
Shutdown of multidrug transporter bmrCD mRNA expression mediated by the ribosome-associated endoribonuclease (Rae1) cleavage in a new cryptic ORF
- Expression Génétique Microbienne (EGM), CNRS, Université Paris Cité, Institut de Biologie Physico-Chimique, 75005 Paris, France
- Corresponding authors: braun{at}ibpc.fr, condon{at}ibpc.fr
Abstract
Rae1 is a well-conserved endoribonuclease among Gram-positive bacteria, cyanobacteria, and the chloroplasts of higher plants. We have previously shown that Rae1 cleaves the Bacillus subtilis yrzI operon mRNA in a translation-dependent manner within a short open reading frame (ORF) called S1025, encoding a 17-amino acid (aa) peptide of unknown function. Here, we map a new Rae1 cleavage site in the bmrBCD operon mRNA encoding a multidrug transporter, within an unannotated 26-aa cryptic ORF that we have named bmrX. Expression of the bmrCD portion of the mRNA is ensured by an antibiotic-dependent ribosome attenuation mechanism within the upstream ORF bmrB. Cleavage by Rae1 within bmrX suppresses bmrCD expression that escapes attenuation control in the absence of antibiotics. Similar to S1025, Rae1 cleavage within bmrX is both translation- and reading frame-dependent. Consistent with this, we show that translation-dependent cleavage by Rae1 promotes ribosome rescue by the tmRNA.
Keywords
INTRODUCTION
In Bacillus subtilis, mRNA degradation is typically catalyzed either by the RNase J1/J2 complex that degrades RNA exoribonucleolytically from the 5′ end, or by endoribonucleases such as the double strand-specific RNase III or the single strand-specific RNase Y, a functional analog of Escherichia coli RNase E (Condon and Bechhofer 2011; Lehnik-Habrink et al. 2011; Durand et al. 2012; Trinquier et al. 2020; Laalami et al. 2021). In these pathways, ribosomes generally protect mRNAs, either by blocking the progression of RNase J1/J2 (Mathy et al. 2007; Daou-Chabo et al. 2009; Braun et al. 2017) or by masking endoribonucleolytic cleavage sites located within open reading frames (ORFs). In contrast to these well-described pathways that are typically inhibited by translation, we have recently identified a new RNase called Rae1 (ribosome-associated endoribonuclease 1) that actually requires translation to cleave mRNA (Lalaouna and Massé 2017; Leroy et al. 2017). We solved the crystal structure of Rae1 and modeled it into the A-site of the B. subtilis ribosome such that it is positioned to cleave the mRNA between the A-site and P-site codons (Condon et al. 2018). Despite the predicted similarity in mechanisms, Rae1 does not share any structural homology with RelE, a member of the type II toxin–antitoxin (TA) family that binds the ribosomal A-site and typically cleaves mRNAs after the second nucleotide of the A-site codon (Hwang and Buskirk 2017), or to yeast Cue2, involved in No Go Decay (NGD), that has also been proposed to act at the A-site (D'Orazio et al. 2019).
Following a global study to identify Rae1 substrates, we previously showed that Rae1 cleaves the yrzI operon mRNA in a translation- and reading frame-dependent manner within a short ORF called S1025 that encodes a 17-aa peptide of unknown function (Leroy et al. 2017). The bmrBCD operon mRNA, encoding a heterodimeric ABC multidrug exporter formerly known as YheHI (Torres et al. 2009; Galián et al. 2011), was predicted as a second direct target of Rae1 in this global study (Leroy et al. 2017). Its expression pattern is highly similar to that of yrzI (correlation coefficient 0.7) in tiling array experiments performed in >100 different growth conditions in B. subtilis (Nicolas et al. 2012), suggesting that these two operons have linked functions. Both mRNAs were also among the six most highly up-regulated mRNAs in the presence of subinhibitory doses of the protein synthesis inhibitor chloramphenicol (Cm) (Lin et al. 2005). Expression of the bmrCD portion of the operon is also up-regulated in the presence of other antibiotics that target the ribosome, including erythromycin and lincomycin, via a transcription attenuation mechanism thought to be triggered by antibiotic-induced ribosome pausing within the bmrB upstream ORF (Reilman et al. 2014).
Here, we examine the role of Rae1 in the degradation of the polycistronic bmrBCD mRNA and map a Rae1 cleavage site just downstream from the attenuator structure of the operon, within a newly identified short ORF, we call bmrX. Cleavage by Rae1 shuts down considerable readthrough expression of bmrCD in the absence of antibiotics. Consistent with our previous results, we show that Rae1 also cleaves within bmrX in a translation- and reading frame-dependent manner. We also show that mRNA cleavage by Rae1 activates the transfer-messenger RNA (tmRNA)-dependent pathway for ribosome rescue.
RESULTS AND DISCUSSION
The polycistronic bmrBCD mRNA is a direct target of Rae1
The polycistronic bmrBCD mRNA (Fig. 1A) encoding the multidrug transporter BmrCD (Torres et al. 2009) was identified as a second potential target by RNA-seq analysis in strains either lacking Rae1 or complemented with a plasmid overexpressing it (Leroy et al. 2017). Because the expression of the bmrBCD operon is repressed by AbrB at the transcriptional level (Reilman et al. 2014), we first wanted to confirm that increased expression observed in a rae1 deletion strain was due to an increase in bmr mRNA stability rather than an indirect effect on abrB. Northern blots of total mRNA isolated at different times after the inhibition of transcription initiation by rifampicin addition showed no impact of the rae1 deletion on abrB mRNA stability. In contrast, the same northern blot probed for bmrC showed that the bmr transcript was highly stabilized in the Δrae1 strain compared to the wild-type (WT), where it was barely visible at the initial time point and disappeared rapidly thereafter (Fig. 1B). The bmr mRNA was also destabilized in a Δrae1 strain ectopically expressing WT Rae1, but not a D7N D81N catalytic mutant, indicating that its destabilization requires Rae1 catalytic activity (Fig. 1C). Furthermore, the bmr mRNA levels were increased in both Δrae1 and ΔabrB cells, and even further in the Δrae1 abrB double mutant consistent with independent modulation of bmrCD expression by AbrB and Rae1 (Supplemental Fig. 1A). These data clearly indicate that Rae1 shuts off bmr operon expression at a post-transcriptional level by accelerating its rate of decay.
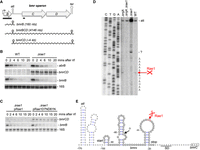
Expression of the bmr operon is extinguished at the post-transcriptional level by Rae1 cleavage downstream from the attenuator. (A) Annotated structure of the bmr operon. ORFs are represented by gray boxes, the promoter by a rightward-pointing arrow, and the transcription attenuator (att) and terminator (ter) by hairpin structures. The two primary transcripts (bmrB and bmBCD) and the processed mRNA (bmrCD) are shown as wavy lines and their approximate sizes are indicated. The position of the probes used is indicated by black bars. (B) Northern blot showing levels of bmr and abrB mRNAs at times after rifampicin addition in WT and Δrae1 strains at mid-log phase. (C) Northern blot showing levels of bmr mRNAs at times after rifampicin addition to the Δrae1 strain containing either a plasmid expressing WT Rae1 (pRae1) or the catalytic mutant (pRae1D7ND81N) at mid-log phase. Blots were probed in a sequential order with oligo CC2344 (bmrC) (B,C), oligo CC2145 (abrB) (B), a riboprobe against bmrB (B,C), and finally with a probe complementary to 16S rRNA (CC058) as a loading control (B,C). (D) Primer extension assay on total RNA isolated from WT and strains lacking Rae1 (Δrae1), RNase J1 (ΔrnjA), and both (Δrae1 ΔrnjA) using oligonucleotide (CC2344) that hybridizes to the early bmrC coding sequence. Sequence lanes are labeled as their reverse complement to facilitate direct read-out. The mapped 5′ ends are shown to the right of the autoradiogram. A reverse transcriptase (RT) stop corresponding to the attenuator (att) is indicated. This experiment was repeated twice. (E) Predicted secondary structure (Mulfold: http://www.unafold.org/mfold/applications/rna-folding-form.php) of the bmrB–bmrC intergenic region. Coordinates are given relative to the AUG start codon of bmrC (boxed). The two 5′ extremities mapped by RT are shown. The predicted attenuator stem–loop (att) is shown and the Shine–Dalgarno (SD) sequence of B. subtilis 16S rRNA is underlined. The start and the stop codon of the predicted bmrX ORF are indicated.
Rae1 cleaves in the intergenic region between bmrB and bmrC
Since we had previously shown that Rae1 cleaves within the short S1025 coding sequence (Leroy et al. 2017), we initially hypothesized that the Rae1 cleavage site might be located within the short bmrB ORF involved in antibiotic-mediated attenuation control (Reilman et al. 2014). In the absence of antibiotics, transcription attenuation results in the production of a 160 nt transcript containing only bmrB, whereas in the presence of antibiotics, an intact readthrough bmrBCD transcript would be predicted to be 4146 nt in length (Fig. 1A). The stability of the 160 nt bmrB transcript was similar in both the WT and the Δrae1 strain (Fig. 1B), as well as in a Δrae1 strain ectopically expressing WT Rae1 or a D7N D81N catalytic mutant (Fig. 1C), indicating that Rae1 does not cleave the attenuated bmrB transcript. Strikingly, we did not detect the full-length polycistronic bmrBCD mRNA with a riboprobe complementary to the bmrB transcript (Supplemental Fig. 1B), suggesting that it is rapidly processed to a shorter form containing only bmrC and D, and further suggesting that the Rae1 cleavage site is located downstream from bmrB, within the ∼4 kb bmrCD portion of the transcript. Experiments from DeLoughery et al. (2018) suggest that the full-length bmr transcript is initially processed by RNase Y upstream of the attenuator hairpin, which likely protects the bmrCD portion of the transcript from 5′-exoribonucleolytic degradation. The strong accumulation of this transcript in the absence of Rae1 suggests that transcriptional attenuation does not efficiently block transcription of the bmrCD portion of the mRNA in the absence of antibiotics. Thus, these two mechanisms together apply “belt and braces” security to fully turn off the expression of the multidrug transporter BmrCD in the absence of substrates.
To narrow down the location of the Rae1 cleavage site, we generated a series of plasmids containing the bmrC ORF with different truncations from the 5′ end (Supplemental Fig. 1C). The pPspac-bmrBC construct begins 5 nt downstream from the native transcription start site, pPspac-hp-bmrC starts 9 nt upstream of the attenuator and pPspac-bmrC starts 40 nt upstream of the bmrC start codon (Supplemental Fig. 1C). All constructs were placed under the control of the IPTG-inducible Pspac promoter to homogenize the 5′ ends of the resulting mRNAs and to avoid potential transcriptional regulation by AbrB. The Rae1 sensitivity of the different mRNAs transcribed from these plasmids was analyzed by northern blot in WT and Δrae1 strains. The mRNAs from the pPspac-bmrBC and pPspac-hp-bmrC constructs both accumulated in the Δrae1 strain, while that from the shorter pPspac-bmrC construct did not (Supplemental Fig. 1D), suggesting that the Rae1 cleavage site is located between the attenuator and the start of bmrC.
To map the Rae1 cleavage site precisely, we performed primer extension assays on total RNA isolated from WT and Δrae1 strains in a ΔrnjA background to protect the downstream cleavage product from 5′–3′ degradation, if necessary. A RT stop at the base of the bmr attenuator structure was detected in all strains (Fig. 1D). This band was stronger in the Δrae1 mutant consistent with the higher levels of the bmrCD mRNA in this strain. In the WT and ΔrnjA strains, we observed an RT stop mapping to 32 nt upstream of the bmrC start codon that was absent in both the Δrae1 and Δrae1 ΔrnjA double mutant (Fig. 1D,E), suggesting that this is the site of Rae1 cleavage. In the Δrae1 strain, we detected an additional strong RT stop 30 nt upstream of the Rae1 site (Fig. 1D,E). It is not clear whether this stop is caused by secondary structure in this location or whether another enzyme cleaves at this site in the absence of efficient degradation initiated by Rae1. Altogether, these data indicate that Rae1 cleaves in the bmrB–bmrC intergenic region.
Rae1 cleaves the polycistronic bmrBCD mRNA within a newly identified small ORF
We had previously shown that Rae1 cleaves within the S1025 ORF in a translation-dependent manner. We thus searched for a possible unannotated ORF in the bmrB-C intergenic region that overlapped the mapped Rae1 cleavage site. We anticipated that this new ORF would be truncated in the pPspac-bmrC (−40) construct to explain why it was insensitive to Rae1 despite containing the cleavage site (Fig. 1E; Supplemental Fig. 1C,D). We first made plasmid-borne fusions of the native bmr promoter (Pbmr) and the first ∼300 nt of the bmrBCD operon to GFP, in all three reading frames (Fig. 2A). These three constructs were introduced into the WT strain and assayed for GFP production directly on agar plates. Only reading frame 0 promoted GFP expression (Fig. 2B, left), corresponding to a potential 26-aa ORF that we named bmrX (Fig. 2C). Previously published ribosome profiling data was consistent with ribosome binding to the predicted GUG start codon of bmrX (Supplemental Fig. 2A; Li et al. 2012). Reinforcing this data, mutation of the GUG to a stop codon (UAG) abolished GFP expression (Fig. 2B, right). Rae1 cleavage occurred precisely between an AAA (Lys) and AUA (Ile) codon at positions 22 and 23 of the bmrX ORF (Fig. 2C). The BmrX peptide is conserved in several other Bacillus species, in particular the last 4 aa, with the EKIEGG codon context of the Rae1 cleavage site conserved in a subset of these peptides (Supplemental Fig. 2B). There is no obvious relationship between the bmrX and S1025 cleavage sites, with the latter occurring between a glutamate codon (GAG) in position 13 and a lysine codon (AAG) in position 14 in a conserved MEKDQV sequence (Leroy et al. 2017).
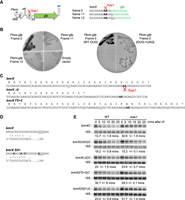
Rae1 cleaves within an unannotated cryptic ORF, bmrX, in a translation-dependent manner. (A) Structure of the pbmrBXgfp plasmid. ORFs are represented by colored boxes, the promoter by a rightward-pointing arrow, the attenuator (att) and transcription terminator (ter) by hairpin structures, and the Rae1 site by a scissors symbol. Coordinates are given relative to the AUG start codon of bmrC. The sequence of the junction of the gfp gene with the bmrBC intergenic region in the three frames is shown. The gfp sequence is indicated in green. Rae1 cleaves between the two A-residues indicated in bold. (B) Fluorescence of WT strains expressing GFP from the fusion constructs pbmrBXgfp in the three different frames (0, +1, or +2) and of the WT containing an empty vector (left panel). Fluorescence of WT strains expressing GFP in frame 0 containing either the GUG start codon or the UAG mutant (right panel). (C) Nucleotide and amino acid sequence of WT and mutant derivatives of bmrX. The Rae1 cleavage site is indicated in bold. The bmrX(Δ2) mutant contains a deletion of two C-residues (underlined in the WT bmrX sequence), creating a premature stop codon. In the bmrX(UAG) mutant, the GUG translation start codon was replaced by UAG. In the bmrX(FS + 2) mutant, 2 nt (underlined) were added to induce a +2 frameshift. (D) Ribosome binding sequence of the WT bmrX and the bmrX(SD+) mutant. The potential base-pairing with the 3′ end of 16S rRNA is indicated. The modified nucleotides in bmrX(SD+) mutant are in bold and the start codon is underlined. (E) Northern blot analysis of WT and Δrae1 strains containing WT or mutant derivatives of Pspac-bmrBXC at times after rifampicin addition, probed with oligo CC2344 (bmrC) and then with a probe complementary to 16S rRNA (CC058) as a loading control. Quantification of the blots presented is given in Supplemental Figure 4. The calculated half-lives are the average of at least three independent experiments.
We had previously shown that insertion of a fragment containing the S1025 ORF into the 3′ UTR of the highly stable hbsΔ reporter mRNA rendered this mRNA sensitive to Rae1 (Leroy et al. 2017). We therefore asked whether exchanging S1025 in the hbsΔ-S1025 construct with the bmrX ORF (Supplemental Fig. 3A) would promote a similar destabilization of the transcript. The hbsΔ-bmrX construct yields two primary transcripts from promoters P3 (P3-ter) and P1 (P1-ter) and a highly stable ribosome-protected species (R-ter). As was observed for hbsΔ-S1025, all three hbsΔ-bmrX transcripts showed increased stability in the Δrae1 strain compared to WT. Indeed, the R-ter species was fully stabilized in the absence of Rae1, while only traces of it were detected in WT cells (Supplemental Fig. 3B) showing that, like S1025, the bmrX ORF can sensitize the hbsΔ mRNA to Rae1. To further corroborate this result, we made additional S1025 and bmrX constructs without the hbsΔ moiety expressed from a constitutive variant of the Pspac promoter lacking the lac operator [Pspac (con)]. The steady-state levels of the short S1025 transcript were highly increased in the Δrae1 strain compared to WT (Supplemental Fig. 3C), as observed with the hbsΔ-S1025 construct (above) and the endogenous polycistronic transcript (Leroy et al. 2017). An accumulation of the short bmrX-containing transcript was also observed in the absence of Rae1, confirming that the presence of the bmrX ORF also sensitizes this mRNA to Rae1, albeit to a lesser degree than S1025 (Supplemental Fig. 3C). These data confirm that the previously unannotated 26-aa bmrX ORF contains a bona fide and transportable Rae1 cleavage site.
Cleavage by Rae1 within the bmrX ORF is translation-dependent
To ask whether Rae1 cleaves within the bmrX ORF in a translation-dependent manner, we made four mutations in the pPspac-bmrBC (−315) construct described above (Supplemental Fig. 1C), which we renamed pPspac-bmrBXC to account for the newly discovered ORF. The mutations consisted of (i) replacing the bmrX GUG start codon by an UAG stop codon [bmrX(UAG)], (ii) deleting two consecutive C-residues in the bmrX coding sequence upstream of the Rae1 cleavage site to create a premature stop codon [bmrX(Δ2)] before the Rae1 cleavage site, (iii) adding 2 nt upstream of the Rae1 cleavage site to modify the reading frame [bmrX(FS+2)], or (iv) optimizing the SD sequence and translation start site from CAGGUGUAUGGGAUGUG to AAGGAGGAUGGGAUAUG to provide a better match to the 3′ end of 16S rRNA [bmrX(SD+)] (Fig. 2C,D). The impact of these four mutations on mRNA stability was analyzed in WT and Δrae1 strains by northern blot (Fig. 2E). Note that, like the mRNA derived from the native bmr locus, the bmrBXC transcript was processed upstream of the attenuator, leaving only the bmrXC portion detectable by northern blot with the bmrC probe.
The WT bmrXC mRNA was stabilized 2.2-fold in the Δrae1 strain (half-life of 13.7 min in the WT vs. 30.6 min in the Δrae1 strain) (Fig. 2E; Supplemental Fig. 4). In contrast, the half-lives of the bmrX(UAG)C, bmrX(Δ2)C and bmrX(FS + 2)C transcripts were all unchanged in the absence of Rae1 (Fig. 2E; Supplemental Fig. 4). The mRNA with the optimized translation start site, bmrX(SD+)C, was only stabilized ∼1.4-fold (23.2 vs. 31.7 min half-life) in the absence of Rae1 (Fig. 2E; Supplemental Fig. 4), suggesting that cleavage by Rae1 becomes less efficient as translation improves. Thus, as previously observed for S1025, destabilization of the bmr mRNA by Rae1 only occurs if the bmrX cleavage site is translated in the correct reading frame.
Cleavage by Rae1 leads to ribosome rescue by the transfer-messenger RNA
Translation-dependent cleavage of the bmrX and S1025 ORFs by Rae1 is predicted to create truncated mRNAs that are covered in ribosomes and potentially lead to the intervention of the tmRNA. The tmRNA enters the A-site of bacterial ribosomes stalled on truncated mRNAs to promote tagging of the truncated peptide for degradation, ribosome rescue, and the turnover of the defective RNA (Himeno et al. 2014). To investigate the involvement of the tmRNA, we used strains expressing a modified tmRNA (ssrA-H6DD) that adds a histidine tag that escapes degradation via two carboxy-terminal D-residues that block the proteolytic pathway (Fujihara et al. 2002). This strain also contained a reporter gene, consisting of GFP fused in frame to the amino terminus of the Rae1 target S1025 (gfp-S1025) integrated into the amyE locus (Fig. 3A). We anticipated that, if the tmRNA intervenes following Rae1 cleavage, a stable His-tag would be added to the GFP-S1025 fusion protein. The ssrA-H6DD strain containing the gfp-S1025 fusion was transformed with either the plasmid expressing Rae1 (pRae1) or the empty vector. We first verified that the gfp-S1025 mRNA was sensitive to Rae1 in this background by showing that it was destabilized in the strain overexpressing Rae1 compared to the empty vector (Fig. 3B). A weak band (*) that migrates just below the full-length gfp-S1025 transcript (940 nt) was detected in cells overexpressing Rae1 that likely corresponds to the cleaved gfp-S1025 species (anticipated size, 774 nt). Consistent with this idea, the shorter species was not detected with a probe that hybridized to the 3′-UTR downstream from the Rae1 cleavage site, indicating that it is truncated from the 3′ end (Fig. 3B). To determine whether a His-tag was added to the carboxyl terminus of the GFP-S1025 fusion, protein extracts were absorbed on Ni2+-NTA agarose beads, washed and eluted with imidazole and the different fractions probed for GFP by western blot (Fig. 3C). The GFP-S1025 fusion protein was strongly enriched in the elution fraction of cells overexpressing Rae1 compared to the empty vector, indicating that the Rae1-cleaved gfp-S1025 mRNA is rescued by the tmRNA.
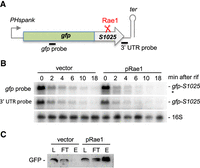
Rae1 cleavage leads to ribosome rescue by tmRNA. (A) Schematic of the gfp-S1025 fusion. The fusion is placed under the control of the IPTG-inducible Hyperspank promoter in vector pDR111. The Rae1 site within S1025 is indicated by red scissors. The position of the probes used is indicated by black bars. (B) Northern blot analysis at times after rifampicin addition strains containing the gfp-S1025 fusion and mutant ssrA (ssrA-H6DD) transformed either as a plasmid expressing the WT protein Rae1 (pRae1) or the empty vector. The blot was probed in a sequential order with oligos complementary to the 3′ UTR (CC429), gfp (CC2422), and then 16S rRNA (CC058) as a loading control. (C) Proteins were extracted from strains used in panel B, absorbed, washed and eluted from Ni2+-NTA agarose beads. The different fractions were assayed by western blot using GFP-specific antibodies. Fraction L corresponds to the extract loaded on the beads, FT to the flow-through fraction, and E to the elution fraction. This experiment was repeated three times.
Rae1 overexpression mitigates the induction of S1025 and bmrX expression in response to chloramphenicol
A DNA microarray analysis identified the two confirmed Rae1 targets, the bmr and yrzI operon mRNAs, among the six most highly up-regulated mRNAs in B. subtilis following the addition of sublethal concentrations of Cm (Lin et al. 2005). We therefore asked whether the increased expression of these operons in the presence of Cm was Rae1-dependent. We extracted RNA from WT and Δrae1 cells 0, 15, 30, or 60 min after the addition of 0.1 or 0.5 times the minimum inhibitory concentration (MIC) of Cm (5 µg/mL). Northern blot analysis showed that both the yrzI and bmrXCD operon mRNAs accumulated upon Cm addition to WT cells (Fig. 4A), in agreement with the published data (Lin et al. 2005). As observed previously (Leroy et al. 2017), three transcripts were detected for yrzI: the two primary transcripts P1-T4 and the P3-T4 from the P1 and P3 promoters to the main operon terminator (T4) and the matured R-T4 transcript extending from 21 nt upstream of the yrzI coding sequence to the T4 terminator. A very similar qualitative and quantitative pattern was observed in the absence of Rae1, suggesting that the increased expression of these operons in the presence of Cm in WT cells is Rae1-independent.

Overexpression of Rae1 mitigates the induction of S1025 and bmrX expression in response to chloramphenicol. (A) Northern blot analysis of total RNA upon addition of subinhibitory concentrations of Cm to exponentially growing cultures at 0.5 µg/mL and 2.5 µg/mL. Chloramphenicol was added for the times indicated. (B) Northern blot analysis of total RNA isolated from a Δrae1 strain containing either an empty vector (pDG) or a plasmid expressing the WT protein Rae1 (pRae1) at mid-log phase in rich medium supplemented, or not, with 0.5 µg/mL chloramphenicol. Quantifications of the impact of Rae1 overexpression on mRNA levels from two independent experiments are indicated. (A,B) Northern blots were probed in a sequential order: oligo CC2344 (bmrC), oligo CC1589 (yrzI), and then with a probe complementary to 16S rRNA (CC058) as a loading control. For yrzI, the correspondence between bands and transcripts is given to the right of the blot: the P1-T4 and the P3-T4 correspond to primary transcripts from the P1 and P3 promoters to the main operon terminator (T4). R-T4 refers to the 521-nt species extending from 21 nt upstream of the yrzI coding sequence to the T4 terminator.
We next asked the reverse question, that is whether the overexpression of Rae1 could interfere with the induction of the expression of these mRNAs by Cm. We compared yrzI and bmrXCD mRNA levels in Δrae1 cells containing either an empty vector (pDG) or a plasmid expressing the WT Rae1 protein (pRae1) in the presence of a sublethal concentration of Cm (0.1 MIC). Northern blot analysis showed that the effect of antibiotic addition was diminished for both mRNAs when Rae1 was overexpressed (Fig. 4B), suggesting that Rae1 can mitigate the induction of yrzI and bmrXCD expression in response to sublethal concentrations of Cm (Fig. 4B). However, the reduction in mRNA levels was weaker in the presence of Cm than in its absence: 1.4-fold versus twofold for the bmrXCD transcript and 1.7-fold versus 7.3-fold for the yrzI R-T4 transcript (Fig. 4B), suggesting that Rae1 is less efficient at promoting the degradation of its targets in the presence of Cm. Whether this is due to a specific effect of Cm on Rae1 binding to the ribosome, or a general effect on translation, remains to be seen (it could, for example, have been anticipated that slowing translation without inhibiting it completely might actually favor Rae1 activity).
Conclusion
In conclusion, we show that Rae1 cleaves the polycistronic bmrXCD transcript within the unannotated 26-aa cryptic ORF bmrX in a translation- and reading frame-dependent manner, as reported previously for the yrzI operon mRNA. This cleavage shuts off leaky readthrough expression at the attenuator that occurs in the absence of antibiotics. In the presence of antibiotics, such as Cm, Rae1 appears to be rapidly overwhelmed by the readthrough transcript and has little impact on its accumulation (Fig. 4A), suggesting Rae1 might be present in only limiting amounts in the cell.
The two characterized Rae1 targets share in common four features: they both encode a small peptide, both are cleaved by Rae1 when they are translated in a specific reading frame, both contain a transportable Rae1 cleavage site within their ORFs and both are up-regulated in response to sublethal concentrations of Cm (Lin et al. 2005). Our current model is that the peptide sequences of BmrX and S1025 permit Rae1 binding to the ribosome, for example, by provoking a ribosome stall, and that mRNA cleavage by Rae1 promotes ribosome rescue by the tmRNA.
Smr-domain-containing proteins have also been shown to cleave mRNAs in a translation-dependent manner by associating with collided ribosomes (D'Orazio et al. 2019; Saito et al. 2022). We therefore considered the possibility that the homologous protein of B. subtilis, MutS2 (Cerullo et al. 2022), could act cooperatively with Rae1. However, the yrzI and bmr mRNAs were similarly unstable in WT and mutS2 deleted strains and stabilized in both rae1 and rae1 mutS2 strains (Supplemental Fig. 5), indicating that Rae1 and MutS2 act independently of each other to promote ribosome rescue of these two substrates.
Although BmrX and S1025 are restricted to the Bacillales, Rae1 is far more widely conserved among Firmicutes, the Cyanobacteria and the chloroplasts of higher plants (Condon et al. 2018). Thus, the physiological importance of Rae1 likely extends well beyond these two operons in B. subtilis. Our working model is that Rae1 plays a more general role in translation quality control and that this activity has been appropriated to keep a tight rein on expression levels in the case of the bmr and yrzI operons.
MATERIALS AND METHODS
Strains and constructs
Strains and oligonucleotides used are shown in Supplemental Tables S1 and S2, respectively. The B. subtilis strains used in this study were derivatives of W168 (laboratory strain SSB1002).
Plasmids containing the bmrC ORF with different truncations from the 5′ end (pPspac-bmrBXC [pl867], pPspac-hp-bmrC [pl828], and pPspac-bmrC [pl829]) were constructed by PCR amplification using the forward primer CC2617, CC2451, or CC2452 and the reverse primer CC2136, containing an E. coli rRNA (rrnG) transcription terminator to limit transcription of downstream plasmid DNA. The PCR fragments were cloned between the XbaI and SphI sites of plasmid pDG148 and placed under the IPTG-inducible Pspac promoter (Stragier et al. 1988).
The pPspac-bmrBXC plasmid was used to construct four plasmids containing mutations in the bmrX ORF: pbmrBX(UAG)C (pl866), pbmrBX(Δ2)C (pl873), pbmrBX(FS + 2)C (pl841), and pbmrBX(SD+)C (pl876). They were obtained by two-fragment overlapping PCR using the pPspac-bmrBXC plasmid as template. The upstream fragment was amplified with the forward primer CC2617 and the reverse primer CC2565, CC2467, CC2774, or CC2698 and the downstream fragment with the forward primer CC2566, CC2468, CC2775, or CC2699 and the reverse primer CC2136. The overlapping fragments were reamplified using oligo pair CC2617/CC2136 and cloned between the XbaI and SphI sites of plasmid pDG148 and placed under the IPTG-inducible Pspac promoter (Stragier et al. 1988).
The pbmrBXgfp fusions containing the native bmr promoter (Pbmr) and the first 301 nt of the bmrBXCD operon in all three reading frames fused to GFP were obtained by two-fragment overlapping PCR. They were referred as pl845 (frame 0), pl844 (frame +1), and pl846 (frame +2). The upstream fragment was amplified with the primer pair CC2132/2419 and pPspac-bmrBC as a template, and the downstream fragment using the forward primers CC2521, CC2522, or CC2523 and the reverse primer CC2421, with pHM2-hbsΔ::gfpSDwt (pl719) as a template (Braun et al. 2017). The overlapping fragments were reamplified using oligo pair CC2132/CC2421 and cloned between the XbaI and SphI sites of plasmid pDG148. The pbmrBXUAGgfp fusion (pl858) containing a UAG stop codon instead of the GUG start codon was obtained by two-fragment overlapping PCR. The upstream fragment was amplified with primer pair CC2132/CC2565 and the downstream fragment with primer pair CC2566/CC2421 with pbmrBXgfp (pl845) as a template. The overlapping fragments were reamplified using oligo pair CC2132/CC2421 and cloned between the XbaI and SphI sites of plasmid pDG148.
The pDR-gfpS1025 (pl875) containing the gfp fused to the S1025 ORF was obtained by subcloning the gfpS1025 insert from the pHM2-gfpS1025 (pl869) between the SalI and HindIII of the integrative the pDR111 vector. The pHM2-gfpS1025 (pl869) plasmid was obtained by two-fragment overlapping PCR. The upstream fragment was amplified with the primer pair CC2634/2635 and pbmrBXgfp (pl845) as a template, and the downstream fragment was used with the primer pair CC2636/CC572, pHM2-hbsΔ-S1025 (pl707) as a template (Leroy et al. 2017). The overlapping fragments were reamplified using oligo pair CC2634/CC572 and cloned between the HindIII and BamHI site of the integrative pHM2 plasmid and placed under the control of a constitutive promoter Pspac(con).
The pHM2-hbsΔ-bmrX plasmid (pl908) was obtained by three-fragment overlapping PCR. The upstream fragment was amplified with primers CC1607 and CC2893, and the downstream fragment with primers CC2894 and CC572, using pHM2-hbsΔ-S1025 (pl707) as a template (Leroy et al. 2017). The third fragment was obtained by hybridizing the oligo pair CC2875/CC2876. The overlapping fragments were reamplified using oligo pair CC1607/CC572 and cloned between the EcoRI and BamHI site of the integrative pHM2 plasmid, placed under the control of the native promoters of hbs gene.
The pHM2-bmrX (pl936) and the pHM2-S1025 (pl925) plasmids were constructed by PCR amplification using the forward primer CC2962 and the reverse primer CC572 with pHM2-hbsΔ-bmrX (pl908) or pHM2-hbsΔ-S1025 (pl707) as a template. The PCR fragments were cloned between the HindIII and BamHI site of the integrative pHM2 plasmid, placed under the control of the constitutive Pspac(con) promoter.
All plasmid constructs were verified by sequencing and transformed into the strains SSB1002 (WT) and CCB375 (Δrae1) (Supplemental Table S1). In all the plasmid constructs using the pDG148 vector, the cloned genes were followed by an E. coli rRNA (rrnG) transcription terminator to limit the transcription of downstream plasmid DNA. The integrative plasmid derived from pHM2 and from pDR111 were linearized with XbaI and SacI, respectively, before transformation, for integration into the amyE locus of SSB1002 (WT) or CCB375 (Δrae1) strains (Supplemental Table S1). The CCB375 (Δrae1) and the CCB942 (ssrADD) strains are resistant to erythromycin and chloramphenicol, respectively. The replicative pDG148 and the integrative pDR111 and the pHM2 vectors confer kanamycin, spectinomycin, and chloramphenicol resistance, respectively.
Strain CCB748 was constructed by transferring the rnjA::spec construct from strain CCB434 into CCB375 (Δrae1) (Supplemental Table S1).
Strains CCB1447 and CC1448 were constructed by transforming strain CCB1444 with the pRae1 plasmid (pl660) that allows Rae1 expression (Leroy et al. 2017) or the empty pDG148 vector. Strain CCB1444 was constructed in two steps: the integrative plasmid pDR-gfpS1025 (pl875) (Supplemental Table S1) was first transformed into the WT strain SSB1002 and the resulting strain (CCB1368) was transformed with the ssrA-H6DD::Cm construct from strain AHMG Pspac-ssrA-H6DD (Fujihara et al. 2002).
GFP detection
GFP fluorescence of bacteria was detected on agar plates using Typhoon scanner (GE) with excitation at 470 nm and emission at 520 nm.
RNA isolation and northern blots
Northern blots were performed on total RNA isolated from B. subtilis cells growing 2xYT medium either by the glass beads/phenol method described in Bechhofer et al. (2008) or by the RNAsnap method described in Stead et al. (2012). Northern blots were performed as described previously (Durand et al. 2012). The bmrB riboprobe was transcribed in vitro using T7 RNA polymerase (Promega) and labeled with [α-32P]-UTP using a PCR fragment amplified with oligo pair CC2195/CC2196 as template. Northern blots were exposed to PhosphorImager screens (GE Healthcare) and the signal was obtained by scanning with a Typhoon scanner (GE Healthcare) and analyzed by Fiji (ImageJ) software.
Primer extension assays
Primer extension assays were performed on glass bead/phenol-extracted RNAs as described previously (Britton et al. 2007). Oligo CC2344 was used to map the Rae1 cleavage site within the bmrX ORF of the bmr polycistronic mRNA.
Ribosome rescue assay
For protein extractions, cells from strains CCB1447 and CC1448 were resuspended in ice-cold Buffer (20 mM Tris–HCl pH 7.5, 1 mM EDTA, 50 mM NaCl) and lysed by sonication (three times 30″). The lysate was cleared at 16,100 g for 10 min at 4°C and protein concentration was determined by the Bradford method. An amount of 400 µg of protein extracts were incubated with 50 µL of Ni-NTA agarose beads overnight, washed twice with 10 volumes of 20 mM imidazole and eluted with one volume of 250 mM imidazole. The different fractions were assayed by western blot (ECL; GE) using GFP-specific mouse antibodies (Sigma).
SUPPLEMENTAL MATERIAL
Supplemental material is available for this article.
ACKNOWLEDGMENTS
We thank laboratory members for helpful discussion. This work was supported by funds from the CNRS and Université Paris Cité (UMR8261), the Agence Nationale de la Recherche (ARNr-QC, Labex Dynamo and Equipex Cacsice). We also thank A. Muto for the gift of the strain AHMG Pspac-ssrA-H6DD.
Footnotes
-
Article is online at http://www.rnajournal.org/cgi/doi/10.1261/rna.079692.123.
- Received April 19, 2023.
- Accepted April 21, 2023.
This article is distributed exclusively by the RNA Society for the first 12 months after the full-issue publication date (see http://rnajournal.cshlp.org/site/misc/terms.xhtml). After 12 months, it is available under a Creative Commons License (Attribution-NonCommercial 4.0 International), as described at http://creativecommons.org/licenses/by-nc/4.0/.








