTHDF2, and vice versa, to help test their functional redundancy and to begin to understand whether there are subtle differences in the individual proteins that may influence the magnitude of their degradation effects. Experiments in this direction have been performed by Hanna and colleagues (Lasman et al. 2020). They could not detect any offspring that were Ythdf2+/− and null in the two other readers (Ythdf1−/−;Ythdf2+/−;Ythdf3−/−). These results suggest that the lack of Ythdf1 or Ythdf3 can be compensated by the two other readers. However, the lack of Ythdf2 cannot be compensated by Ythdf1 or Ythdf3. The fact that the partial expression of Ythdf2 in the heterozygous lineage requires the expression of at least one other Ythdf protein suggests that the function of the readers is dosage-dependent and that cumulative expression of Ythdf readers beyond a certain threshold is required for embryonic viability.
CONCLUSIONS
Because there is still debate about the function of YTHDF proteins (Zou et al. 2023a), the goal of this Perspective is to clarify the reasons why the original data and conclusions were questioned. The field of m6A is a fast-moving field, in particular with regard to the function of m6A and its reader proteins. After the discovery of YTHDF proteins as m6A readers in 2012 (Dominissini et al. 2012), studies rapidly appeared that described the functions of these proteins. However, in many cases technical issues have led to flawed data sets, such as the CLIP data sets where reads were 5–7 nt in length or the ribosome profiling data sets which were thought to be YTHDF1 knockdown but clearly have high levels of YTHDF1. Additionally, we highlight how even high-quality data can be analyzed in different ways resulting in opposite conclusions about YTHDF proteins and their identical or divergent binding sites. We also show that relatively simple approaches, like correlation plots examining YTHDF binding at each m6A site, or presenting CLIP data for visual inspection, can clearly show the similar binding patterns of the YTHDF proteins.
Our Perspective also highlights how models of YTHDF function were previously proposed that seem to be inconsistent with basic principles of RNA binding or mRNA translation. Since the YTHDF proteins are so similar in sequence and structure, a model that involves different functions for YTHDF proteins should also be accompanied by experimentally validated molecular mechanisms to explain how similar proteins have highly divergent functions. Similarly, translational regulation is thought to occur in the 5′ UTR, so a model proposing that YTHDF1 at the stop codon promotes translation should be accompanied by a mechanism that can support this somewhat counterintuitive claim. Newer data and new analysis from multiple independent groups now point to a more intuitive model of YTHDF function as a mediator of m6A-mediated mRNA degradation, which is the primary effect of m6A in mRNA.
COMPETING INTEREST STATEMENT
S.R.J. is the cofounder, advisor, and/or has equity in Chimerna Therapeutics, 858 Therapeutics, and Lucerna Technologies.
ACKNOWLEDGMENTS
This work was supported by the National Institutes of Health (NIH) grants R35NS111631 and RM1HG011563 to S.R.J. and K22CA258954 to S.Z.
Footnotes
-
Article is online at http://www.rnajournal.org/cgi/doi/10.1261/rna.079988.124.
-
Freely available online through the RNA Open Access option.
This article, published in RNA, is available under a Creative Commons License (Attribution-NonCommercial 4.0 International), as described at http://creativecommons.org/licenses/by-nc/4.0/.








