
The Bacillus subtilis ywbD gene encodes RlmQ, the 23S rRNA methyltransferase forming m7G2574 in the A-site of the peptidyl transferase center
- Philippe Wolff1,5,
- Geoffray Labar2,5,
- Antony Lechner1,
- Dany Van Elder3,
- Romuald Soin4,
- Cyril Gueydan4,
- Véronique Kruys4,
- Louis Droogmans3 and
- Martine Roovers2
- 1Architecture et Réactivité de l'ARN, Institut de Biologie Moléculaire et Cellulaire du CNRS, Université de Strasbourg, F-67084 Strasbourg, France
- 2Labiris, B-1070 Bruxelles, Belgium
- 3Laboratoire de Chimie Biologique, Université Libre de Bruxelles (ULB), Labiris, B-1070 Bruxelles, Belgium
- 4Laboratoire de Biologie Moléculaire du Gène, Institut de Biologie et de Médecine Moléculaires, Université Libre de Bruxelles (ULB), B-6041 Gosselies, Belgium
- Corresponding author: mroovers{at}spfb.brussels
-
↵5 These authors contributed equally to this work.
Abstract
Ribosomal RNA contains many posttranscriptionally modified nucleosides, particularly in the functional parts of the ribosome. The distribution of these modifications varies from one organism to another. In Bacillus subtilis, the model organism for Gram-positive bacteria, mass spectrometry experiments revealed the presence of 7-methylguanosine (m7G) at position 2574 of the 23S rRNA, which lies in the A-site of the peptidyl transferase center of the large ribosomal subunit. Testing several m7G methyltransferase candidates allowed us to identify the RlmQ enzyme, encoded by the ywbD open reading frame, as the MTase responsible for this modification. The enzyme methylates free RNA and not ribosomal 50S or 70S particles, suggesting that modification occurs in the early steps of ribosome biogenesis.
Keywords
INTRODUCTION
RNA molecules from organisms belonging to the three domains of life (Archaea, Eukarya, and Bacteria) contain posttranscriptionally modified nucleosides. The modification density is the highest for transfer RNA (tRNA) followed by ribosomal RNA (rRNA). More than one hundred different modified nucleosides have been discovered (Boccaletto et al. 2022), among which methylation and uridine isomerization (pseudouridylation) are the most common. Methylations are encountered on the 2′-O-position of the ribose or on the nucleobases.
Most of the RNA methylations depend on S-adenosylmethionine (SAM) as the methyl donor and are catalyzed by two out of five classes of SAM-dependent methyltransferases (MTases). This classification is based on the fold of the core of the enzyme (Schubert et al. 2003). The Rossmann fold-like RNA MTases (RFM) belong to the first class of MTases, and the SPOUT RNA MTases belong to the fourth class of this sorting. Most of the RNA MTases belong to class I but share only a fair degree of primary sequence similarity (Ofengand and Del Campo 2004).
The methylations found in rRNA are not evenly distributed over the ribosome. Most of them are grouped in regions important for ribosome function such as the decoding center (DC), the peptidyl transferase center (PTC), the tRNA binding sites (Aminoacyl- and Peptidyl-sites), and the interface between the small and large ribosomal subunit (Sloan et al. 2017). A noteworthy example is the A-loop of the 23S rRNA, which contains 2′-O-methylated nucleosides (Um2552 and/or Gm2553). Guanosine 2553 makes a base pair with C75 of the A-site bound tRNA (Kim and Green 1999), stimulating peptidyl transferase activity (Brunelle et al. 2006). Besides, the universally 2′-O-methylated G2251 residue is essential for positioning of tRNA at the P-site during peptidyl transfer (Green et al. 1997). The neighboring residue G2252 base pairs with C74 of P-site bound tRNA (Samaha et al. 1995).
Methylations of the 16S rRNA in the small ribosomal subunit are generally formed during late steps of ribosome assembly, while those of the 23S rRNA in the large subunit occur more often in early assembly steps (Sergeeva et al. 2015; Sergiev et al. 2018).
Although modifications cluster mainly in positions that are involved in key functions of the ribosome, all organisms display a distinct repertoire of methylated rRNA residues (Sergiev et al. 2018). This is consistent with the phylogenetic distribution of RNA MTases across bacterial phyla (Mosquera-Rendón et al. 2014). Some rRNA MTases are present in a large variety of bacteria, like RlmH, RlmB, and RlmN catalyzing, respectively, m3Ψ1915, Gm2251, and m2A2503 in the 23S rRNA, whereas others are poorly conserved and can be rather unique to a phylogenetic lineage, as is the case for RlmM and RlmK, catalyzing, respectively, Cm2498 and m7G2069 in 23S rRNA. This diverse distribution of modifications is well illustrated when the modification pattern is compared between Bacillus subtilis (Gram positive) and Escherichia coli (Gram negative). The RlmA enzyme forms m1G745 in E. coli, whereas the corresponding enzyme forms m1G748 in B. subtilis (Liu and Douthwaite 2002). In addition, several differences were observed when comparing the modification present in helices 90, 91, and 92 of the A-site of 23S rRNA of E. coli and B. subtilis. U2552 is methylated in E. coli but not in B. subtilis, whereas Gm2553 is absent in E. coli and present in B. subtilis (Hansen et al. 2002). In the same paper, the authors pinpoint an unidentified methylated guanosine at position 2574 of the 23S rRNA of B. subtilis, which is absent in E. coli (Fig. 1). In this work, we show that this methylated guanosine is 7-methylguanosine (m7G) and we identify the MTase involved in its formation.
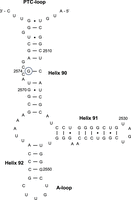
Secondary structure of part of domain V of B. subtilis 23S rRNA. Helices 90–92 and the PTC- and A-loop are indicated. Guanosine 2574 is encircled. E. coli numbering is applied.
RESULTS AND DISCUSSION
Identification of m7G at position 2574 of B. subtilis 23S rRNA by mass spectrometry analysis
To characterize the modified guanosine at position 2574 of B. subtilis 23S rRNA by mass spectrometry, a fragment of the rRNA containing this residue was isolated. For this purpose, the two DNA oligonucleotides MS1 and MS2, formerly used to detect Gm2553 in the same rRNA (Roovers et al. 2022), with sequences complementary to the rRNA on either side of the position of interest, were hybridized to 23S rRNA of B. subtilis, and RNase H was used to digest the two duplexes (DNA/RNA). Position 2574 is covered by oligonucleotide MS2, but the cleavage occurred a few nucleotides 3′ to this position. The RNase H fragment was purified by gel electrophoresis and the corresponding gel band was treated by RNase T1. LC–MS/MS analysis of the hydrolysate revealed the presence of the sequence UAC[m7G]CGp (mass of 1966.3 Da), and MS/MS fragmentation confirmed methylation at position 2574 with the characteristic neutral loss of 165Da corresponding to the fragmentation of the 7-methylguanine (Fig. 2A).
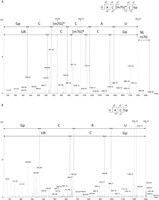
Identification of m7G at position 2574 of B. subtilis 23S rRNA using mass spectrometry. (A) MS/MS sequencing spectra of 1966.3 Da (m/z 982.63, z = 2−), sequence UAC[m7G]CGp, obtained from the analysis of B. subtilis WT 23S RNase H fragment after T1 digestion. Collision-induced dissociation (CID) fragmentation shows a neutral loss of 165 Da, which is specific to m7G and corresponds to the loss of the methylated base. (B) MS/MS sequencing spectra of 1302.2 Da (m/z 650.58, z = 2−), sequence UACGp, obtained from the analysis of B. subtilis ΔywbD 23S RNase H fragment after T1 digestion. The 3′Gp corresponds to the position 2574.
YwbD catalyzes m7G formation at position 2574 of 23S rRNA
To identify the enzyme responsible for the formation of m7G2574 in the 23S rRNA of B. subtilis, different m7G-MTases listed in Modomics (Boccaletto et al. 2022) were used as queries for Blast searches on B. subtilis open reading frames (ORFs). Three candidates came forward from this search: the B. subtilis TrmB, RsmG, and YwbD proteins belonging to the cluster of orthologous groups (COG)s 0220, 0357, and 1092, respectively. YwbD is a homolog to the RlmK moiety of RlmKL of E. coli. RlmKL is a bifunctional enzyme with an m2G2445 (RlmL) and an m7G2069 (RlmK) forming activity in the 23S rRNA (Wang et al. 2012). YwbD belongs to a family of proteins with m5C and m7G forming activity, but its function has not been established yet.
TrmB and RsmG are well-known MTases forming m7G, respectively, in tRNA and 16S rRNA (Zegers et al. 2006; Nishimura et al. 2007). Nevertheless, they were worth testing for having m7G2574 activity since modification enzymes with dual RNA substrate specificity have been reported. The TlyA MTase of Mycobacterium tuberculosis 2′-O-methylates C1409 of 16S rRNA and C1920 of 23S rRNA (Johansen et al. 2006). A slightly shorter version of this TlyA methylates only C1920 of 23S rRNA in Campylobacter jejuni (Monshupanee et al. 2012; Sałamaszyńska-Guz et al. 2018). The RlmN MTase modifies both rRNA (m2A2503 in 23S RNA) and tRNA (m2A37) (Benítez-Páez et al. 2012). Similarly, the pseudouridine synthases RluA and RluF of E. coli act on both 23S rRNA and tRNA (Raychaudhuri et al. 1999; Addepalli and Limbach 2016).
The genes of the three B. subtilis candidates (trmB, rsmG, and ywbD) were PCR amplified and cloned in the pET28 plasmid for expression in E. coli. The recombinant C-terminally His-tagged proteins were purified to quasi-homogeneity by immobilized metal ion affinity chromatography. These candidates were tested in an RNA methyltransferase assay using unfractionated (total) RNA preparation from E. coli as substrate. The rationale of using E. coli RNA is because this bacterium lacks m7G2574 and has an unmodified guanosine at the corresponding position. As shown in Figure 3A, only recombinant YwbD methylates E. coli RNA, indicating that this homolog of the E. coli RlmK enzyme methylates a position different from G2069 in B. subtilis. Subsequently, RNA from B. subtilis ΔywbD (in which the methylated nucleoside formed by YwbD should be lacking) and from the B. subtilis wild-type (WT) strain were tested as substrates for the enzyme. As shown in Figure 3B, total (unfractionated) RNA and rRNA from B. subtilis ΔywbD were substrates of YwbD, whereas neither tRNA fraction (which comprises also 5S rRNA) from the same mutant strain nor total RNA from B. subtilis WT were substrates of the enzyme. This observation confirms that YwbD is a 16S or 23S rRNA methyltransferase and does not methylate tRNA or 5S rRNA.
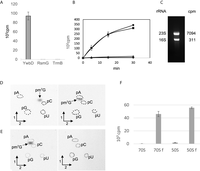
YwbD is the 23S rRNA methyltransferase forming m7G2574. (A) Fifty micrograms of total RNA extracted from E. coli WT was incubated with 1 µCi [methyl-3H] SAM and 100 ng of purified YwbD, RsmG, or TrmB of B. subtilis. After incubation, the samples were processed as described in Materials and Methods. (B) Twenty micrograms of total RNA extracted from B. subtilis WT (▴) or ΔywbD (●) cells, or rRNA (▪) and tRNA (♦) extracted from B. subtilis ΔywbD cells were incubated with 1 µCi [methyl-3H] SAM and 100 ng of purified YwbD. After incubation, reaction mixtures were processed as described in Materials and Methods, and the radioactivity incorporated in the different RNA preparations was measured using a scintillation counter. (C) B. subtilis ΔywbD rRNA (10 µg) was incubated with 1 µg of purified YwbD and 1 µCi [methyl-3H] SAM at 37°C. After 30 min incubation, the reaction mixture was loaded onto a 1% agarose gel. After migration, the bands corresponding to 23S and 16S rRNA were cut out of the gel and melted by heating. Scintillation cocktail was added, and radioactivity was measured in a scintillation counter. The number of cpm present in 23S and 16S rRNAs is indicated. (D) Autoradiography of 2D chromatograms of P1 hydrolysates of rRNA extracted from B. subtilis ΔywbD, in vitro methylated using YwbD and [methyl-14C] SAM. Modified nucleotides were analyzed by two-dimensional thin-layer chromatography (2D-TLC) on cellulose plates (Merck). The first dimension was developed with solvent A and the second dimension was developed with solvent B (left panel) or with solvent C (right panel). The radioactive spots were visualized by autoradiography. The nucleotides were identified using reference maps (Grosjean et al. 2007). Dotted circles show the migration of pA, pC, pG, and pU nucleotides used as ultraviolet markers. The spot corresponding to pm7G is indicated. (E) Autoradiography of 2D chromatograms of P1 hydrolysates of methylated WT 23S rRNA transcript (left panel) or (G2574A) 23S rRNA transcript (right panel) by YwbD in the presence of [methyl-14C] SAM. The second chromatography dimension was developed with solvent C. (F) Purified 70S and 50S ribosomal particles are poor substrates of YwbD. A total of 0.5 A260 units of 70S or 50S ribosomal particles were incubated for 30 min with 1 µg of purified YwbD and 1 µCi [methyl-3H] SAM. Incorporated radioactivity was measured by scintillation counting. RNA fr70S or fr50S are for RNA freed from, respectively, 70S or 50S ribosomal particles.
To determine whether 16S or 23S rRNA is modified by YwbD, rRNA from B. subtilis ΔywbD was methylated in vitro by the recombinant enzyme in the presence of 3H-SAM. After incubation, RNA was separated on a 1% agarose gel. The gel blocks containing the 16S or 23S rRNA were excised, melted by heating at 100°C, and radioactivity was measured by scintillation counting. Figure 3C shows that YwbD methylates the 23S rRNA and not the 16S rRNA.
To identify the nature of the modification catalyzed by YwbD, rRNA from B. subtilis ΔywbD was methylated in vitro by the recombinant enzyme in the presence of 14C-SAM. After the reaction, RNA was precipitated and hydrolyzed by nuclease P1. The resulting 5′-phosphate-nucleosides were separated by 2D-TLC followed by autoradiography. Figure 3D shows the formation of a radioactive nucleotide with the migration characteristics of pm7G.
To determine whether YwbD can form m7G at position 2574 of 23S rRNA, two T7 transcripts were tested in vitro as substrates of the recombinant enzyme. The first transcript corresponds to the B. subtilis WT 23S rRNA, and the second is a mutant in which G2574 was replaced by A using site-directed mutagenesis. The T7 transcripts were incubated in the presence of YwbD and 14C-SAM. After ethanol precipitation and P1 nuclease hydrolysis, the nucleotides were analyzed by 2D-TLC followed by autoradiography. As shown in Figure 3E, m7G was formed when the reaction was performed with the WT transcript, whereas no reaction was observed with the G2574A mutant, showing that YwbD is specific for position 2574 of the 23S rRNA. Consistent with this finding, MS/MS analysis showed that after RNase T1 hydrolysis of the MS1/MS2 RNase H fragment from B. subtilis ΔywbD, the fragment UACGp was found instead of UAC[m7G]CGp, indicating the absence of m7G in the 23S rRNA of the mutant (Fig. 2B). Hence YwbD is renamed as RlmQ, according to the literature relative to bacterial rRNA MTases.
To determine whether RlmQ modifies guanosine 2574 in 23S rRNA of the assembled ribosome, E. coli ribosomes were isolated from the JE28 strain in which the ribosomal L12 protein bears a hexa-histidine affinity tag at its carboxy terminus (Ederth et al. 2009). Assays were performed with the fully assembled 70S ribosome, as well as with the isolated 50S ribosomal subunit. For comparison, rRNA freed from the ribosomal particles by phenol extraction was also tested. The results presented in Figure 3F show that 70S and 50S particles are very poor substrates, whereas freed rRNA is efficiently modified by RlmQ, suggesting that modification occurs in the early steps of ribosome biogenesis.
The loss of RlmQ does not lead to growth defect, ribosome assembly intermediates accumulation, or changes in antibiotic sensibility
In E. coli, only a very small number of rRNA MTases are essential for correct ribosome assembly. Only the absence of RlmE (also called FtsJ or RrmJ), the enzyme forming Um2552, leads to accumulation of a 50S ribosomal subunit assembly intermediate (45S intermediate) (Arai et al. 2015; Pletnev et al. 2020). Because position 2574 takes part of the A-site of the essential PTC, the absence of m7G2574 could have an impact on growth and ribosome assembly. Growth rates and ribosomal subunit profiles of B. subtilis WT and ΔywbD strains were compared. Both strains were grown in the same conditions as described in Roovers et al. (2022). The doubling times for both strains were similar in rich medium (WT, 24.6 ± 3.1 min and ΔywbD, 24.0 ± 1.5 min) as well as in minimal medium (WT, 105 ± 8 min and ΔywbD, 104 ± 10 min). For ribosome profiling, cells were lysed and ribosome subunits were separated by sucrose gradient centrifugation in the presence of low (0.5 mM) and high (10 mM) Mg2+ concentrations. Figure 4 shows that the ribosomal subunit profile is the same for the two strains.
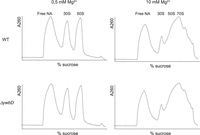
Sucrose density gradient profile of ribosomal particles from B. subtilis WT and ΔywbD grown at 37°C. Free NA signifies free nucleic acids. The peaks corresponding to 30S and 50S ribosomal subunits are indicated. The Mg2+ concentration used for ribosome particle isolation is indicated.
Various antibiotics target the ribosome thereby affecting mRNA translation or peptide bond formation at the PTC. The antibiotics chloramphenicol, lincomycin, puromycin, and linezolid target this PTC (Wilson 2014). Since m7G2574 is located in the A-site of the PTC the sensibility to these four drugs was measured for the B. subtilis WT and ΔywbD strains. The results presented in Table 1 show that the sensibility is not changed in the mutant compared to the WT strain.
Minimal inhibitory concentration (µg/mL) of different antibiotics on B. subtilis WT and ΔywbD strains at 37°C
Conclusions
In this work, we show that the modified nucleoside at position 2574 of the 23S rRNA of B. subtilis is m7G, and the enzyme responsible for its formation is encoded by the ORF ywbD. The enzyme takes part of COG1092 and is named RlmQ, according to the literature relative to rRNA MTases.
The members of COG1092 comprise m5C- and m7G-MTases as well as several uncharacterized putative MTases. While the binding domain for SAM is preserved among these members, motifs VI and VIII of the Rossmann-like fold catalytic domain diverge more strongly (Sunita et al. 2008). The cluster is divided into five distinct groups based on domain organization, oligomerization, presence of a PUA domain, or presence of a cysteine residue (or two) in the catalytic domain of the enzyme (Kita et al. 2013). Enzymes belonging to the third group are only found among the Firmicutes. These enzymes are monomeric, have a PUA RNA-binding domain, and have no cysteine in the catalytic site. These typical traits are present in B. subtilis RlmQ and in the putative MTases SAV1081 and Smu776 of Staphylococcus aureus and Streptococcus mutans, respectively. The 3D structure of these two latter MTases has been determined but their function has remained unknown (Wang et al. 2012; Kita et al. 2013). Since these enzymes are close homologs of RlmQ, it is highly probable that they also catalyze the formation of m7G2574 in 23S rRNA.
MATERIALS AND METHODS
Bacterial strains
Bacillus subtilis 168 was used as the WT strain. The ΔywbD strain derived from strain 168, was obtained from the Bacillus Genetic Stock Center (ref. BKK38361[ywbD::kan]). E. coli JE28 was a kind gift of Suparna Sanyal (Uppsala University, Sweden).
Cloning of the B. subtilis ywbD open reading frame
The ywbD ORF was amplified by PCR using Phusion polymerase (ThermoFisher Scientific) and oligonucleotides with the restriction sites NcoI and XhoI to facilitate cloning into the pET28 expression vector. This plasmid allows T7-dependent expression in E. coli of the B. subtilis YwbD protein bearing a C-terminal His-tag.
Expression and purification of YwbD
The E. coli BL21(DE3) strain was transformed with the expression vector and grown in 1 L of Luria broth at 37°C until an optical density of 0.6 was reached. Then 0.2 mM isopropyl-β-d-thiogalactopyranoside was added to the culture which was maintained at 37°C for 3 h. The cells were harvested by centrifugation. Cell lysis and recombinant YwbD purification were performed as described previously for RlmP (Roovers et al. 2022).
Preparation of B. subtilis total RNA, rRNA, and tRNA
The procedure for total RNA, rRNA, and tRNA isolation was as in Roovers et al. (2022).
Mutation and T7 in vitro transcription of the B. subtilis 23S rRNA gene
The rrnO gene encoding 23S rRNA was cloned in the pJET2.1 vector (Roovers et al. 2022). The G2574A mutant was generated by site-directed mutagenesis. T7 in vitro transcription was performed according to the instructions of the RiboMAX Kit (Ambion). The 23S rRNA transcripts were purified using illustra MicroSpin G25 columns (GE Healthcare).
Purification of His-tagged ribosomes from E. coli JE28
The procedure for His-tagged ribosome and 50S particle purification was as in Roovers et al. (2022).
RNA methyltransferase assays
A semiquantitative method to follow RNA methylation in vitro consisted in measuring the amount of 3H or 14C transferred to B. subtilis or E. coli RNA, or to rRNA transcripts, using [methyl-3H or 14C] SAM as the methyl donor. The reaction mixture (200 µL) consisted of 50 mM Tris-HCl, 5 mM MgCl2, 1 µCi [methyl-3H] SAM (82 Ci/mmol) or 20 nCi [methyl-14C] SAM (58 mCi/mmol), pH 8.0 and the amount of RNA and enzyme specified in the legend of the figures. The mixture was incubated for 30 min at 37°C. The reaction was stopped by phenol extraction and the nucleic acids were TCA-precipitated. Radioactive methylated RNA was captured on a Whatman GF/C filter and washed three times with ethanol prior to the measurement of radioactivity in a scintillation counter.
For modified nucleotide identification, the in vitro methylated RNA was ethanol precipitated after phenol extraction and thereafter hydrolyzed by nuclease P1. Modified nucleotides were analyzed by 2D-TLC on cellulose plates (Merck). The first dimension was developed with solvent A (isobutyric acid/concentrated NH4OH/water; 66/1/33; v/v/v); the second dimension was developed with solvent B (0.1M sodium phosphate at pH 6.8/solid (NH4)2SO4/n-propanol; 100/60/2; v/w/v) or solvent C (concentrated HCl/2-propanol/water; 17.6/68/14.4; v/v/v). The migration pattern was visualized by autoradiography. The nucleotides were identified using a reference map (Grosjean et al. 2007).
Mass spectrometry
Isolation of a specific RNA fragment
For LC–MS/MS analysis, a fragment of B. subtilis 23S rRNA containing G2574 was obtained by RNase H (Thermo Fisher Scientific) cleavage of RNA regions complementary to the DNA oligonucleotides MS-1 and MS-2 (Roovers et al. 2022). The 23S rRNA digestion was performed in RNase H buffer (20 mM Tris-HCl, 40 mM KCl, 8mM MgCl2, 1 mM DTT, pH 7.8) for 2 min at 80°C followed by slow cooling to 50°C and incubation with 0.5 U of RNase H for 20 min at 50°C. The RNase H fragment was isolated by denaturing (8 M urea) 10% polyacrylamide gel electrophoresis. The corresponding band was excised under UV light for LC–MS/MS analysis.
T1 digestion and mass spectrometry analysis
LC–MS/MS analysis was done as previously described (Antoine and Wolff 2020). Briefly, gel pieces containing the RNase H fragment were digested by 20 µL of 0.1 U/µL RNase T1 (Thermo Fisher Scientific) during 4 h at 50°C. Samples were desalted using ZipTip C18 (Millipore) by several washes with 200 mM ammonium acetate and elution with 50% acetonitrile in milli-Q water and finally dried under vacuum. The pellet containing RNase digestion products was resuspended in 3 µL of milli-Q water. The products were separated on an Acquity peptide BEH C18 column (130 Å, 1.7 µm, 75 µm × 200 mm) using a nanoAcquity system (Waters). The column was equilibrated in a buffer containing 7.5 mM triethylammonium acetate, 7.0 mM triethylamine, and 200 mM hexafluoroisopropanol at a flow rate of 300 nL/min. The column was achieved using a gradient from 15% to 35% methanol for 2 min and the oligonucleotides were eluted with an increase of methanol up to 50% in 20 min. MS and MS/MS analysis was performed using a SYNAPT G2-S instrument (Waters). All experiments were performed in negative mode with a capillary voltage set at 2.6 kV and a sample cone voltage set at 30 V. The source was heated to 130°C. Samples were analyzed over an m/z range from 500 to 1500 for the full scan, followed by a fast data direct acquisition scan (Fast DDA). CID spectra were deconvoluted using MassLynx software (Waters) and manually sequenced by following the y and/or c series.
Analysis of ribosomal subunits by sucrose density gradient
The analysis of ribosomal subunits was performed by sucrose density gradient (Arai et al. 2015; Pletnev et al. 2020). B. subtilis cells were cultivated in 1 L of LB medium at 22°C and 37°C until A660 reached 0.5, chilled on ice, and harvested by centrifugation. The cells were suspended in 20 mL of buffer A (20 mM Hepes-KOH, 0.5 or 10 mM Mg(OAc)2, 200 mM NH4Cl, 6 mM β-mercaptoethanol, pH 7.6). Cells were sonicated at 4°C using a Branson 250 sonicator. The lysate was cleared by centrifugation (16,000g for 30 min at 4°C), and 15 A260 units were layered on top of a sucrose gradient (10%–40% [w/v]) in buffer A supplemented with 13 U/µL heparin and separated by ultracentrifugation in a Beckman SW-41Ti Rotor at 35,000 rpm for 5 h at 4°C. Ribosomal subunits were fractionated on a Piston Gradient Fractionator, and the A260 was measured using a UV monitor.
Minimal inhibitory concentration determination
To determine the minimal inhibitory concentration (MIC) of antibiotics for B. subtilis WT and ΔywbD strains, 50 µL of exponentially grown cultures were inoculated in 3 mL of medium supplemented with different concentrations of antibiotic and incubated overnight at 37°C. The minimal antibiotic concentration that completely inhibited growth was defined as MIC.
ACKNOWLEDGMENTS
The authors thank Suparna Sanyal for providing the JE28 strain. C.G., V.K., and L.D. were supported by a grant from the Fonds J. Brachet. P.W. was supported by IdEx Unistra (ANR-10-IDEX-0002), the SFRI-STRAT'US project (ANR 20-SFRI-0012), and EUR IMCBio (ANR-17-EURE-0023) under the framework of the French Investments for the Future Program, as part of the Interdisciplinary Thematic Institute IMCBio (ITI 2021-2028 program of the University of Strasbourg, CNRS, and Inserm). A.L. was supported by the Fonds Régional de Coopération pour la Recherche (Region Grand Est, EpiRNA).
Footnotes
-
Article is online at http://www.rnajournal.org/cgi/doi/10.1261/rna.079853.123.
- Received October 2, 2023.
- Accepted December 3, 2023.
This article is distributed exclusively by the RNA Society for the first 12 months after the full-issue publication date (see http://rnajournal.cshlp.org/site/misc/terms.xhtml). After 12 months, it is available under a Creative Commons License (Attribution-NonCommercial 4.0 International), as described at http://creativecommons.org/licenses/by-nc/4.0/.








