
The life and times of a tRNA
- 1Department of Biochemistry and Biophysics and Center for RNA Biology, University of Rochester School of Medicine, Rochester, New York 14642, USA
- 2Department of Molecular Genetics and Center for RNA Biology, Ohio State University, Columbus, Ohio 43235, USA
- Corresponding authors: eric_phizicky{at}urmc.rochester.edu, hopper.64{at}osu.edu
Abstract
The study of eukaryotic tRNA processing has given rise to an explosion of new information and insights in the last several years. We now have unprecedented knowledge of each step in the tRNA processing pathway, revealing unexpected twists in biochemical pathways, multiple new connections with regulatory pathways, and numerous biological effects of defects in processing steps that have profound consequences throughout eukaryotes, leading to growth phenotypes in the yeast Saccharomyces cerevisiae and to neurological and other disorders in humans. This review highlights seminal new results within the pathways that comprise the life of a tRNA, from its birth after transcription until its death by decay. We focus on new findings and revelations in each step of the pathway including the end-processing and splicing steps, many of the numerous modifications throughout the main body and anticodon loop of tRNA that are so crucial for tRNA function, the intricate tRNA trafficking pathways, and the quality control decay pathways, as well as the biogenesis and biology of tRNA-derived fragments. We also describe the many interactions of these pathways with signaling and other pathways in the cell.
Keywords
INTRODUCTION
The elemental steps of eukaryotic tRNA biogenesis have been known for some time. After transcription by RNA polymerase III, pre-tRNA maturation involves a number of size-altering steps, including endonucleolytic removal of the 5′ leader, endonucleolytic and/or exonucleolytic removal of the 3′ trailer, untemplated CCA addition to the 3′ end, untemplated addition of a G−1 residue to the 5′ end of tRNAHis, and enzymatic splicing of the introns found between N37 and N38 in a subset of tRNAs. Each of these stages also involves the formation of modifications, ∼13 in the typical cytoplasmic tRNA from the budding yeast Saccharomyces cerevisiae, with each tRNA having its own specific combination of the 25 chemically distinct modifications that occur in 36 different locations in the tRNA. In addition, each tRNA is subject to a number of intracellular trafficking steps, which themselves may differ among different tRNAs (Fig. 1).
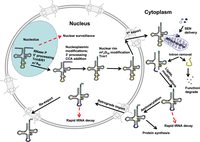
Schematic of tRNA biogenesis, subcellular dynamics, and quality control turnover pathways in S. cerevisiae. tRNAs are transcribed in the nucleolus where the 5′ leader (left purple circles) of the initial transcript is removed by RNase P and likely where m1A58 is modified (black circle) by Trm6/61. About half of the known modifications (examples, orange circles) occur in the nucleoplasm where 3′ CCA nucleotides (green circles) are also added. Dimethylation of G26 (magenta circle) is catalyzed by Trm1, which is located on the inner nuclear membrane, prior to nuclear export of the end-matured, partially processed, intron-containing (yellow circles) pre-tRNAs; end-processed, partially modified tRNAs encoded by genes lacking introns are also exported to the cytoplasm. Introns are removed on the mitochondrial cytoplasmic surface. After/during splicing, additional modifications are added in the cytoplasm (examples, blue circles), and the freed introns are destroyed. Processed/modified cytoplasmic tRNAs return to the nucleoplasm via retrograde tRNA nuclear import and under stress conditions accumulate there; in favorable conditions the tRNAs return to the cytoplasm via reexport where they participate in protein synthesis. There are quality control steps, indicated by red dashed arrows, that destroy tRNAs that have not undergone the canonical (black arrows) steps appropriately. Further details of the cell biology and quality control pathways are provided in the text and Figures 7 and 8.
The tRNA that emerges after this processing pathway has the canonical cloverleaf secondary structure, which is folded into the classical L-shape by a combination of stacking interactions and conserved tertiary interactions (Fig. 2; Kim et al. 1974a,b; Giege et al. 2012). The resulting tRNA has its acceptor stem stacked on the T-stem to form an extended helix with the 3′-CCAOH end protruding from the acceptor stem, and at approximately right angles, the D-stem is weakly stacked on the anticodon stem (ACS), with the anticodon loop (ACL) protruding. Subsequently, the tRNA is charged at the CCA end by its cognate aminoacyl tRNA synthetase (aaRS) to form the corresponding aminoacyl-tRNA (aa-tRNA), which is now ready for its crucial role in translation.
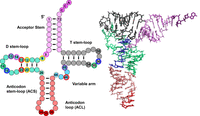
tRNA structure. A schematic of tRNA structure. tRNA is shown in its usual secondary structure, with colored circles representing nucleotides in and adjacent to the acceptor stem (pink), D stem–loop (green), anticodon stem–loop (red), variable arm (aqua) and T-stem–loop (gray), and lines representing base pairs. The 3′ CCA residues N74–N76 are shown in dark pink, and the anticodon residues N34–N36 are dark red. Outer disks of circles are colored to indicate common tertiary interactions, as first detailed for tRNAPhe from yeast (Kim et al. 1974b) (8–14, dark pink; 9–12–23, yellow; 13–22–46, red; 15–48, purple; 18–55, green; 19–56, blue; 26–44, light gray; 54–58, dark gray). Note that different tRNA species can have a D-stem with only 3 bp, a D-loop of variable length, a variable arm with 4 nt or a longer variable arm comprising a stem–loop. Note that tRNA residues are numbered so as to conserve constant numbering of major structural and functional elements, with the anticodon as N34–N36 and the CCA end as N74–N76 (Sprinzl et al. 1998). To this end, additional residues in the D-loop and variable arm have specialized names, and missing residues in some tRNA species are designated by gaps in the numbering for the appropriate residues. On the right is the corresponding crystal structure of tRNAPhe (1EHZ) (Shi and Moore 2000), with residues colored to match the schematic.
Research in the last several years has enormously increased our understanding of almost every step in the eukaryotic tRNA processing pathway in the budding yeast Saccharomyces cerevisiae, and in many cases in other eukaryotic systems, revealing a number of surprises and insights. It is now known that failure of any of a number of the processing steps can lead to tRNA with defects in charging, decoding, or stability, resulting in a number of distinct growth defects in S. cerevisiae and neurological and/or mitochondrial disorders in humans. It has also become apparent that there are multiple points at which tRNA processing intersects with regulatory pathways that respond to nutrients and other environmental factors, stress response pathways, and signaling pathways, to mediate cell growth and translation.
This review aims to capture some of the seminal findings in the biology of eukaryotic tRNA processing during the past several years, with a focus on cytoplasmic tRNAs of S. cerevisiae and other well-studied eukaryotic systems. In the review, we first discuss each tRNA processing step in end maturation and splicing, in their usual in vivo order. This is followed by a discussion of the biology of modifications in and around the ACL, and then the biology of modifications in the main tRNA body, after which there is a discussion of tRNA decay pathways, tRNA nuclear cytoplasmic subcellular dynamics, and tRNA fragments. Along the way, we discuss the intersection of all of these pathways with stress and regulatory pathways. We do not focus on the rich biology of tRNA transcription, aminoacyl tRNA synthetases, and mRNA decoding in the ribosome, as these are covered by numerous other reviews (for example, see Rozov et al. 2016; Graczyk et al. 2018; Rubio Gomez and Ibba 2020).
END PROCESSING AND SPLICING STEPS OF THE tRNA BIOGENESIS PATHWAY
Unexpected finding of frequent 5′ capping of pre-tRNAs
It is now known that Pol III transcription of tRNA is frequently followed by 5′ end capping of the pre-tRNA transcript in S. cerevisiae and human cells, albeit not as frequently as for mRNAs (Ohira and Suzuki 2016). The discovery of pre-tRNA capping was surprising because no interaction exists between the capping machinery and the Pol III transcription machinery, as is well established for the Pol II transcription machinery (for review, see Bentley 2014). Nonetheless, mass spectrometry analysis of pre-tRNAs shows that capping occurs between 5% and 22% of the time on different pre-tRNAs in wild-type (WT) cells, including each of several tRNAs examined from intron-containing and intronless genes. Furthermore, pre-tRNA capping appears to occur by the same mechanism as that for mRNA capping, based on genetic depletion experiments and analysis of intermediates. Moreover, capped pre-tRNAs accumulate to a greater extent when removal of the pre-tRNA 5′ leader by RNase P is inhibited, suggesting that pre-tRNA capping frequency is based on availability of the pre-tRNA (Ohira and Suzuki 2016).
5′ end removal catalyzed by RNAs of RNase P RNPs and protein-only RNase P (PRORP) enzymes
Following the paradigm-breaking discovery that endonucleolytic removal of the tRNA 5′ leader was catalyzed by the RNA component of bacterial RNase P ribonucleoprotein (RNP) (Guerrier-Takada et al. 1983), subsequent work extended RNA catalysis of 5′ leader removal to archaea (Pannucci et al. 1999) and eukaryotes (Kikovska et al. 2007), even as the number of protein subunits of the RNPs increased from one in bacteria to four to five in archaea, and nine to ten in eukaryotes (Supplemental Table S1; Chamberlain et al. 1998; for reviews, see Walker and Engelke 2006; Jarrous and Gopalan 2010). Although the protein subunits do not participate directly in catalysis, they are all essential in yeast (Chamberlain et al. 1998), and cryoEM structures of the human holoenzyme, and the yeast holoenzyme with and without bound pre-tRNA, revealed that the protein subunits stabilize the RNA subunit for catalysis and substrate recognition and participate in recognition of the tRNA 5′ leader (Lan et al. 2018; Wu et al. 2018; see Phan et al. 2021).
One intriguing aspect of RNase P biology is that many of its protein subunits are also part of other essential RNPs (for review, see Jarrous 2017). Indeed, all but one of the subunits of yeast RNase P are shared with the essential and highly conserved RNase MRP (Chamberlain et al. 1998), which has a role in maturation of rRNA and specific mRNAs, but remarkably, a recent cryoEM structure of yeast RNase MRP revealed that several of the shared protein subunits undergo remodeling driven by its distinct RNA subunit (Perederina et al. 2020). In addition, several RNase P subunits are implicated in different roles: three subunits are part of the telomerase complex, helping to stabilize the complex and promoting nuclear localization (Lemieux et al. 2016; Garcia et al. 2020); and another subunit is implicated in different organisms in female gametophyte development and sterility, piRNA synthesis, or fungal resistance (Wang et al. 2012; Molla-Herman et al. 2015; Li et al. 2021). Although these additional functions of RNase P subunits make it more difficult to untangle auxiliary roles of subunits from their specific roles in 5′ leader removal, reconstitution experiments may clarify this (Perederina et al. 2018).
Because of the well-established role of RNA catalysis in RNase P function, it was a distinct surprise to discover that removal of pre-tRNA 5′ leaders was catalyzed by a protein-only RNase P (PRORP) of three subunits in human mitochondria (Holzmann et al. 2008) and a single subunit PRORP in the plant Arabidopsis thaliana (Gobert et al. 2010). Indeed, Arabidopsis PRORPs likely catalyze all 5′ leader removal from tRNAs in vivo in each of the nuclear/cytoplasmic, mitochondrial, and chloroplast compartments (Gutmann et al. 2012). Remarkably, the yeast RNase P function can be replaced by the single subunit nuclear PRORP of Trypanosoma brucei or Arabidopsis, without an obvious growth defect in the latter case (Taschner et al. 2012; Weber et al. 2014).
Subsequent phylogenetic analysis indicates that PRORPs and RNase P RNAs are each widely found in distinct clades within the subgroups of eukaryotes, and in distinct nuclear, mitochondrial, or chloroplast compartments in subsets of these organisms (Lechner et al. 2015), as well as in a small number of bacterial and archaeal phyla (Nickel et al. 2017; Daniels et al. 2019). One unexplained curiosity is why in two cases, examined bacteria sometimes have both a functional PRORP and a functional RNase P RNA (Nickel et al. 2017; Daniels et al. 2019).
Recent structural analysis shows that the human three subunit PRORP binds and positions the pre-tRNA through a subcomplex of two subunits including the TRM10C tRNA methyltransferase, which then recruits the endonuclease PRORP catalytic subunit (Bhatta et al. 2021), and the bacterial single subunit PRORP binds the pre-tRNA with one subunit of the homodimer, to catalyze cleavage by the other subunit (Li et al. 2022).
3′ trailer removal catalyzed by different exonucleases and endonucleases
The processing machinery that removes the 3′ trailer from pre-tRNA in eukaryotes is now understood to result from a combination of nucleases. For most tRNAs, removal of the 3′ trailer sequence occurs after removal of the 5′ leader by RNase P (Fig. 1; Lee et al. 1991; O'Connor and Peebles 1991). As in E. coli (Li and Deutscher 1996), removal of the 3′ trailer sequence in eukaryotes is catalyzed by a combination of endonucleases and exonucleases. Trz1 catalyzes endonucleolytic removal of the 3′ trailer of a number of pre-tRNAs (Schiffer et al. 2002; Takaku et al. 2003), and is known to play a prominent role in 3′ trailer removal in vivo, based on northern analysis after siRNA depletion in Drosophila (Dubrovsky et al. 2004), temperature shift experiments in conditional mutants of the fission yeast Schizosaccharomyces pombe (Zhang et al. 2013), and promoter shut-off experiments in S. cerevisiae (Skowronek et al. 2014). In addition, the 3′ exonuclease Rex1 has a prominent role in 3′ trailer removal of pre-tRNAs in S. cerevisiae. Rex1 was initially implicated in tRNAArg maturation of the tandemly transcribed tRNAArg–Asp genes of S. cerevisiae (van Hoof et al. 2000). Subsequent northern analysis showed that Rex1 had a significant role in 3′ trailer removal in pre-tRNAs with slightly longer 3′ trailers, including two of the four pre-tRNAiMet species and two pre-tRNAVal(CAC) species (Ozanick et al. 2009). Additional experiments showed clear evidence for collaboration in 3′ trailer removal, with Trz1 playing a major role in conjunction with Rex1, with minor additional contributions from Rrp6 and Rex2 (Copela et al. 2008; Skowronek et al. 2014).
The La protein also has a major noncatalytic role in affecting the pathways of 3′ end formation of pre-tRNAs. La protein is an abundant nuclear protein, which binds pre-tRNAs (Rinke and Steitz 1982) at their 3′ oligo(U) ends (Stefano 1984; Teplova et al. 2006; for reviews, see Wolin and Cedervall 2002; Maraia and Bayfield 2006; Porat et al. 2021). La binding leads to endonucleolytic cleavage of the 3′ trailer sequence of the pre-tRNA, and protects the 3′ end of the pre-tRNA from exonucleases in S. cerevisiae (Yoo and Wolin 1997). Thus, in an S. cerevisiae strain lacking La protein (Lhp1), Rex1 acts in conjunction with the 3′ exonuclease Rrp6 to process the 3′ end of the tRNA (Copela et al. 2008) and mutations in La expose tRNAs to Rrp6 in S. pombe (Huang et al. 2006).
CCA addition and removal
The CCA sequence is found at the 3′ ends of all functional tRNAs in all organisms, comprising residues N74–N76, with one of the A76 ribose hydroxyls (2′ or 3′) serving as the covalent attachment site of the cognate amino acid during tRNA charging. The CCA sequence must be added during processing in all eukaryotes and most other organisms, as they lack encoded CCA in their tRNA genes, although some archaea and bacteria (such as E. coli) have encoded CCA in some or all of their tRNA genes. Remarkably, CCA addition is an untemplated addition reaction. In most organisms, CCA addition is catalyzed by a single tRNA nucleotidyl transferase (also known as the CCA-adding enzyme) (Supplemental Table S1), which catalyzes three successive nucleotide additions, although in some ancient bacteria such as Aquifex aeolicus, and in some eukaryotes such as S. pombe, there are separate C74C75-adding and A76-adding enzymes (Tomita and Weiner 2001; Preston et al. 2019). CCA adding enzymes are divided into two classes, each with a similar catalytic domain but with different sequences and overall structures, with class I CCA-adding enzymes in archaea, and class II enzymes in bacteria and eukaryotes (for review, see Xiong and Steitz 2006). Both S. cerevisiae and humans have a single CCA-adding enzyme acting on both nuclear-encoded and mitochondrial-encoded tRNAs (Wolfe et al. 1994; Sasarman et al. 2015).
Previous seminal work elucidated the biochemical gymnastics used by CCA-adding enzymes to precisely add CMP, CMP, and then AMP to the N73 residue of tRNAs without the aid of a template. Both class I and class II CCA-adding enzymes successively add the three NTPs in a single active site, by fixing the acceptor stem through a set of charge and shape interactions with the protein, followed by presentation of the incoming CTP or ATP at each step through interactions that exclude GTP or UTP (Tomita et al. 2004; Xiong and Steitz 2004, 2006). The class I A. fulgidus CCA enzyme features a refolded tRNA 3′ end at each step to position the growing 3′ end at the same location, and to position the incoming CTP or ATP identically, with size discrimination at steps 1 and 2 to exclude ATP, and selection against CTP during step 3 due to incorrect positioning of its α-phosphate (Xiong and Steitz 2004; Pan et al. 2010).
Prior work also revealed that the CCA-adding enzyme has a crucial function in repair of frayed CCA ends of tRNA, in addition to its de novo CCA-addition activity. Thus, although E. coli tRNA genes all have encoded CCA ends, mutants lacking CCA-adding enzyme have reduced growth due to partial removal of some of the ends by RNase T (Zhu and Deutscher 1987). Similarly, while S. cerevisiae cca1 mutants lacking the enzyme are inviable due to the lack of encoded CCA ends in its tRNA genes, mutants lacking Cca1 in the cytoplasmic compartment, but retaining Cca1 in the nucleus and mitochondria, have a similar growth defect and accumulate tRNAs with shortened ends (Wolfe et al. 1996).
It is now clear that the CCA end is implicated in at least four regulatory pathways. First, the CCA end has an important role in the stress response of cells. Thus, oxidative stress treatment of mammalian cells results in shortening of ∼30 of the tRNA 3′ CCA ends, ascribed to angiogenin (ANG), resulting in reduced cap-dependent translation before recovery (Czech et al. 2013) and the accumulation of the truncated tRNAs in nuclei (Schwenzer et al. 2019), discussed further below. Similarly, nutritional stress in T. brucei results in massive removal of ∼70% of the 3′ CCA ends of tRNAs by the conserved Ccr4 homolog LCCR4, which is rapidly reversed by the CCA-adding enzyme when the stress is removed (Cristodero et al. 2021). Second, the CCA end of the peptidyl tRNA has an important role in ribosome-associated quality control triggered by aberrantly stalled ribosomes. During the response, incomplete polypeptides are degraded after release of the peptidyl tRNA from the ribosomal P site by mammalian ANKZF1 (Vms1 in S. cerevisiae), which precisely cleaves the CCA end from the tRNA, leaving a tRNA ending in a 2′–3′ cyclic phosphate at N73. This tRNA is subsequently recycled by removal of the cyclic phosphate by the Trz1 homolog ELAC1, which is found primarily in vertebrates and plants, followed by CCA addition by the CCA-adding enzyme TRNT1 (Yip et al. 2019, 2020). Third, the CCA end of certain tRNAs is subject to a decay pathway triggered by addition of a second CCA repeat. Thus, the instability of the mouse MEN β tRNA-like small cytoplasmic RNA was found to be due to the combination of an unstable acceptor stem and a 5′ end starting with two successive G residues, which leads to aberrant CCACCA addition, and S. cerevisiae tRNASer(CGA) variants with reduced stability that are targeted for the rapid tRNA decay (RTD) pathway (discussed further below) are subject to similar CCACCA addition (Wilusz et al. 2011). Subsequent analysis showed that unstable tRNAs that elicited the aberrant CCACCA addition had refolded on the enzyme after the initial CCA addition so as to loop out three residues and pair C74 and C75 with G2 and G1, setting up a new round of CCA addition (Kuhn et al. 2015; for review, see Wilusz 2015). This pathway of tRNA quality control mediated by CCACCA addition is also found in E. coli cells, likely leading to decay mediated by RNase R (Wellner et al. 2018). Fourth, it is possible that CCA addition is itself regulatory, as initial evidence indicates that CCA addition becomes limiting in S. cerevisiae when tRNA expression is unchecked due to lack of the repressor Maf1 (Foretek et al. 2017), and wild-type E. coli cells have significant amounts of tRNAs with incomplete CCA ends during exponential growth (Czech 2020). In this regard, two independent likely hypomorphic mutations of the human homolog TRNT1 have been associated with multiple clinical manifestations and early death and defective CCA levels in the noncanonical mitochondrial tRNASer(AGY) (Sasarman et al. 2015).
G−1 addition to tRNAHis and reverse polymerization by Thg1 family proteins
The biology of the tRNAHis guanylyltransferase Thg1 and its related proteins continues to reveal surprises (for reviews, see Jackman et al. 2012; Chen et al. 2019).
Virtually all tRNAHis species have an additional G−1 residue (Fig. 3), which is a critical determinant for tRNAHis recognition and charging by HisRS (Rudinger et al. 1994; Nameki et al. 1995), and arises by two very different mechanisms. In bacteria, the G−1 residue is encoded in the genome, and remains at the 5′ end of tRNAHis due to noncanonical processing by RNase P (Orellana et al. 1986). In contrast, in eukaryotes the G−1 residue is added posttranscriptionally opposite A73 by the essential tRNAHis guanylyltransferase Thg1 (Fig. 3; Supplemental Table S1), which catalyzes an unusual 3′–5′ nt addition reaction involving adenylylation of the 5′-phosphate of tRNAHis to activate it; nucleophilic attack of the 3′-OH of GTP to add the G−1 residue to the 5′-phosphate while displacing the adenylate; and pyrophosphatase to generate the mature G−1 monophosphate 5′ end (Cooley et al. 1982; Jahn and Pande 1991; Gu et al. 2003). Thg1 recognizes the GUG anticodon of tRNAHis as a unique determinant (Jackman and Phizicky 2006a), and biochemical evidence suggests that during tRNAHis maturation CCA is added before G−1 addition (Pohler et al. 2019).
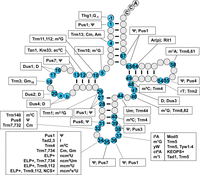
A schematic of modifications and the corresponding genes found in cytoplasmic tRNA in S. cerevisiae. The tRNA secondary structure has gray circles indicating residues without known modifications and blue numbered circles indicating residues with modifications, for each of which the boxed text indicates the corresponding modification and the required gene products. a and b represent nucleotides N20a and N20b, which are found in some tRNAs. Names in all caps (ELP+, NCS+, KEOPS+) refer to the main text for the corresponding genes involved in modification. Conventional abbreviations are used; they are described in the Modomics database (https://genesilico.pl/modomics/) (Boccaletto et al. 2022). (Ψ) pseudouridine, (Am) 2′-O-methyladenosine, (Cm) 2′-O-methylcytidine, (m1G) 1-methylguanosine, (m2G) 2-methylguanosine, (ac4C) 4-acetylcytidine, (D) dihydrouridine, (Gm) 2′-O-methylguanosine, (m2,2G) N2,N2-dimethylguanosine, (m3C) 3-methylcytidine, (I) inosine, (m5C) 5-methylcytidine, (mcm5U) 5-methoxycarbonylmethyluridine, (mcm5s2U) 5-methoxycarbonylmethyl-2-thiouridine, (ncm5U) 5-carbamoylmethyluridine, (ncm5Um) 5-carbamoylmethyl-2′-O-methyluridine, (m1I) 1-methylinosine, (i6A) N6-isopentenyl adenosine, (yW) wybutosine, (t6A) N6-threonylcarbamoyladenosine, (ct6A) cyclic form of t6A, (Um) 2′-O-methyluridine, (m7G) 7-methylguanosine, (rT) ribothymidine, [Ar(p)] 2′-O-ribosyladenosine (phosphate).
Remarkably, Thg1 also catalyzes a true reverse polymerization reaction, involving the template-dependent addition of multiple nucleotides to the 5′ end of tRNAHis variants bearing C73, G73, or U73 instead of A73 (Jackman and Phizicky 2006b), and this reverse polymerization was readily detected in vivo on an S. cerevisiae tRNAHis variant bearing C73 (Preston and Phizicky 2010).
Structural analysis of Thg1 led to a surprise as, despite the lack of sequence similarity, Thg1 was structurally similar to canonical 5′–3′ DNA polymerases, with a palm domain, conserved carboxylates, and two Me++ ions in the active site, suggesting a canonical two-metal ion catalytic mechanism (Hyde et al. 2010; Nakamura et al. 2013). Additional mechanistic analysis showed critical roles for the two conserved aspartate residues that coordinate the Me++ ions for each of the three reaction steps (Smith and Jackman 2012), and showed that reduced pyrophosphatase activity was correlated with increased reverse polymerization, consistent with competition between the two reaction pathways (Smith and Jackman 2014; Desai et al. 2018).
After the unexpected discovery of organisms with tRNAHis species lacking G−1 in a clade within alphaproteobacteria (Wang et al. 2007), tRNAHis species lacking G−1 were also found in several eukaryotes. For example, T. brucei and A. castellanii were found to have tRNAHis lacking G−1 and multiple organisms have no recognizable Thg1 homolog, suggesting that this is much more general (Rao et al. 2013; Rao and Jackman 2015). Remarkably also, the lethality of an S. cerevisiae thg1Δ strain could be suppressed by expression of the corresponding noncanonical HisRS and companion tRNAHis species from T. brucei, A. castellanii, and C. elegans (Rao and Jackman 2015; Lee et al. 2019). Moreover, the virtually normal growth of the S. cerevisiae thg1Δ strain expressing A. castellanii HisRS and tRNAHis essentially proved that the only important role of the G−1 residue of tRNAHis in S. cerevisiae is as an identity element for charging by HisRS (Rao and Jackman 2015).
It is now known that Thg1 is part of the Thg1 superfamily, comprised of a clade of Thg1 orthologs that are widely found in eukaryotes, and a clade of Thg1-like proteins (TLPs) that are found in some archaea, bacteria, and eukaryotes (Heinemann et al. 2009, 2010; Jackman et al. 2012). The bacterial TLPs from Bacillus thuringiensis and Myxococcus xanthus and each of four archaeal TLPs tested all catalyzed templated addition of nucleotides to tRNAs in vitro, and expression of the B. thurigiensis and the four archaeal TLPs each complemented the lethality of an S. cerevisiae thg1Δ strain (Abad et al. 2010; Heinemann et al. 2010; Rao et al. 2011), through U−1 addition to tRNAHis across from A73 in the case of the B. thuringiensis TLP (Dodbele et al. 2019).
Nonetheless, the biochemical activity of TLPs suggests that their primary role is in tRNA editing, in which tRNAs missing one or more 5′ nt are 5′ end-repaired by templated reverse polymerization. This 5′ end repair activity was first inferred by comparison of the sequences of tRNAs and their corresponding genes in mitochondria of the eukaryotic microbe Acanthamoeiba castellanii (Lonergan and Gray 1993). In support of this editing function of TLPs, the B. thuringiensis TLP has increased kcat/KM values for addition of nucleotides to 5′ truncated tRNAs, compared to that for G−1 addition to the mature tRNAHis (Rao et al. 2011); two of the four Thg1/TLPs (TLP3 and TLP4) from Dictyostelium discoideum have substantial kcat/KM values for templated nucleotide addition to 5′ truncated tRNAs (Abad et al. 2011); and depletion of D. discoideum TLP3 results in a severe growth defect and decreased mitochondrial tRNA 5′ editing (Long et al. 2016).
Dictyostelium discoideum TLP4 has a critical but as yet unknown role (Long et al. 2016). Whereas depletion of D. discoideum Thg1 leads to the expected severe growth defect and cytoplasmic tRNAHis lacking G−1, and knockout of TLP2 leads to a minor but distinct growth defect and mitochondrial tRNAHis lacking G−1, depletion of TLP4 leads to a severe growth defect for unknown reasons. Although the function of TLP4 is not yet known, its cytoplasmic location suggests a nonorganellar role, and its biochemical activity on 5S RNAs and a ncRNA emphasizes the potential for TLP4 to act naturally on non-tRNA substrates (Long et al. 2016).
It is also not fully understood how Thg1 acts to regulate mitochondrial function. A V55A mutation in the human Thg1 ortholog THG1L (Supplemental Table S1) is associated with cerebellar ataxia and decreased mitochondrial fusion (Fig. 4; Edvardson et al. 2016). Furthermore, reduced expression of human THG1L (also called IHG-1, induced in high glucose-1) leads to reduced mitochondrial respiration and mitochondrial fusion, linked to reduced interaction with Mfn1 and Mfn2, which mediate mitochondrial fusion (Hickey et al. 2014). It is unknown how THG1L interacts with Mfn1 and Mfn2 and if this interaction is perturbed in the V55A variant as part of a moonlighting role of THG1L, or if the V55A variant has reduced G−1 addition activity on mitochondrial tRNAHis (Suzuki and Suzuki 2014; Nakamura et al. 2018). The THG1L–V55A variant has normal activity in vitro, but its expression in an S. cerevisiae thg1Δ strain results in a growth defect, unlike for the WT THG1L protein (Edvardson et al. 2016).
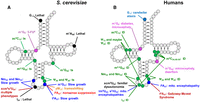
Effect of lack of tRNA modifications in S. cerevisiae and humans. (A) Prominent phenotypes resulting from mutations in S. cerevisiae modification genes. 5-FUs, sensitivity to 5-fluorouracil; ts, temperature sensitivity. (B) Prominent diseases and disorders resulting from mutations in human modification genes. (ID) Intellectual disability.
Pre-tRNA splicing
The discovery of pre-tRNAs with transcribed introns in budding yeast and vertebrate cells (Hopper et al. 1978; Knapp et al. 1978; O'Farrell et al. 1978; De Robertis and Olson 1979) occurred nearly simultaneously with the discovery of mRNA introns in Drosophila and vertebrate cells (Berget et al. 1977; Chow et al. 1977; White and Hogness 1977). However, unlike pre-mRNA splicing, which involves two RNA catalyzed phosphoester transfer reactions occurring in a large RNP complex called the spliceosome to remove the intron in circular form, pre-tRNA splicing is catalyzed by a small endonuclease complex that generates two exons and a linear, or in some cases a circular (Lu et al. 2015), excised intron, followed by exon joining by a ligase enzyme and a small cast of additional proteins.
tRNA introns: characteristics and functions
In all eukaryotes examined (http://gtrnadb.ucsc.edu; Chan and Lowe 2016), a subset of tRNAs is encoded by intron-containing genes. Eukaryotic tRNA introns are located 1 nt 3′ of the anticodon, between N37 and N38, and are generally short, ranging from 14–60 nt in budding yeast, to as long as 133 nt for some introns in other organisms (Chan and Lowe 2016; for reviews, see Yoshihisa 2014; Schmidt and Matera 2020). The percentage of intron-containing tRNA genes differs among organisms, with ∼6% in mouse, rat, and humans and 24% in budding yeast, but their occurrence is clustered in specific gene families (Chan and Lowe 2016; for review, see Schmidt and Matera 2020). For example, every gene member in each of the 10 intron-containing gene families in budding yeast and the 16 intron-containing families in fission yeast contains an intron, and all eukaryotic tRNATyr genes in studied organisms have an intron. However, in mouse and humans, introns are not always found in all members of isoacceptor gene families with introns. For example, although in humans all 13 tRNATyr(GUA) genes and all five tRNAIle(UAU) genes contain an intron, only 5/6 of the tRNAArg(UCU), and 5/7 tRNALeu(CAA) gene family members contain introns (Chan and Lowe 2016). It is also interesting to note that in budding yeast, the intron sequences within each intron-containing gene family are either identical or very similar to each other, but for fission yeast and vertebrates the sequences of introns vary among the family members (Chan and Lowe 2016). A number of archaeal tRNA genes also have introns which, as in eukaryotes, are generally relatively short, occur mostly between N37 and N38, and are generally clustered in all the genes of each isoacceptor gene family member with introns (Yoshihisa 2014).
Although tRNA introns do not possess conserved sequence motifs at their termini, they generally have structure (Fig. 5). The classical archaeal exon–intron structure is a bulge-helix-bulge (BHB) RNA structure, in which nucleotides N32–N35 form a helix with corresponding residues in the intron, with cleavage sites in the adjacent 3 nt single-stranded bulges (Thompson and Daniels 1988, 1990; Yoshihisa 2014). Eukaryotic tRNA introns have a similar, but less well-defined structure, generally with nucleotide sequences that are complementary with N33–N35 or sometimes N34–N36 or other combinations of nucleotides within the ACL, extended by an additional base pair, called the anticodon–intron (A–I) base pair between C32/U32 of the ACL and the antepenultimate A/G of the intron. This results in a bulge-helix-loop (BHL) exon–intron structure (Fig. 5), with the cleavage sites in the single-stranded regions comprising the bulge and the loop (Lee and Knapp 1985; Baldi et al. 1992; Di Nicola Negri et al. 1997; Schmidt and Matera 2020).
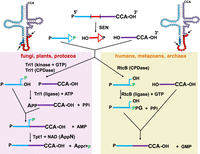
Schematic of tRNA splicing pathways in different eukaryotes. (Top left and right) A typical unspliced pre-tRNA is shown in its accepted secondary structure, with the intron residues indicated by red circles except for the antepenultimate intron residue (dark red); residues N1–N37 of the 5′ exon indicated by light blue circles, except for N32 (white) and anticodon residues N34–N36, (dark blue); and residues N38–N73 indicated by purple residues. The antepenultimate intron residue pairs with N32 in the pre-tRNA. Arrows indicate sites of endonucleolytic cleavage of the pre-tRNA by the SEN/TSEN splicing complex. (Top left) Canonical pre-tRNA with a well-defined BHL motif. (Top right) One of several pre-tRNAs with a slightly different BHL motif. (Top center) A typical unspliced pre-tRNA is shown in linear form with the 5′ exon in blue, the intron in red, and the 3′ exon in purple. Endonucleolytic cleavage of the pre-tRNA results in formation of a 2′–3′-cyclic phosphate at the 3′ end of both the 5′ exon and and the intron, leaving a 5′-OH at the 5′ end of both the 3′ exon and the intron. (Left panel) In fungi, plants, and protozoa, the RNA 5′-kinase activity of the ligase Trl1 phosphorylates the 5′-OH end of the 3′-half-molecule using GTP, and the cyclic phosphodiesterase (CPDase) activity of Trl1 opens the 2′–3′ cyclic phosphate to a 2′-phosphate (green). Then the ligase activity of Trl1 joins the half-molecules by adenylylation of the 5′-phosphate of the 3′ exon and ligation to the 3′-OH of the 5′ exon, leaving a 2′ phosphate (green) at the splice junction. This 2′-phosphate is subsequently transferred to NAD by the 2′-phosphotransferase (Tpt1). (Right panel) In humans and metazoans, as well as in some archaea, the CPDase activity of the ligase RtcB opens the 2′–3′ cyclic phosphate of the 5′ exon to form a 3′-phosphate (green). Then, the ligase activity of RtcB joins the half-molecules by guanylylation of the 3′-phosphate of the 5′ exon and ligation to the 5′-OH of the 3′ exon, releasing GMP.
Splicing is essential in all studied eukaryotes because in each organism for at least one intron-containing isoacceptor gene family, all of the genes possess introns. Intron-containing tRNAs cannot function in protein synthesis prior to splicing because tRNA introns disrupt the ACL and there is at least one report that documented intron-containing tRNAs cannot be aminoacylated (O'Farrell et al. 1978; for reviews, see Phizicky and Hopper 2010; Yoshihisa 2014; Chan and Lowe 2016; Schmidt and Matera 2020).
Although tRNA splicing is essential, the reverse is not the case; that is, the presence of introns in any tRNA gene family is not essential. Early studies from the Abelson laboratory demonstrated that a budding yeast strain possessing a deletion of the intron from the single copy essential tRNASer(CGA) gene was viable, thereby documenting that the intron in this tRNA gene is unessential (Ho and Abelson 1988). Subsequent studies by the Yoshihisa group created 10 yeast strains, each missing the intron from every member of the corresponding intron-containing isoaccepter gene family; this tour-de-force report documented that introns for all budding yeast tRNA genes are unessential (Mori et al. 2011; Hayashi et al. 2019). In fact, each of the 10 yeast deletion strains had rather few growth defects (Hayashi et al. 2019).
That tRNA introns are unessential for life (at least for budding yeast) raises the very interesting question as to why the presence of tRNA introns has been conserved throughout eukaryotes. Surprisingly, multiple roles for tRNA introns have been documented, thereby providing selection pressure for their conservation. These roles include: the efficiency of tRNA genes in functioning as transcription barriers for local ORFs (Donze and Kamakaka 2001); posttranscriptional regulation of mature tRNA levels via the Met22-dependent pre-tRNA decay (MPD) turnover pathway (discussed below), which has specificity for intron-containing pre-tRNAs (Payea et al. 2020); the presence of particular tRNA modifications; and the altered modification pattern of tRNAs. It is well documented that particular tRNA modification enzymes have specificity for intron-containing pre-tRNAs. For example, if an intron is removed from one of the eight genes encoding tRNATyr(GUA) (SUP6), the resulting tRNATyr lacks Ψ at anticodon residue U35 and its function as a suppressor tRNA is reduced (Johnson and Abelson 1983). Similarly, intron removal from tRNALeu(CAA) genes results in lack of m5C at C34 in the anticodon (Strobel and Abelson 1986; Hayashi et al. 2019). In each of these cases, removal of the intron eliminates the specificity of the corresponding modification enzymes, Pus7 and Trm4, respectively, for the corresponding tRNAs (Behm-Ansmant et al. 2003; for review, see Grosjean et al. 1997). Introns in pre-tRNAs also have been shown to dictate modification fidelity; for example, intron deletions of the tRNAIle(UAU) genes in budding yeast result in a lack of Ψ34 and instead U34 is erroneously modified with 5-carbamoylmethyluridine (ncm5U) (Hayashi et al. 2019).
Eukaryotic tRNA splicing endonucleases
Intron removal from eukaryotic pre-tRNAs is catalyzed by a heterotetramic protein endonuclease complex called tRNA splicing endonuclease (SEN in budding yeast or TSEN vertebrate cells, Supplemental Table S1; Peebles et al. 1979, 1983; Trotta et al. 1997; Paushkin et al. 2004; Hayne et al. 2022). Two of the four subunits (Sen2 and Sen34) of the SEN/TSEN complexes are conserved and possess catalytic activity, while the remaining two subunits (Sen15 and Sen54) are not conserved (Paushkin et al. 2004). Genes that encode proteins similar to the SEN catalytic subunits that function in pre-tRNA splicing have been discovered in plants, trypanosomes, and Drosophila (Akama et al. 2000; Rubio et al. 2013; Schmidt et al. 2019; for reviews, see Fabbri et al. 1998; Phizicky and Hopper 2010; Yoshihisa 2014; Schmidt and Matera 2020). Since pre-tRNA splicing is necessary to generate the entire cadre of tRNAs required to decode the genome, it is not surprising that each of the SEN and TSEN subunits is essential for life in budding and fission yeast and in human cell lines (Giaever et al. 2002; Kim et al. 2010a; Wang et al. 2015). Interestingly, autosomal recessive mutations in each of the TSEN subunits cause subclasses of Pontocerebellar Hypoplasia (PCH), congenital neurodegenerative diseases (Budde et al. 2008; Cassandrini et al. 2010; Breuss et al. 2016; for review, see Sekulovski and Trowitzsch 2022). It is unclear why the TSEN mutations preferentially affect neuronal tissues, but this is a common phenomenon in tRNA processing biology in humans, as mutations in human modification genes are often linked to neurological disorders (Fig. 4).
Prior results described crucial similarities and differences between the structure and substrate recognition properties of TSEN and the equivalent archaeal tRNA splicing endonuclease. Whereas the archaeal tRNA splicing endonuclease from H. volcanii recognizes an isolated BHB RNA structure (Thompson and Daniels 1988, 1990), the S. cerevisiae SEN complex recognizes a combination of features, including the mature domain of the intron-containing pre-tRNA, the distance from the mature domain to the splice sites, and the A–I base pair in the context of the BHL structure found in eukaryotic pre-tRNAs (Reyes and Abelson 1988; Baldi et al. 1992; Di Nicola Negri et al. 1997). Structural analysis of the archaeal endonuclease from Methanococcus jannaschii and a co-crystal structure of the Archaeoglobus fulgidus enzyme with a BHB RNA substrate revealed an active site His-Tyr-Lys triad that is conserved between eukaryotes and archaea, with substrate bulge recognition aided by two nearby arginines, which originate in the other subunit of the homodimer, and form a cation-π sandwich with one of the substrate adenine residues in the bulge (Li et al. 1998; Xue et al. 2006; Calvin et al. 2008). Remarkably this cross-subunit interaction is functionally conserved for the corresponding arginine and tryptophan residues of the S. cerevisiae Sen34 subunit, as these residues are required for cleavage of the 5′ splice site by the Sen2 subunit of TSEN (Trotta et al. 2006).
Additional work has added substantially to our understanding of how the different recognition properties of the archaeal and eukaryotic endonucleases evolved. It was initially found that the A. fulgidus endonuclease recognized an isolated BHB motif RNA substrate, but could only recognize the more relaxed BHL motif in the context of a pre-tRNA containing the mature domain (Tocchini-Valentini et al. 2007). Subsequently, it was found that the crenarchaeal endonuclease from Aeropyrum pernix had specificity for both the BHB and the BHL structural motifs, and that this was due to a crenarchaeal specific loop (CSL) which, when inserted into the A. fulgidus enzyme, converted it to an enzyme that recognized the BHL structural motif (Hirata et al. 2011; Kaneta et al. 2018; for review, see Hirata 2019).
Until recently, it was not possible to understand the biochemical functions of the two eukaryote-specific noncatalytic SEN15 and SEN54 subunits of the heterotetrameric SENs, because efforts to reconstitute functional SEN or TSEN complexes from purified recombinant subunits had failed for decades. However, in vitro reconstitutions have now succeeded (Hayne et al. 2020, 2022; Sekulovski et al. 2021). Hayne et al. obtained functional human endonuclease expressed in E. coli or HEK cells when all four TSEN (2, 15, 34, and 54) subunits were coexpressed, whereas Sekulovski et al. were able to reconstitute human endonuclease activity from recombinant TSEN15–34 and TSEN2–54 heterodimers expressed in insect or mammalian cells (Hayne et al. 2020, 2022; Sekulovski et al. 2021). Success in reconstitution of TSEN provided the opportunity for structural and biochemical analysis.
Recently, three groups (Hayne et al. 2023; Sekulovski et al. 2023; Zhang et al. 2023) obtained high resolution (2.9–3.9 Å) cryo-EM structures of the human TSEN heterotetramer enzyme in complex with intron-containing pre-tRNAs. The enzyme-substrate complexes were trapped by either modifying the RNA cleavage sites and/or by utilizing enzyme with alterations of catalytic amino acids. Overall, the resolved heterotetrameric enzyme-tRNA complexes are structurally similar to the archaeal enzymes, documenting their evolutionary relationship. However, the human TSEN subunits contain extensions and insertions that provide additional enzyme-substrate interactions. Importantly, although the reconstituted human TSEN complex can utilize short RNA sequences containing just the intron and the anticodon stem–loop as substrates (albeit with low kinetic activity), the structural analyses show that TSEN54 has extensive interactions with the mature tRNA domain, supporting the earlier model that TSEN54 acts as a molecular ruler to regulate cutting at the appropriate splice sites (Trotta et al. 1997). TSEN15 interactions with tRNAs were not resolved, but it is predicted that TSEN15 “mediates interactions with the intron surrounding the 5′ splice site” (Hayne et al. 2023).
The high-resolution TSEN structures provide further information regarding how the TSEN mutations that cause PCH may affect TSEN structure/function. None of the causative alterations lie within catalytically important locations, but rather they disrupt subunit interactions (Hayne et al. 2023; Sekulovski et al. 2023). Thus, the TSEN mutations likely affect the SEN complex stoichiometry, thermostability of the heterotetramer, and/or efficiency of tRNA splicing (Breuss et al. 2016; Sekulovski et al. 2021).
Clp1 and TSEN
The role of the human RNA kinase hsClp1 in tRNA splicing continues to be an enigma. Clp1 functions in mRNA 3′ end processing (de Vries et al. 2000), but also copurifies with the TSEN isolated from human 293 cell lines (Paushkin et al. 2004) and coexpressed recombinant hsClp1 copurifies with TSEN (Hayne et al. 2020; Sekulovski et al. 2021). Similarly to mutations of the TSEN subunits, autosomal recessive mutations in CLP1 genes are linked to PCH-like disorders in human patients, as well as in zebrafish and mouse models (Schaffer et al. 2014). Moreover, the CLP1 mutations were reported to affect the endonuclease subunit stoichiometry and tRNA splicing in vitro activity (Hanada et al. 2013; Karaca et al. 2014; Schaffer et al. 2014). Therefore, it was surprising that Clp1 is not required for either TSEN complex in vitro assembly or for pre-tRNA splicing by the reconstituted human complex from either E. coli or mammalian cells (Hayne et al. 2020; Sekulovski et al. 2021). Moreover, in vivo studies of the Drosophila Clp1 ortholog provided evidence that Clp1 may instead function to negatively regulate the ligation step of pre-tRNA splicing (Hayne et al. 2020). Further, a recent study that created mouse models with the PCH relevant CLP1 mutations documented changes in tRNA processing intermediates, but these tRNA processing alterations did not correlate with pathogenicity; rather, the pathogenicity correlated with alterations of 3′ poly(A) site selection of particular RNAs; thus, the authors suggest that PCH due to CLP1 mutations may result from defects in RNA 3′ processing instead of tRNA biology (Monaghan et al. 2021). Nevertheless, cryo-EM structures of TSEN in complex with hsClp1 document that TSEN54 interacts with Clp1 (Hayne et al. 2023; Sekulovski et al. 2023). Future studies are required to resolve the functional relationship of TSEN and Clp1.
Subcellular location for tRNA splicing
The subcellular location of pre-tRNA splicing differs among organisms. Early studies using Xenopus oocytes reported that pre-tRNA splicing occurs in the nucleus (Melton et al. 1980; De Robertis et al. 1981). Later studies verified this nuclear location in human cells (Paushkin et al. 2004). In contrast, the budding and fission yeast SEN complexes are not located in the nucleus, but rather are peripherally associated on the cytoplasmic surface of mitochondria (Fig. 1; Yoshihisa et al. 2003, 2007; Wan and Hopper 2018). For both budding and fission yeast, a conserved mitochondrial membrane protein component of the mitochondrial import machinery, Tom70, is important for tethering of the SEN complexes to mitochondria (Wan and Hopper 2018), documenting conservation for the location of, and the mechanism of, achieving SEN location to mitochondria for at least 600 million years (Parfrey et al. 2011). tRNA splicing in the protozoan Trypanosome brucei (Lopes et al. 2016) and, likely, in the plant Arabidopsis thaliana (Park et al. 2005) also occurs after pre-tRNAs are exported from the nucleus to the cytoplasm; however, there is no evidence that either the Trypanosome or Arabidopsis TSEN localize at the mitochondrial surface (Englert et al. 2007; Lopes et al. 2016). It would be very interesting to discern the subcellular location of TSEN in other eukaryotic organisms to learn when and why the split from cytoplasmic to nuclear pre-tRNA splicing occurred.
Additional SEN RNA substrates
Since the preponderance of studies of eukaryotic SEN indicated that it interacted with the mature tRNA anticodon stem–loop rather than the splice junctions or intron sequences (Reyes and Abelson 1988; Sekulovski et al. 2021, 2023; Hayne et al. 2023), and that accurate pre-tRNA cleavage proceeds by a mechanism measuring the length of the anticodon stem (Reyes and Abelson 1988), it was not anticipated that there would be SEN substrates in addition to tRNAs. However, the Xenopus, budding yeast, and reconstituted human TSEN can cleave mini substrates in vitro that contain tRNA stem–loop structures (Fabbri et al. 1998; Hayne et al. 2020), and there is in vivo evidence suggesting that the budding yeast SEN complex has substrates in addition to intron-containing pre-tRNAs. Two studies generated yeast strains that were able to bypass the requirement for SEN to generate mature tRNAs and the results demonstrated that even though cells possessed normal levels of mature, spliced tRNAs they nevertheless required all four functional SEN subunits for viability (Dhungel and Hopper 2012; Cherry et al. 2018), and cells with nuclear SEN and defective mitochondrially located SEN have defects in an unessential step in pre-rRNA processing (Volta et al. 2005; Dhungel and Hopper 2012). These data support the hypothesis that there are essential cytoplasmic non-tRNA substrates for the SEN complex.
Additional SEN substrates have been identified. Budding yeast SEN complex functions in cleavage/turnover of mRNAs encoding proteins that are imported into mitochondria such as CBP1 mRNA, encoding an unessential mitochondrial protein, at the boundary of a stem–loop structure (Tsuboi et al. 2015). Most recently, the van Hoof laboratory, using an unbiased bioinformatics approach, identified several budding yeast mRNAs that encode additional essential and unessential mitochondrial proteins, which are cleaved by SEN (Hurtig et al. 2021). Interestingly, although there is no known sequence specificity of SEN required for removal of tRNA introns, mRNA cleavage by the SEN complex appears to require an A nucleotide located at the −1 position of the mRNA cleavage sites (Hurtig et al. 2021). This newly discovered tRNA endonuclease-initiated decay (TED) role for the SEN complex likely functions in quality control of mRNAs encoding mitochondrial proteins that are located at the mitochondrial cytoplasmic surface (Hurtig et al. 2021).
The two eukaryotic pathways for ligation of tRNA exons
The ligation step of eukaryotic tRNA splicing proceeds by two very different mechanisms to join the 5′ exon, which terminates with a 2′–3′ cyclic phosphate, to the 3′ exon, which initiates with a 5′-OH end (Fig. 5).
In S. cerevisiae, the single subunit ligase Trl1 (also called Rlg1) first heals the ends, using its cyclic phosphodiesterase (CPDase) activity to open the 2′–3′ cyclic phosphate to a 2′-phosphate and its polynucleotide kinase (PNK) to phosphorylate the 5′-OH (Fig. 5; Supplemental Table S1). Then Trl1 joins the ends with its ligase activity, first activating the 5′-phosphate by formation of an adenylylated intermediate (Greer et al. 1983; Phizicky et al. 1986). The resulting ligated RNA has a 2′-phosphate at the splice junction, which is transferred to NAD by the 2′-phosphotransferase Tpt1 to form ADP-ribose 1″–2″-cyclic phosphate (McCraith and Phizicky 1991; Culver et al. 1993, 1997; Spinelli et al. 1997). This ligation mechanism is conserved in fungi (Remus et al. 2017; Banerjee et al. 2019a; Peschek and Walter 2019), protozoa (Lopes et al. 2016), and plants (Englert and Beier 2005; Wang et al. 2006), and also in several metazoan species, albeit through a separate ligase and PNK/CPDase and/or Clp1 (Englert et al. 2010).
In contrast, in most metazoans and archaea, the RNA ligase joins the two exons by incorporating the phosphate from the 2′–3′ cyclic phosphate of the 5′ exon in the junction (Nishikura and De Robertis 1981; Filipowicz and Shatkin 1983; Laski et al. 1983; Zofallova et al. 2000), a reaction that is catalyzed by RtcB (Supplemental Table S1; Englert et al. 2011; Popow et al. 2011; Tanaka and Shuman 2011; for review, see Popow et al. 2012). The biochemical reaction of RtcB is unusual (Fig. 5). Although joining of the 2′–3′ cyclic phosphate to a 5′-OH should in principle be isoenergetic, the RtcB reaction uses a more circuitous route to ligation. RtcB first uses its CPDase activity to generate a 3′-phosphate, which is then followed by guanylylation of RtcB using GTP, transfer of the guanylate to the RNA-3′-p to generate the activated RNA-p-pG intermediate, and then ligation by attack by the 5′-OH of the 3′ exon to generate the products RNA and GMP (Tanaka et al. 2011a; Chakravarty and Shuman 2012; Chakravarty et al. 2012; Englert et al. 2012; Desai et al. 2013; Banerjee et al. 2021).
RtcB has additional partners that affect its activity. Although E. coli RtcB can function independently to replace Trl1 function in S. cerevisiae for both tRNA splicing and HAC1 mRNA splicing (Tanaka et al. 2011b), the activity of Pyrococcus horikoshii RtcB is stimulated by Archease, a member of the same operon (Desai et al. 2014), and remarkably, Archease stimulates some RtcB orthologs from single turnover to multiple turnover enzymes (Desai et al. 2015). Moreover, in humans the Archease ortholog ARCH (ZBTB8OS) interacts with RTCB and is crucial for tRNA splicing in vivo and in vitro, and the RTCB guanylation step is stimulated in vitro and in vivo by the DEAD box helicase DDX1 (Popow et al. 2014). In contrast, overexpression of the 2′–3′-cyclic phosphatase activity of ANGEL2 (which completely removes the phosphate) can compete with tRNA ligase and inhibit mammalian tRNA splicing (Pinto et al. 2020).
In both S. cerevisiae and mammalian cells, the respective ligase pathways also participate in the ligation step of the noncanonical mRNA splicing of HAC1/XBP1, encoding a crucial transcription factor in the unfolded protein response (UPR) pathway, after endonucleolytic excision of the HAC1/XBP1 intron by Ire1 (Sidrauski et al. 1996; Sidrauski and Walter 1997; Jurkin et al. 2014; Lu et al. 2014).
Mechanistic studies of the Trl1 ligation step and the Tpt1 2′-phosphotransferase step
A series of elegant papers have illuminated the unique biochemical properties of the single subunit funga/plant Trl1 ligase. The modular nature of the Trl1 domains has been well documented by showing functional complementation of S. cerevisiae trl1 mutants lacking complete Trl1 function by individual kinase (Ramirez et al. 2008) and CPDase subunits (Schwer et al. 2008). The unique GTP specificity of the Trl1 kinase activity in Trl1 is accounted for by a unique G-loop and extensive guanine-specific interactions with residues in the G-loop (Remus et al. 2017), and the unique 2′-phosphate specificity of the Trl1 ligase activity (Remus and Shuman 2013) is plausibly explained by a sulfate binding site in the structure of the ligase domain (Banerjee et al. 2019a).
Much has been learned about the unusual mechanism by which Tpt1 catalyzes transfer of the 2′-phosphate from the splice junction of ligated tRNA to NAD to form ADP-ribose 1″–2″-cyclic phosphate. Prior biochemical analysis showed that Tpt1 catalyzes nucleophilic attack by the RNA-2′-phosphate oxygen at the 1″-position of NAD+ to displace nicotinamide and form an ADP-ribosylated RNA covalent intermediate, followed by cyclization catalyzed by the neighboring 2′-OH to form the product ADP-ribose 1″–2″-cyclic phosphate, with concomitant release of the dephosphorylated RNA (Spinelli et al. 1999; Sawaya et al. 2005; Steiger et al. 2005). Subsequent kinetic analysis of variants in Runella slithyformis Tpt1 revealed that the R68A variant was completely unaffected in the rate of formation of the covalent intermediate (step 1) but had a severe 200-fold reduction in the rate of step 2, in which the dephosphorylated RNA was released during cyclization (Munir et al. 2018a). The crystal structure of the Clostridium thermocellum Tpt1 showed four critical residues in the active site (Banerjee et al. 2019b), which had been previously implicated in catalysis (Steiger et al. 2005; Munir et al. 2018a), and revealed two highly informative bound ligands: ADP-ribose 1″-phosphate, mimicking the ADP-ribose 1″–2″-cyclic phosphate product of step 2, after subsequent CPDase activity in the crystal; and acetyl-coA, with its adenosine 3′,5′ bis-phosphate (pAp) moiety mimicking the substrate RNA after dephosphorylation of the 2′-phosphate (Banerjee et al. 2019b).
The puzzle of Tpt1 in other organisms
One major unanswered question regarding the Tpt1 protein family is why its members are found widely in bacterial, archaeal, and eukaryotic organisms that do not apparently require its enzymatic activity. For example, mouse, E. coli, and R. slithyformis each have a functional Tpt1 ortholog that complements the lethality of an S. cerevisiae tpt1Δ mutant (Spinelli et al. 1998; Munir et al. 2018a), although mammals use the metazoan/archaeal RtcB pathway for splicing of both tRNA and HAC1/XBP1, which does not generate a 2′-phosphate (Popow et al. 2011; Jurkin et al. 2014), and bacteria such as E. coli do not undergo tRNA splicing or have a known pathway that generates an RNA with internal 2′-phosphate. Indeed, neither the E. coli nor the mouse Tpt1 ortholog is essential in their respective organisms (Spinelli et al. 1998; Harding et al. 2008).
Recent experiments suggest that Tpt1 could have other biochemical functions in some of these and other organisms. Remarkably, a subset of Tpt1 enzymes can catalyze NAD-dependent ADP-ribosylation of the 5′-phosphate of RNA or DNA, with an oxygen of the 5′-phosphate of the oligonucleotide acting as nucleophile (like the 2′-phosphate of ligated tRNA during the canonical Tpt1 reaction), forming an ADP-ribosyl cap on the nucleotide (Munir et al. 2018b). This RNA and DNA ADP-ribosylation activity extends to the human TRPT1 ortholog and, although the product 5′ capped oligonucleotide cannot be resolved by the Tpt1 reaction to release the RNA or DNA, the product can be reversed by a number of ADP-ribosylhydrolases as well as by some macrodomains (Munnur et al. 2019). Other results show that C. thermocellum and A. pernix Tpt1 proteins can catalyze removal of terminal 2′-phosphates, and to some extent 3′-phosphates, from RNA (Munir et al. 2019). It thus seems likely that one or more noncanonical Tpt1 reactions like these could explain the evolutionarily widespread occurrence of Tpt1 in organisms that do not generate RNA with an internal 2′-phosphate, or that have a functional RtcB. One of these noncanonical Tpt1 activities might also explain the puzzling result that overexpression of S. cerevisiae TPT1 rescues the synthetic lethality of S. cerevisiae elg1Δ srs2Δ mutants, which are defective in the repair of DNA damage, as does overexpression of the CPDase Cpd1 (Gazy et al. 2013), which generates Appr-1″-p from ADP-ribose 1″–2″-cyclic phosphate (Martzen et al. 1999; Nasr and Filipowicz 2000).
tRNA intron turnover
A quantitatively important by-product of tRNA splicing is the excised introns, which are produced in equimolar amounts to spliced tRNA, at the rate of ∼600,000 times a generation in budding yeast (Wu and Hopper 2014). Even though eukaryotic cells generate these enormous quantities of freed linear introns during tRNA splicing, these introns are rarely detected under normal physiological conditions, because the excised introns are either converted to more stable molecules or are rapidly and efficiently destroyed.
The excised introns derived from budding yeast pre-tRNAs remain as linear RNAs (Knapp et al. 1979; Wu and Hopper 2014), which are subject to decay. One pathway by which yeast linear tRNA introns are degraded is the kinase-mediated decay pathway, in which the RNA kinase activity of the Trl1/Rlg1 ligase phosphorylates the 5′ terminus of the linear excised intron, rendering the intron as a substrate for the cytoplasmic 5′ to 3′ exoribonuclease, Xrn1 (Wu and Hopper 2014). Curiously, the kinase-mediated decay pathway functions in the turnover of only a subset of the budding yeast tRNA introns (Wu and Hopper 2014); the gene products involved in the turnover of other excised tRNA introns have not yet been delineated. In addition, the kinase-mediated decay pathway acts at two points during the UPR pathway: to degrade the 3′ exon and therefore inhibit HAC1 ligation by competition with Trl1 (Cherry et al. 2019; Peschek and Walter 2019), and to degrade the HAC1 intron and activate HAC1 translation by relieving an inhibiting interaction with the HAC1 mRNA (Mori et al. 2010; Cherry et al. 2019).
In contrast, the excised tRNA introns in Drosophila exist as circular molecules (Lu et al. 2015; for review, see Schmidt and Matera 2020), arising from RtcB-mediated direct ligation of the 5′ and 3′ termini (Schmidt et al. 2019), as introduction of RtcB to yeast also efficiently converts the introns to circular molecules (Schmidt et al. 2019). How these circular introns are turned over remains unknown.
tRNA MODIFICATIONS
It is well known that tRNAs are by far the most modified class of RNAs in the cell. A total of 155 nucleoside or base modifications are currently listed in the Modomics database (Boccaletto et al. 2022), the vast majority of which are found in tRNAs (Grosjean 2015). These modifications (Fig. 3; Supplemental Table S1) provide substantial chemical diversity to tRNAs (Helm and Alfonzo 2014), and their lack frequently leads to growth defects in S. cerevisiae and neurological or mitochondrial disorders in humans (Fig. 4; for reviews, see Hopper 2013; Ramos and Fu 2019; Suzuki 2021). Previous analysis of a database of 561 sequenced tRNAs from bacteria, archaea, eukaryotes, mitochondria, and chloroplasts (Sprinzl and Vassilenko 2005, now Juhling et al. 2009) found chemical modifications on 11.9% of tRNA residues, with a median of eight modifications per tRNA species (Phizicky and Alfonzo 2010; Phizicky and Hopper 2010), and a range of average modification frequencies from 6.5% to 16.5% in different subgroups of species and from 8.6% to 10.2% in organelles (Machnicka et al. 2014). Among 34 S. cerevisiae cytoplasmic sequenced tRNA species, there are 25 chemically distinct modifications, which are found at 36 different residues, with an average of 12.6 modifications per species, ranging from 7 to 17 modifications per tRNA; and for 17 sequenced S. cerevisiae mitochondrial tRNAs, 9.5% of the residues have modifications, with six to nine modifications per tRNA (Phizicky and Alfonzo 2010; Phizicky and Hopper 2010).
Modifications within the main tRNA body and outside the ACL region (i.e., N1–N30 and N40–N76, and not N31–N39) comprise the majority of tRNA modifications found in tRNAs (Figs. 2, 3). For example, of the ∼12.6 modifications found in a typical cytoplasmic tRNA in S. cerevisiae, 10 are body modifications, and comprise 14 of the 25 different modifications (Fig. 3; Supplemental Table S1). As described in detail later in this review, the body modifications have important roles in tRNA folding and/or stability, and their biology intersects several other important cellular pathways.
ACL modifications play major roles in decoding mRNA at the A-site of the ribosome. Most of the diversity in tRNA modifications occurs within the ACL region (Machnicka et al. 2014). Indeed, the ACL region contains 15 of the 25 distinct modifications in S. cerevisiae, 17 of the 28 in humans, and 21 of the 28 in E. coli, and on average ∼30% of the residues in this region are modified in eukaryotes (Han and Phizicky 2018).
Below, we provide highlights in the biology of modifications, starting with modifications in the ACL region.
MODIFICATIONS IN THE ANTICODON LOOP REGION
Of the modifications in the ACL region of tRNAs, the N34 and N37 modifications are by far the most commonly modified and have the most variety. Among ∼600 completely analyzed tRNAs, N34 modifications are found in 255 tRNAs and N37 modifications occur 426 times, and remarkably, the 29 chemically distinct modifications at N34 and 13 at N37 together comprise 70% of the chemically different modifications in this data set (Machnicka et al. 2014).
The major driving forces for N34 and N37 modifications are to stabilize codon–anticodon interactions that have multiple A–U pairs, and to properly discriminate pairing between the wobble nucleotide (N34) and the third nucleotide of the codon (B3) at the A-site of the ribosome (for review, see Grosjean and Westhof 2016). The N34–B3 interactions at the A-site require structural accommodation within the codon–anticodon helix (Demeshkina et al. 2012). N34 modifications help achieve this accommodation by the changes in chemical properties of N34, which can alter the population of tautomeric forms of the base, orientation of the base about the glycosidic bond, hydrogen bonding, or the sugar pucker, to allow a proper fit of N34–B3 within the decoding site of the ribosome (Murphy and Ramakrishnan 2004; Murphy et al. 2004; Weixlbaumer et al. 2007; Kurata et al. 2008; Vendeix et al. 2012; Grosjean and Westhof 2016). N37 modifications improve stacking. For example, t6A37 in E. coli tRNALys(UUU) promotes stacking interactions with A38 in the ACL, and a cross-strand stacking interaction with the B1 base of the codon in the decoding center of the ribosome A site (Murphy et al. 2004), and the ms2 moiety of ms2t6A37 found in human tRNALys(UUU) enhances the stacking interactions with the B1 base (Vendeix et al. 2012).
In the sections below, we elaborate on specific examples of the biology of ACL region modifications (Fig. 3; Supplemental Table S1), with emphasis on relatively recent discoveries.
The essential deamination of adenosine to form I34 by the Tad2:Tad3 (ADAT2:ADAT3) complex
In all eukaryotes, tRNAs with A34 are deaminated to form I34, allowing decoding of codons ending in U, C, or A. This reaction is catalyzed by the essential Tad2:Tad3 complex in S. cerevisiae, with Tad2 as the catalytic subunit, requiring formation of the complex with Tad3 for activity (Auxilien et al. 1996; Gerber and Keller 1999; Liu et al. 2020), and by the homologous ADAT2:ADAT3 complex in mouse (Ramos-Morales et al. 2021). Although lack of I34 is lethal due to lack of proper decoding by tRNAs with A34, the ADAT3–V144M mutation is linked to severe intellectual disability (Fig. 4) and strabismus in patients from multiple families (Alazami et al. 2013). Analysis of tRNAs from lymphoblastoid cell lines (LCLs) derived from patients showed reduced levels of I34 modification for each of several tRNAs, which was associated with an increased frequency of aggregates in the corresponding ADAT2:ADAT3–V144M complex, relative to the WT complex (Ramos et al. 2019).
Modification of U34 by elongator and other proteins to form mcm5U34, mcm5s2U34, ncm5U34, and ncm5Um34
Of the 29 chemically distinct N34 modifications in different organisms (Machnicka et al. 2014), nine are found in S. cerevisiae tRNAs, four of which contain the carboxymethyluridine moiety (xcm5U34), with x representing an attached methyl (m) or amino (n) group (Karlsborn et al. 2014b). These xcm5U modifications are found in S cerevisiae on 11 of the 13 tRNA species with U34, including five with ncm5U, one with ncm5Um, two with mcm5U, and three with mcm5s2U (Fig. 6). As elaborated below, these modifications are subject to complex biochemistry, and have major regulatory roles.
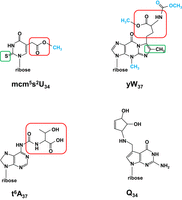
Schematic of complex modifications. All modifications are shown as nucleosides. (Top left) The mcm5s2U34 modification. The schematic is shown with the 2-thio moiety s2 boxed in green, the 5-carboxymethyl moiety cm5 boxed in red, and the terminal methyl group colored blue. In ncm5U, the terminal methyl group would be an amino group, and the sulfur in the 2-thio moiety would be an oxygen. (Top right) The yW37 modification. The schematic is shown with the methyl/methylene residues added to m1G to form the additional ring of imG14 boxed in green, the α-amino-α-carboxypropyl group added from S-adenosylmethionine boxed in red, and other added groups colored blue. (Bottom left) The t6A37 modification. The schematic is shown with the threonylcarbamoyl group boxed in red. (Bottom right) The Q34 modification.
The apparatus required for xcm5U34 modification in S. cerevisiae is enormous, as 15 genes are required for formation of mcm5U, 11 genes are required for formation of the s2U group of mcm5s2U (for review, see Karlsborn et al. 2014b), and two are required for formation of the Um moiety of ncm5Um (Pintard et al. 2002; Guy et al. 2012). Three findings led the way in identifying the components required for these modifications. First, Bystrom and coworkers cloned the S. pombe sin3+ gene (Huang et al. 2005) by screening for complementation of a sin3 mutant, previously shown to have reduced nonsense suppression and reduced mcm5s2U in tRNAs (Heyer et al. 1984), and sequencing revealed that it encoded the S. cerevisiae ortholog of Elp3, a subunit of the elongator complex, previously implicated in other functions. Consistent with a requirement for the elongator complex in xcm5U34 modification, mutations in each of the six elongator genes eliminated the modification and reduced nonsense suppression in S. cerevisiae (Huang et al. 2005). Second, the connection was made between resistance to γ-toxin from Kluyveronmyces lactis and lack of mcm5s2U34 in tRNAs. Thus, S. cerevisiae kti11, kti12, and kti13 mutants, like elp mutants, were each resistant to K. lactis γ-toxin produced by the zymocin complex (Frohloff et al. 2001; Jablonowski et al. 2001; Fichtner et al. 2002), and had reduced xcm5U in tRNAs and reduced nonsense suppression (Huang et al. 2005). As K. lactis γ-toxin was shown to encode an endonuclease that cleaved S. cerevisiae tRNAs with mcm5s2U34 at U34 (Lu et al. 2005), other genes important for mcm5s2U34 formation were identified by screening for γ-toxin resistance (Mehlgarten and Schaffrath 2003; Huang et al. 2008). Third, several laboratories uncovered the biochemical pathway by which the s2U tRNA modification was made in a sulfur relay from cysteine to tRNA, via Nfs1, Tum1, Uba4, and then Urm1, followed by thiolation of tRNA by Ncs2–Ncs6 using the Urm1-activated thiol (Nakai et al. 2008; Leidel et al. 2009; Noma et al. 2009).
The biochemical function/activity of some of these proteins has become clearer. Elp3 is known to be the catalytic component for carboxymethylation of U34, generating a 5′-deoxyadenosyl radical from S-adenosylmethionine that is used to generate an acetyl coA radical, to catalyze formation of cm5U34 (Selvadurai et al. 2014). The external methyl group of mcm5U and mcm5s2U is known to be attached by a complex of Trm9 (human ALKBH8) and Trm112 in yeast and humans (Kalhor and Clarke 2003; Fu et al. 2010; Mazauric et al. 2010; Songe-Moller et al. 2010). Surprisingly, however, yeast trm9Δ and trm112Δ mutants accumulate the corresponding ncm5U and ncm5s2U modifications, rather than the anticipated cm5U and cm5s2U modifications, suggesting either that ncm5U (ncm5s2U) is the precursor for mcm5U (mcm5s2U), rather than cm5U, or that ncm5U (ncm5s2U) is a default modification in the absence of the corresponding methyl modification (Mazauric et al. 2010; Chen et al. 2011a). An additional surprise was the discovery of mammalian tRNAs with a hydroxylated version of mcm5U, with tRNAGly(UCC) bearing (S)-mchm5U, catalyzed in vivo and in vitro by the AlkB domain of ALKBH8, and tRNAArg(UCG) bearing the (R)-diastereomer (van den Born et al. 2011).
Remarkably, the multiple phenotypic consequences of elongator mutants in S. cerevisiae (Fig. 4) are almost all due to reduced function of some combination of the three tRNAs with the mcm5s2U modification (tRNAGln(UUG), tRNALys(UUU), and tRNAGlu(UUC)) (for review, see Karlsborn et al. 2014b). Although the elongator complex was initially implicated in Pol II transcription elongation due to chromatin remodeling by histone acetylation (Otero et al. 1999; Wittschieben et al. 1999; Kim et al. 2002; Winkler et al. 2002), and in polarized transport of secretory vesicles to the bud tip (Rahl et al. 2005), these phenotypes are all due to reduced function of tRNAGln(UUG) and tRNALys(UUU), as they were completely rescued by overexpression of the two tRNA species (Esberg et al. 2006). Moreover, many of the phenotypes of elongator mutants were also found in an ncs2Δ mutant strain, which lacks the s2U moiety of mcm5s2U34 (Esberg et al. 2006). Similarly, defects in telomeric gene silencing and the DNA damage response that were ascribed to elongator mutants (Li et al. 2009) were rescued by overexpression of all three tRNAs with mcm5s2U, and tuc2Δ (ncs2Δ) mutants, lacking the s2U moiety, had the same phenotypes as the elongator mutants (Chen et al. 2011b).
The xcm5U and mcm5s2U modifications are found widely in eukaryotes (Karlsborn et al. 2014b), in which mutants invariably have significant defects. In S. pombe, lack of the conserved Ctu1–Ctu2 (Ncs6–Ncs2) complex resulted in loss of s2U from mcm5s2U in tRNAs, associated with temperature sensitivity and a septation defect leading to aberrant ploidy, and the temperature sensitivity was rescued by overproduction of tRNALys(UUU) and tRNAGlu(UUC) (Dewez et al. 2008). Similarly, S. pombe elp3 mutants, which have U34 instead of mcm5s2U34, are sensitive to H2O2 stress due to reduced function of tRNALys(UUU) (Fernandez-Vazquez et al. 2013). In metazoans and plants, mutations in the mcm5s2U modification components have distinct phenotypes, but the corresponding tRNA rescue experiments to directly link the effects to tRNA biology have not been reported. In C. elegans, mcm5s2U is implicated in neurological and developmental defects, based on analysis of five mutants with reduced levels of the mcm5U or s2U moieties (Dewez et al. 2008; Chen et al. 2009a; Kim et al. 2010b), accompanied by a temperature sensitive germ-line maturation defect for three tested mutants (Dewez et al. 2008; Chen et al. 2009a), and a temperature sensitive defect in a chemotaxis learning assay for two elongator mutants (Chen et al. 2009a). In humans, an intronic mutation in the ELP1 ortholog IKBKAP has been linked to the recessive neurodegenerative genetic disease familial dysautonomia (Fig. 4; Slaugenhaupt et al. 2001) and is associated with reduced mcm5s2U (Karlsborn et al. 2014a). In Drosophila and mouse, the corresponding elp3 mutants and null IKBKAP (ELP1) mutants are embryonic lethal, with vascular and neural development defects in the mouse (Chen et al. 2009b; Walker et al. 2011), and germ-line-specific conditional mutants cause male infertility, associated with defective chromosome synapsis and meiotic recombination, and reduced xcm5U modification (Lin et al. 2013).
There has been substantial progress in understanding the precise translation defect due to lack of mcm5U or s2U in S. cerevisiae. It was noted previously that the rescue of xcm5U mutant phenotypes by overexpression of unmodified tRNAs implied reduced tRNA function at or before the A-site decoding step, rather than a miscoding defect arising from the lack of modifications (Bjork et al. 2007). A subsequent seminal study measured decoding potential by examining growth of mutants lacking xcm5U or s2U groups, in combination with deletions of selected nonessential tRNA genes with C34. As C34-containing tRNAs can only decode G ending codons, whereas U34-containing tRNAs can in principle decode both A-ending and G-ending codons, this strategy allowed for decoding analysis in codon boxes that are decoded with both U34-containing tRNAs and nonessential C34-containing tRNAs (Johansson et al. 2008). For example, it was found that deletion of both copies of tRNAGly(CCC) resulted in little growth defect, but was nearly lethal in combination with an elp3Δ mutation, providing strong evidence that mcm5U34 in tRNAGly(UCC) was important for reading GGG proline codons. This and other similar experiments suggested that mcm5U and ncm5U improve decoding of G-ending codons, and that in tRNAs with mcm5s2U34, both mcm5U and s2U cooperate to improve decoding of G-ending codons (Johansson et al. 2008), although in this study only a few of the tRNAs with xcm5U or mcm5s2U could be examined.
Subsequent examination of mcm5s2U and xcm5U modifications by mass spectrometry and ribosome footprint profiling techniques substantially enhanced understanding of their translation roles. A mass spectrometry study in S. cerevisiae showed that lack of either xcm5U or s2U led to underrepresentation of proteins with a high abundance of AAA, CAA, and GAA codons, decoded by tRNALys(UUU), tRNAGln(UUG), and tRNAGlu(UUC), respectively, the three tRNAs with mcm5s2U (Rezgui et al. 2013). Ribosome profiling analysis extended these results. Ribosome profiling of elp3Δ mutants lacking mcm5U showed a minor but distinct increased occupancy of CAA and GAA codons, and several mutants lacking s2U had increased occupancy of CAA and AAA, codons, with little effect on the corresponding G-ending codons (Zinshteyn and Gilbert 2013). In addition, elp3Δ and mutants lacking s2U had low level induction of GCN4 translation that was independent of Gcn2 (the eIF2α kinase), suggesting constitutive activation of the general amino acid control (GAAC) signaling pathway (Natarajan et al. 2001; Hinnebusch 2005; Castilho et al. 2014; Wu et al. 2020) as a consequence of a lack of mcm5s2U (Zinshteyn and Gilbert 2013).
A major breakthrough in understanding of the translation defect due to lack of xcm5U and/or s2U modifications came from further ribosome profiling, combined with gene expression analysis using RNA seq, which revealed a prominent proteotoxic stress defect associated with lack of the modifications (Nedialkova and Leidel 2015). Thus, ribosomes in S. cerevisiae ncs2Δ and elp6Δ mutants (lacking s2U and mcm5U, respectively) had increased occupancy of CAA and AAA codons accompanied by the accumulation of protein aggregates, which was linked to poor clearance of stress induced protein aggregates, and both phenotypes were suppressed by overexpression of the three tRNA species with mcm5s2U. Remarkably, these functions of mcm5s2U are conserved, as ribosomes in C. elegans ncs2−/− mutants also had increased occupancy of CAA and GAA codons and a similar protein aggregation phenotype (Nedialkova and Leidel 2015).
Several studies have described how the xcm5U and s2U modifications contribute to decoding interactions in the context of human tRNALys(UUU) anticodon stem–loops with mcm5s2U34, ms2t6A37, and Ψ39. Physical and NMR analysis showed that mcm5U34 has virtually no effect on the structure of U34 or of the ACL, whereas the s2U modification promotes stacking of U34 and U35 and modestly increases the 3′ endo conformation of U34 (Durant et al. 2005). Further studies of the human tRNALys(UUU) ASL at the ribosome A-site showed that mcm5s2U34 has normal Watson–Crick pairing with the corresponding B3 nucleotide, with mcm5s2U34 shifting to the enol form (Vendeix et al. 2012). Analysis of translation showed that lack of s2U in yeast tRNALys(UUU) with mcm5U results in reduced tRNA binding at the A-site due to increased off-rate, a reduced rate of conformational changes necessary for the translation cycle, and a greater rate of rejection before peptide formation (Ranjan and Rodnina 2017).
The xcm5U modifications are subject to multiple levels of regulation. Intriguingly, it appears that proper elongator modification function requires intermediate levels of Elp1 phosphorylation. Thus, zymocin resistance (reduced mcm5s2U modification) in S. cerevisiae is associated with mutations in the protein kinase Kti14 (Hrr25) that result in reduced Elp1 phosphorylation, as well as with a sit4Δ mutation, which is associated with increased Elp1 phosphorylation; and related experiments show that a sit4Δ mutation in an hrr25-3 mutant restores the normal moderate Elp1 phosphorylation levels of WT cells and normal zymocin sensitivity (Mehlgarten et al. 2009). In support of a direct effect of phosphorylation, subsequent experiments provided evidence that Hrr25 directly phosphorylated Elp1, and showed that the Elp1 phosphorylation state was important for interactions with Kti12 (Abdel-Fattah et al. 2015). Kti12 is structurally similar to O-phosphoseryl tRNA kinase (PSTK), a protein involved in biosynthesis of tRNASec, and like PSTK, binds efficiently to bulk tRNAs, has a tRNA-dependent ATPase activity that is essential for xcm5U modification, and binds directly to Elp1 and stimulates the ATPase (Krutyholowa et al. 2019). However, it is not known how this ATPase activity is linked to elongator modification function.
Furthermore, it is now known that elongator modifications reciprocally regulate TOR signaling in S. pombe. Thus, elongator mutants up-regulate TORC1 by down-regulating inhibitors such as Tsc2, and down-regulate TORC2 by inhibiting expression of positive effectors such as Ste20, resulting in increased TORC1 signaling, unbalanced TORC signaling, and sensitivity to the TORC1 regulator rapamycin (Candiracci et al. 2019). Conversely, TOR also regulates elongator function, as overexpression of the TORC1 kinase Tor2 leads to reduced levels of xcm5U and xcm5Um modifications, whereas rapamycin treatment of WT cells (inhibiting TORC1), or overexpression of the TORC2 kinase Tor1, increased the amount of xcm5U and xcm5Um. Activation of elongator by the TORC2 pathway in this set of experiments appeared to occur by down-regulation of glycogen synthase kinase (Gsk3), which phosphorylates and inhibits Elp4 function in the elongator complex (Candiracci et al. 2019).
Levels of s2U34 are also subject to environmental regulation. In WT S. cerevisiae strains, the s2U modification is significantly reduced at temperatures higher than 30°C, due to lack of formation of the modification at higher temperatures (Alings et al. 2015; Damon et al. 2015; Han et al. 2015). Study of the effects of growth conditions showed that s2U modification is significantly reduced in synthetic medium containing glucose (fermenting sugar) or lactate (nonfermenting), due to reduced intracellular methionine and cysteine, and that sulfur starvation leads to reduced expression of Uba4 in the s2U pathway and reduced s2U in tRNA, resulting in increased expression of genes involved in biosynthesis of methionine and cysteine (Laxman et al. 2013). Subsequent metabolic analysis showed that tRNA thiolation mutants reroute carbon flux as if the cells are starved, down-regulating the pentose phosphate pathway and nucleotide biosynthesis pathway and up-regulating pathways leading to storage carbohydrates trehalose and glycogen, and that this is due to down-regulation of phosphate homeostasis and reduced intracellular phosphate (Gupta et al. 2019; for review, see Gupta and Laxman 2020).
Trm7/FTSJ1, partners Trm732/THADA and Trm734/WDR6, and Nm32 and Nm34 modification
The importance of ribose 2′-O-methylation (Nm) in the ACL of eukaryotes has been evident since the identification of S. cerevisiae Trm7 as the 2′-O-methyltransferase responsible for Nm32 and Nm34 formation of its three substrate tRNAs (tRNAPhe, tRNATrp, and tRNALeu(UAA)), and the finding that mutants had a severe growth defect (Fig. 4) linked to poor translation (Pintard et al. 2002).
It is now known that S. cerevisiae Trm7 requires interaction partners Trm732 and Trm734 for formation of Nm32 and Nm34, respectively, on each Trm7 substrate tRNA, and that the critical tRNA substrate is tRNAPhe, as overproduction of tRNAPhe almost completely suppresses the S. cerevisiae trm7Δ growth defect (Guy et al. 2012). Moreover, the Cm32 and Gm34 modifications of tRNAPhe(GAA) each help direct formation of wybutosine (yW) from 1-methylguanosine at G37 (m1G37) (discussed further below), as tRNAPhe from trm7Δ mutants has m1G37 instead of yW37, and tRNAPhe from trm732Δ and trm734Δ mutants each has only partial yW37 modification. Thus, in a trm7Δ mutant the entire ACL of tRNAPhe is undermodified at C32, G32, and G37. In addition, the severe growth defect of trm7Δ mutants is known to be due to loss of both the Cm32 and Gm34 modifications, as trm732Δ trm734Δ mutants phenocopy the severe growth defect of trm7Δ mutants and completely lack yW37, whereas trm732Δ and trm734Δ mutants each grow normally, as do trm732Δ tyw1Δ and trm734Δ tyw1Δ double mutants (Guy et al. 2012), which also have m1G37 instead of yW37 due to lack of Tyw1 (Waas et al. 2005; Noma et al. 2006).
This modification circuitry for Nm32 and Nm34 formation in tRNA substrates and yW formation in tRNAPhe is conserved widely through eukaryotes. Thus, tRNAPhe from S. pombe trm7Δ mutants and from patients with null mutations in the human TRM7 ortholog FTSJ1 each lack detectable Cm32 and Gm34, and have m1G37 instead of yW37 or the human yW derivative peroxywybutosine (Guy and Phizicky 2015; Guy et al. 2015). Furthermore, S. pombe trm732Δ and trm734Δ mutants each lack the corresponding Cm32 and Gm34 modifications, and expression of S. pombe Trm732 or its human ortholog THADA complements the growth defect of an S. cerevisiae trm732Δ trm734Δ mutant and restores Cm32 formation in tRNAPhe (Guy and Phizicky 2015; Guy et al. 2015). As Trm7, Trm732, and Trm734 orthologs are found in diverse eukaryotes, as is yW37 or its derivatives, it seems likely that the Trm7 modification circuitry is widely conserved in eukaryotes (Guy and Phizicky 2015).
Intriguingly, there are two Trm7 paralogs in D. melanogaster and related genus members, one of which (CG5220, dTrm7_32) is required for Nm32 modification, and the other (CG7009, dTrm7_34) for Nm34 modification (Angelova et al. 2020). Nonetheless, it seems likely that the Trm732 ortholog (CG15618, DmTHADA) and Trm734 ortholog WDR6 (CG33172) are also required in Drosophila for formation of Nm32 and Nm34, respectively, as knockdowns of these genes each have a similar phenotype as knockdowns of Trm7 orthologs, and the dTrm7_34 protein physically interacts with the Drosophila Trm734/WDR6 (Angelova et al. 2020).
TRM7/FTSJ1 is important in all eukaryotes examined, although the biological manifestations of mutations differ in different eukaryotes. In both S. cerevisiae and S. pombe, trm7Δ mutants have a severe growth defect due to reduced tRNAPhe function (Pintard et al. 2002; Guy et al. 2012; Guy and Phizicky 2015). Moreover, both S. cerevisiae and S. pombe trm7Δ mutants constitutively and robustly activate the GAAC pathway in the absence of an apparent charging defect, presumably due to increased ribosome collisions (Chou et al. 2017; Han et al. 2018). Remarkably, the constitutive GAAC activation is itself part of the reason for the severe growth defect of S. cerevisiae trm7Δ mutants, as their severe growth defect is partially alleviated by mutation of the GAAC pathway (Han et al. 2018).
In multicellular organisms, lack of TRM7 is manifested by distinct phenotypes. In Drosophila, homozygous null mutants of either dTrm7_34 or dTrm7_32 do not have a noticeable growth defect but each mutant (or dsRNA knockdown), as well as the corresponding DmTHADA or WDR6 knockdowns, inhibit Ago2-dependent silencing by the siRNA pathway and piRNA-mediated silencing (Angelova et al. 2020). Furthermore, homozygous double mutant flies lacking both dTrm7_32 and dTrm7_34 have modestly reduced size and weight, reduced life spans, and some locomotion defects (Angelova et al. 2020). In A. thaliana, a trm7 mutant (scs9) has reduced resistance to a bacterial infection and a mild growth defect (Ramirez et al. 2018). In humans, FTSJ1 mutations lead to nonsyndromic X-linked intellectual disability (Fig. 4, NSXLID; Freude et al. 2004; Ramser et al. 2004; Froyen et al. 2007; Takano et al. 2008; Guy et al. 2015), suggesting little other obvious abnormality, whereas mouse Ftsj1−/− males have impaired learning as well as several phenotypes related to metabolism (Jensen et al. 2019).
As speculated by Carré and colleagues (Angelova et al. 2020), it is intriguing to note the possible connection between Trm7 biology and transposable elements in Drosophila and S. cerevisiae. Lack of either Drosophila TRM7 ortholog, or their partner proteins DmTHADA or WDR6 results in derepression of expression of the retrotransponon gypsy in ovarian follicle cells, while lack of TRM7 or TRM734 in S. cerevisiae results in expression of the Ty1 transposable element (Nyswaner et al. 2008). These phenomena might reflect a common theme.
It is not known in multicellular animals and plants if a single tRNA is responsible for the different phenotypes of TRM7/FTSJ1 mutants, as in fungi for tRNAPhe(GAA). In this connection, it is intriguing to note that 2,6-diaminopurine binds to and inhibits FTSJ1 in human Calu-6 cancer cells, resulting in reduced Cm34 modification of tRNATrp(CCA) and increased readthrough of UGA stop codons (Trzaska et al. 2020). It is also not known if other phenotypes attributed to lack of TRM732/THADA or TRM734/WDR6 are related to tRNA biology. S. cerevisiae Trm734 was previously identified as Ere2, a protein that interacts with Ere1 in the retromer-mediated pathway to recycle cell membrane proteins back to the cell surface after internalization (Shi et al. 2011). In addition, Drosophila THADA mutants are cold sensitive and obese, with elevated triglycerides, and THADA was shown to interact with the sarco/ER Ca2+ ATPase as an uncoupler (Moraru et al. 2017). As both TRM732/THADA and TRM734/WDR6 are large proteins with relatively small highly conserved domains (Guy and Phizicky 2015; Hirata et al. 2019; Funk et al. 2022), it is possible that these functions of Trm732 and Trm734 are unrelated to tRNA biology.
The crucial and universal m1G37 modification and its tRNAPhe derivative wybutosine, yW37
The highly conserved m1G37 modification is crucially important for tRNA function in organisms in all domains of life. An early biochemical study showed that m1G37 on tRNAAsp protects the tRNA from mischarging by yeast arginyl-tRNA synthetase (Putz et al. 1994). Subsequent seminal studies showed that lack of m1G37 due to null or near null mutations in S. cerevisiae TRM5 or S. typhimurium trmD, respectively, leads to a severe growth defect, consistent with the widespread occurrence of m1G37 in tRNAs in organisms (Bjork et al. 2001) and showed a prominent role of m1G37 in preventing +1 frameshifting (Bjork et al. 1989; Urbonavicius et al. 2001, 2003).
Recent results have substantially increased our knowledge of the role of m1G37. Biochemical analysis with ASL's based on tRNAPro(CGG) show that m1G37 improves the binding constant for RNA binding to a cognate CCG codon by threefold (relative to the ASL with G37) and weakens the binding constant by ninefold to a +1 CCC-U frameshifting codon (Nguyen et al. 2019). While it is not known how lack of m1G37 affects eukaryotic translation, ribosome profiling in E. coli shows that lack of m1G37 leads to ribosome stalling at the A-site of a subset of codons (Pro CCN, Arg CGG, and Leu CUA codons), showing a direct role for m1G37 in decoding that is distinct from frameshifting, attributed to reduced charging of tRNAPro and tRNAArg(CCG), and reduced peptide bond formation for some of the tRNAs (Masuda et al. 2021).
Wybutosine, yW37 (Fig. 6), and its various derivatives are found ubiquitously on tRNAPhe in eukaryotes and archaea and is formed from m1G37 (Droogmans and Grosjean 1987). Although the biogenesis pathway of wybutosine and derivatives varies in different organisms (discussed in Sample et al. 2015), these pathways always involve formation of wyosine (imG-14) by methylation of m1G37 and ring closure on the WC face of G37 (Fig. 6), catalyzed by Tyw1/Taw1, followed by various further maturation steps, which in S. cerevisiae involves addition of the main body of methionine from S-adenosylmethionine to the Hoogsteen side of the third ring by Tyw2, methylation at N3 of the guanosine moiety by Tyw3/Taw3, and esterification and amidation of the carboxyl and amino groups of the methionine moiety by Tyw4 (Kalhor et al. 2005; Waas et al. 2005; Noma et al. 2006). tRNAPhe with m1G37 instead of yW37 stimulated frameshifting in vitro (Carlson et al. 2001), and each of the successive maturation steps in wybutosine in S. cerevisiae further reduced −1 frameshifting of a test sequence in a reporter in vivo (Waas et al. 2007).
Recent developments have emphasized the crucial importance of m1G37 and yW37 in mitochondrial function. Prior work in S. cerevisiae showed that m1G37 is found on at least eight mitochondrially encoded tRNA species, including tRNAfMet (Canaday et al. 1980), that Trm5 is localized to mitochondria in addition to the nuclear/cytoplasmic compartment, and that specific loss of mitochondrial Trm5 significantly reduces oxygen consumption, albeit with little evident growth phenotype on media containing the nonfermentable carbon source glycerol (Lee et al. 2007). Curiously, although T. brucei TRM5 is also localized to both the mitochondrial and the nuclear/cytoplasmic compartments, and down-regulation of TRM5 leads to reduced m1G37 in both mitochondrial and cytoplasmic tRNAs, translation is reduced in mitochondria but not in the cytoplasm (Paris et al. 2013). The importance of m1G37 in T. brucei mitochondrial tRNAs also extends to the imG-14 derivatives unexpectedly found in T. brucei mitochondrial tRNAPhe (Sample et al. 2015). T brucei has a nuclear Tyw1 paralog that is responsible for imG-14 formation in cytoplasmic tRNAPhe, some of which is imported into the mitochondria, and a mitochondrial Tyw1 paralog that is responsible for imG-14 formation of mitochondrial tRNAPhe, which is imported before modification. Down-regulation of either paralog resulted in little growth defect in normal growth media but resulted in reduced growth in low-glucose media, in which cells need full mitochondrial function. This result suggests an important unexpected role for the imG-14 or derivative modification on mitochondrial tRNAPhe. Although other explanations are possible, one attractive explanation advanced by the authors was that the extensive U-rich mRNAs that arise from pan-editing in kinetoplasmid mitochondria might impose strict requirements that all tRNAs are modified as completely as possible to prevent frameshifting (Sample et al. 2015).
A similar apparent mitochondrial bias for TRM5 function has also been found in humans. Thus, each of two unrelated patients with compound heterozygous TRMT5 mutations had lactic acidosis and multiple deficiencies in mitochondrial function in skeletal muscle, accompanied by reduced m1G37 in mitochondrial tRNA. Moreover, analysis in yeast showed that the mutations were functionally hypomorphic as expression of the corresponding trm5 variants as the only source of mitochondrial TRM5 led to partially reduced oxygen consumption and respiratory function (Powell et al. 2015).
The biology of the universally important N6-threonylcarbamoyladenosine modification, t6A37
Much has been learned about the occurrence, biosynthesis, and function of t6A since its original discovery in tRNA (Schweizer et al. 1969). The t6A modification, or derivatives of it, is found in all organisms examined in all domains of life, and is invariably found at residue A37, immediately 3′ of U36 of tRNAs with NNU anticodons. Indeed, only a few tRNAs with the NNU anticodon do not have the t6A37 modification including, most prominently, initiator tRNA in prokaryotes, organelles, and archaea, which often harbor unmodified A37 (summarized in Morin et al. 1998). Initial studies showed that formation of t6A required ATP to incorporate a one-carbon group and threonine at N6 of A37 (Elkins and Keller 1974; Korner and Soll 1974), and physical studies showed that t6A37 of the tRNALys(UUU) ASL unexpectedly decreased stacking in the ACL, bulging out the adjacent U36 residue, and forming a cross-strand stack with N1 of the codon (Murphy et al. 2004; Durant et al. 2005).
Discovery of the genes involved in t6A biosynthesis facilitated study of its biology. Bioinformatic analysis, coupled with biochemical analysis, showed that the highly conserved Sua5 (S. cerevisiae)/YrdC (E. coli) family of proteins is directly involved in t6A biosynthesis (El Yacoubi et al. 2009), and genetic analysis established that lack of the gene results in a severe slow growth phenotype in S. cerevisiae (Na et al. 1992), and lethality in E. coli (El Yacoubi et al. 2009). Subsequent experiments in S. cerevisiae showed that Kae1, Bud32, and Pcc1 of the highly conserved KEOPS complex are involved in t6A biosynthesis, and that mutants have very similar slow growth phenotypes; in contrast, the KEOPS complex member Cgi121 is not involved in t6A biosynthesis and mutants grow nearly normally (El Yacoubi et al. 2011; Srinivasan et al. 2011).
Biochemical experiments with purified B. subtilis proteins established a synthetic route (for review, see Thiaville et al. 2014) involving direct carboxylation of threonine with bicarbonate (or CO2) and transfer to ATP, displacing PPi to form the intermediate threonylcarbamoyl AMP (TC-AMP), which was then transferred to N6 of A37 to form t6A (Fig. 6; Lauhon 2012). Subsequent experiments with purified yeast components likewise showed that Sua5 catalyzes TC-AMP formation, and that Bud32, Kae1, and Pcc1 of the KEOPS complex catalyzes its transfer to N6A37 of substrate tRNAs, requiring the Bud32 (TsaE in bacteria) ATPase (Perrochia et al. 2013a,b). Remarkably, the Kae1 paralog Qri7 of S. cerevisiae mitochondria can replace the entire KEOPS complex in vivo and in vitro (Wan et al. 2013). Additional structural and functional analysis showed that the TC-transfer step is catalyzed by the TsaD (Kae1) subunit in complex with TsaB, and elaborated the role of TsaE (Bud32) in regulation of the TsaD catalytic activity, allowing multiple turnover reactions through its ATPase activity (Luthra et al. 2018, 2019; Missoury et al. 2018).
Unexpectedly, t6A37 is not the final modification product in E. coli and S. cerevisiae, and based on phylogenetic analysis, likely also in most proteobacteria and bacteroidetes, most fungi, and several protists and plants. In these organisms, the normal t6A modification is converted to the cyclic derivative ct6A by TcdA dehydratase and related family members, in which one of the hydroxyls from the terminal carboxyl group of the threonine moiety is lost during a condensation reaction that results in an oxazolone ring instead of the more customary linear threonine adduct (Miyauchi et al. 2013). Although the corresponding E. coli tcdA mutant grows at a normal rate, it does not compete with WT, and the corresponding S. cerevisiae tcd1Δ and tcd2Δ mutants each lack ct6A and grow poorly on glycerol-containing media, which require respiration. Consistent with a mild translation defect, tRNALys(UUU) lacking ct6A in E. coli tcdA mutants have reduced ability to decode near cognate AGA and noncognate UAG codons (Miyauchi et al. 2013). Intriguingly, echinoderm mitochondria decode AAA as asparagine instead of lysine, and their tRNALys(CUU) has hydroxy-t6A (ht6A) instead of t6A and binds more poorly to ribosomes with AAA codons, in principle allowing for AAA decoding by tRNAAsn(GUU) (Nagao et al. 2017).
Consistent with early analysis, t6A is generally essential in bacteria and archaea, whereas mutants in eukaryotes are slow growing but viable (Fig. 4; El Yacoubi et al. 2009, 2011; Srinivasan et al. 2011; Naor et al. 2012); however, the source of this discrepancy is unknown (for review, see Thiaville et al. 2015).
In S. cerevisiae, there are a number of consequences of lack of t6A modification. Temperature sensitive mutants in the KEOPS complex components trigger translation of GCN4, the transcriptional activator of the GAAC pathway, attributed to defective recognition of the AUG codons of the normally inhibitory upstream open reading frames of GCN4 by tRNAiMet(CAU) lacking t6A (Daugeron et al. 2011). Additional experiments showed that null mutants in the t6A pathway accumulate aggregates indicative of proteotoxic stress (Thiaville et al. 2016), particularly in the absence of both mcm5U and t6A (Pollo-Oliveira et al. 2020), similar to the accumulation of aggregates previously observed in mutants lacking mcm5U and/or s2U (Nedialkova and Leidel 2015). In addition, null mutants in the t6A pathway were sensitive to stresses such as temperature, ethanol, and rapamycin, and had ribosome occupancy profiles suggesting that t6A acts to homogenize translation rates across codons and to prevent increased translation initiation at non-AUG codons (Thiaville et al. 2016).
In metazoans, reduced levels of t6A also have dramatic effects. In Drosophila, hemizygous kae1 larvae have reduced t6A that is correlated with a characteristic mass called a Black spot phenotype, an extended larval period, defective imaginal discs, and reduced proliferation of mitotic vs. nonproliferating tissues (Lin et al. 2015). In humans, pedigree analysis linked a homozygous kae1 missense mutation to a global developmental delay and renal defects, and the corresponding yeast mutant had reduced t6A (Edvardson et al. 2017). Furthermore, mutations in each of the gene products of the KEOPS complex are associated with Galloway–Mowat syndrome (GAMOS) (Fig. 4; Braun et al. 2017). A patient with early onset nephrotic syndrome associated with microcephaly and developmental delays had a mutation in OSGEP (KAE1, TsaD), the catalytic subunit of the TC-AMP transfer step, and whole exome sequencing of a panel of 907 patients with nephrotic syndrome, including 91 with GAMOS, revealed 32 familes with mutations in this or other subunits of the KEOPS complex (LAGE3, TP53RK, and TPRKB; orthologs of yeast Pcc1, Bud32, and Cgi121) (Braun et al. 2017). Moreover, corresponding knockouts (OSGEP and TPRKB) in zebrafish recapitulated the microcephaly with marked apoptosis in the brain; mouse knockouts (OSGEP, TPRKB, and LAGE3) reproduced the microcephaly; and human podocyte cell lines expressing shRNAs directed against two of these genes had reduced t6A, accompanied by increased apoptosis and decreased cell survival (Braun et al. 2017). More recently, examination of 14 GAMOS-affected patients revealed mutations in the remaining two genes associated with t6A biosynthesis, human GON7 (also called C14orf142) and YRDC (ortholog of yeast SUA5). Consistent with their yeast growth phenotypes, the patients with GON7 mutations had a milder disease presentation than those with YRDC mutations (Arrondel et al. 2019).
The intriguing biology of N6-isopentenyl-adenosine, i6A37, and Mod5/TRIT1
The N6-isopentenyl-adenosine (i6A) modification was first linked to tRNA function by the isolation of an S. cerevisiae mod5 mutant that reduced nonsense suppression by the tyrosine-inserting nonsense suppressor SUP7 and had tRNA with reduced amounts of i6A37 (Laten et al. 1978). The i6A37 modification occurs widely in tRNA from bacteria and eukaryotes, and its formation is catalyzed by the isopentenyl transferase (dimethylallyl transferase), called Mod5 in S. cerevisiae, miaA in E. coli, Tit1 in S. pombe, GRO-1 in C. elegans, and TRIT1 in humans (Dihanich et al. 1987; Lemieux et al. 2001; Soderberg and Poulter 2001; Spinola et al. 2005). Mod5 catalyzes isopentenylation at the N6 position of A37 of tRNAs with A36–A37–A38 in their ACLs (Motorin et al. 1997; Soderberg and Poulter 2000), with additional recognition of N34 for some orthologs (Lamichhane et al. 2011). In S. cerevisiae, the single MOD5 gene is responsible for the modification of both cytoplasmic and mitochondrial tRNA substrates (Dihanich et al. 1987) due to separate translation starts (Gillman et al. 1991; Slusher et al. 1991), and Mod5 is found in the nucleus, cytoplasm, and mitochondria (Boguta et al. 1994).
Mutants lacking i6A37 have several additional phenotypes. S. pombe tit1Δ mutants, like S. cerevisiae SUP7 mod5 mutants, have reduced decoding, as shown for each of two tested tRNAs on a reporter, suggesting a common role for i6A37 in decoding efficiency (Lamichhane et al. 2013). In addition, tit1Δ mutants have a growth defect in media containing the TOR inhibitor rapamycin that is almost completely rescued by overexpression of cytoplasmic tRNATyr(GUA) and tRNATrp(CCA), and a growth defect in media containing the nonfermentable carbon source glycerol that is unexpectedly partially rescued by these same two tRNAs, suggesting that the growth defect is not entirely due to a mitochondrial defect (Lamichhane et al. 2013, 2016). In contrast, a human TRIT1-R323Q mutation, in a highly conserved residue near the active site (Zhou and Huang 2008), was linked to encephalopathy and myoclinc epilepsy, and to multiple defects in oxidative phosphorylation that were attributed to reduced i6A in mitochondrial tRNASer (Yarham et al. 2014). Likewise, a pleiotropic C. elegans gro-1 mutant had developmental, behavioral, and reproductive defects, and increased life span, and these were all rescued by expression of a mitochondrial GRO-1 but not cytoplasmic GRO-1 (Lemieux et al. 2001).
Intriguingly, Mod5 is also required in S. cerevisiae for tRNA-mediated gene silencing of neighboring genes due to the nucleolar localization of tRNA genes being transcribed, and expression of human TRIT1 in S. cerevisiae confers this same property (Thompson et al. 2003; Wang et al. 2005; Pratt-Hyatt et al. 2013). As this silencing function is not due to the catalytic activity of Mod5, it is presumably a conserved moonlighting function of Mod5/TRIT1, which may impact human biology (Pratt-Hyatt et al. 2013).
The most intriguing aspect of i6A biology is its prion-related functions in S. cerevisiae, linked to Mod5 aggregation using a novel aggregation domain (Suzuki et al. 2012). Like other genetically characterized prions such as [PSI+], [MOD5+] cells have reduced function, which is mitotically stable over multiple generations, but can be reversed by chemical or genetic treatment (in this case by inhibition of the Hsp104 chaperone); is genetically dominant; and can be transmitted to a cell that is not [MOD5+] by introduction of the [MOD5+] protein. Moreover, consistent with previous results with S. cerevisiae mod5 hypomorphic mutants (Benko et al. 2000), [MOD5+] cells had reduced i6A in their tRNA and increased ergosterol due to increased activity of Erg20, which competes with Mod5 for the same dimethylallyl pyrophosphate substrate. Of special note, [MOD5+] yeast were resistant to several antifungal agents, likely due to increased ergosterol, and treatment with antifungal agents in wild-type cells triggered the generation of [MOD5+] cells, which was reversible upon removal of the drugs (Suzuki et al. 2012).
Although there is currently no detailed structural information of the effect of i6A on individual translation steps, the ASL structures show that i6A37 disrupts C32–A+38 base pairing and U33–A37 base–base interactions and increases dynamics in the loop (Denmon et al. 2011).
The biology of Dnmt2 and m5C38 and queueosine at N34 (Q34)
The m5C38 modification, catalyzed by Dnmt2 (Goll et al. 2006), and the Q34 modification (Fig. 6) are considered together here because of the partial overlap of their biology in eukaryotes (for review, see Ehrenhofer-Murray 2017). Dnmt2 enzymes and m5C38 are widely found in eukaryotes, but not in S. cerevisiae. Q34 is widely found in bacteria and eukaryotes (but not in S. cerevisiae), where it is found in tRNAs with GUN anticodons (tRNATyr(GUA), tRNAHis(GUG), tRNAAsn(GUU), and tRNAAsp(GUC)). Whereas bacteria form Q34 in a complicated pathway involving biosynthesis of pre-Q1, exchange of pre-Q1 for G34 by a tRNA-guanine transglycosylase (TGT), and further processing, eukaryotes form Q34 by direct transfer of free queuine found in cells (derived from bacteria) to the tRNA by a eukaryotic TGT, displacing guanosine at G34 (for review, see El Yacoubi et al. 2012).
There has been intensive study of the role of Dnmt2 in tRNA since the discovery that Dnmt2 from mouse, Arabidopsis, Drosophila, and humans was not a DNA methyltransferase, as anticipated based on phylogenetic analysis (Goll and Bestor 2005), but was instead a tRNA methyltransferase that catalyzed formation of m5C38 on tRNAAsp (Goll et al. 2006; Jurkowski et al. 2008; for review, see Jeltsch et al. 2017). Mapping of m5C sites in WT and mutant strains (Schaefer et al. 2009) showed that Drosophila and mouse Dnmt1 modifies tRNAAsp, tRNAVal(AAC), and tRNAGly(GCC) (Schaefer et al. 2010; Tuorto et al. 2012), whereas the S. pombe Dnmt2 ortholog Pmt1 primarily targets tRNAAsp, with partial modification of tRNAGlu (Becker et al. 2012). Moreover, m5C38 modification of tRNAAsp in S. pombe, Dictyostelium, and mouse is strongly dependent on prior Q34 modification in vivo, and in vitro for S. pombe Pmt1, thus linking cellular queuine and Q34 modification to m5C38 modification of tRNAAsp (Muller et al. 2015; Tuorto et al. 2018).
Although S. pombe pmt1Δ (dnmt2) mutants have no obvious growth or stress phenotype (Becker et al. 2012), several important Dnmt2 roles have emerged from study in other organisms. Thus, Drosophila Dnmt2−/− mutants are sensitive to growth at high temperature and to oxidative stress, which is correlated with stress-induced tRNA cleavage due to lack of m5C38 (Schaefer et al. 2010), which in turn leads to significant changes in dsRNAs, siRNAs and viral sensitivity, as discussed further below (Durdevic et al. 2013a). In mouse, Dnmt2−/− mutants have delayed endochondral ossification in newborns and reduced populations of hematopoietic stem cells and progenitor cells, accompanied by increased mistranslation of Asp codons by tRNAGlu and of Glu codons by tRNAAsp (Tuorto et al. 2015). In addition, the mouse Dnmt2 gene is required for epigenetic regulation of the Kit gene, responsible for altered fur color, and of Sox9, resulting in excess growth of the embryo and adult body (Kiani et al. 2013).
All of these phenotypes are highly likely to be due to tRNA m5C38 modification, as no other RNA substrates have been validated, and reported DNA substrates have not been supported by more rigorous analysis (Schaefer and Lyko 2010; Raddatz et al. 2013). However, it is possible that some of the phenotypes of mutants can be explained by other properties of the Dnmt2 protein, such as chaperone effects or binding effects (Jeltsch et al. 2017).
Intriguingly, Q34 has slightly different effects on translation in mouse and S. pombe. In mammalian cells, lack of Q34 in tRNA resulted in increased ribosome occupancy and reduced translation of codons for all four tRNAs that normally have the queuosine modification, with the U-ending codons reduced more than the C-ending codons (except for tRNAAsp(GUC), which also lack m5C38). The reduced translation through these codons was reflected in reduced amounts of proteins richer in these codons, an increase in unfolded proteins, and activation of the UPR (Tuorto et al. 2018). However, in S. pombe strains lacking queuosine, ribosome profiling showed a reduced decoding rate for C-ending codons for tRNAAsp and tRNAHis, and faster decoding of U-ending codons for tRNAAsn and tRNATyr, the effect of which was to even translation rates of the synonymous codons with Q34 (Muller et al. 2019a).
Recent results also show that Q34 levels are regulated by oxidative stress in HepG2 cells, altering translation and gene expression (Huber et al. 2022). Thus, each of three different oxidative stress treatments, including arsenite treatment, resulted in increased Q34 modification of tRNAs in HepG2 cells, resulting in codon bias-linked up-regulation of proteins involved in glycolysis, and down-regulation of oxidative phosphorylation. In contrast, queuine limitation resulted in increased arsenite sensitivity and increased levels of reactive oxygen species, linked to mitochondrial dysfunction.
m3C32 modification by Trm140 family members
The m3C modification is frequently found in tRNAs at three locations in cytoplasmic tRNAs: at C32 in the ACL of eukaryotic tRNAThr and tRNASer isoacceptors, and mammalian tRNAArg(CCU) and tRNAArg(UCU) isoacceptors; at either e1 or e4 within the variable arm of metazoan tRNASer isoacceptors; and at N20 of human tRNAeMet(CAU) (Clark et al. 2016; Boccaletto et al. 2018). In addition, m3C32 is found in Bos taurus mitochondrial tRNASer(UGA) (Boccaletto et al. 2018), and has been found in mRNA in mammalian cells (Xu et al. 2017). These m3C modifications are catalyzed by Trm140 family members. In S. cerevisiae, Trm140 protein has m3C32 methyltransferase activity and is required in vivo for m3C32 modification of all three tRNAThr isoacceptors and all three tRNASer isoacceptors with C32 (D'Silva et al. 2011; Noma et al. 2011).
Trm140 family members have very different mechanisms for substrate recognition and m3C modification in different fungi and metazoans. For example, in S. pombe, Trm140 is required for m3C32 modification of tRNAThr substrates and Trm141 for m3C32 modification of tRNASer substrates (Arimbasseri et al. 2016). Furthermore, m3C modification of S. pombe tRNASer substrates requires prior i6A37 formation by Tit1/Mod5 (Arimbasseri et al. 2016), suggesting a similar modification circuitry to that observed for other ACL modifications (Guy et al. 2012; Guy and Phizicky 2015; Muller et al. 2015; for review, see Han and Phizicky 2018). Moreover, phylogenetic analysis showed that the TRM140 (METTL2) and TRM141 (METTL6) family members are widely distributed in fission yeasts and metazoans, and extend to a third family member (METTL8) in vertebrates (Arimbasseri et al. 2016).
In contrast, S. cerevisiae Trm140 has two seemingly distinct recognition modes, enabling modification of both tRNAThr and tRNASer substrates (Han et al. 2017). For tRNAThr isoacceptors, the ACL residues G35–U36–t6A37 are both necessary and sufficient for Trm140 recognition and m3C32 modification; whereas for tRNASer, m3C modification is stimulated in vivo and in vitro by seryl-tRNA synthetase and the distinctive tRNASer variable arm that SerRS recognizes, as well as by t6A37 or i6A37. As i6A and t6A are not chemically related, it is not clear why both modifications stimulate m3C formation, although it seems plausible that they each expose C32 for modification. The presence of a single Trm140 family member is conserved through the Saccharomycotina and Pezizomycotina subdivisions of the phylum Ascomycota, and to a more limited extent, in Basidiomycota, implying that this dual tRNAThr and tRNASer recognition mechanism is retained in these organisms (Han et al. 2017).
In other eukaryotes, the theme of interacting proteins required for m3C modification recurs. Thus, METTL2 is required in humans and mouse for m3C32 modification of both tRNAThr(UGU) and tRNAArg(CCU) (Noma et al. 2011; Xu et al. 2017), and subsequent experiments show that m3C32 modification of tRNAArg substrates in humans requires interaction of METTL2 with DALRD3, based on complex formation in lysates, copurification of tRNAArg(CCU) and tRNAArg(UCU) with the complex, and the loss of the m3C modification in tRNAArg species in a human DALRD3 knockout cell line (Lentini et al. 2020). Similarly, METTL6 targets tRNASer, likely due to its interaction with seryl-tRNA synthetase (Xu et al. 2017). Furthermore, T. brucei TRM140 forms a complex with ADAT2/ADAT3, which normally deaminates A34 to I34, and all three proteins are required to catalyze m3C32 modification of tRNAThr(IGU) and for subsequent m3C32 deamination to form m3U32 (Rubio et al. 2017; McKenney et al. 2018). Intriguingly, the human METTL8 Trm140 paralog targets mitochondrial tRNAs (tRNASer(UGA) and tRNAThr(UGU) for m3C32 modification (Lentini et al. 2022), and may also target mRNA (Xu et al. 2017).
The biological role of m3C modification and Trm140 family members is not clear. There is no obvious growth defect in S. cerevisiae trm140Δ mutants and in S. pombe trm140Δ trm141Δ mutants in a variety of conditions (D'Silva et al. 2011; Arimbasseri et al. 2016), although S. cerevisiae trm140Δ trm1Δ strains, lacking both m3C32 and m2,2G26, have a mild, but distinct, growth defect in low concentrations of cycloheximide, suggesting a translation defect (D'Silva et al. 2011). In contrast, a homozygous human mutation in DALRD3 is associated with a developmental delay and infantile epilepsy in patients and lack of m3C32 in tRNAArg(CCU) and tRNAArg(UCU) (Lentini et al. 2020), and in T. brucei, METTL6 depletion results in reduced ribosome stability and a cytokinesis defect, although it is not known which RNAs are modified by T. brucei METTL6 (Fleming et al. 2016).
It is also not clear how m3C32 affects tRNA function. The pKa of 8.7 for 3-methylcytidine (Brookes and Lawley 1962; Ueda and Fox 1963) suggests a positive charge in the tRNA ACL, which likely also affects noncanonical N32–N38 interactions that are commonly found in tRNAs (Auffinger and Westhof 1999). As N32–N38 interactions are known to modulate the binding of tRNA to the A-site (Olejniczak and Uhlenbeck 2006; Olejniczak et al. 2005) and the fidelity of translation (Ledoux et al. 2009; Pernod et al. 2020), it is possible that m3C has similar or related effects.
Ψ38,39,40 and Pus3
The pseudouridine (Ψ) modification of U38 and U39 in tRNAs is found in all domains of life, including in the streamlined bacterial genome of Mycoplasma capricolum (Andachi et al. 1989), and the TruA/Pus3 family of pseudouridylases that catalyzes formation of Ψ38 Ψ39 is similarly highly conserved (Koonin 1996; Mueller and Ferre-D'Amare 2009; de Crecy-Lagard et al. 2012). Bacterial TruA from E. coli and Salmonella typhimurium catalyze formation of Ψ40 in addition to Ψ38 and Ψ39 (Singer et al. 1972; Hur and Stroud 2007), whereas the eukaryotic Pus3 (Deg1) ortholog from S. cerevisiae catalyzes only Ψ38 and Ψ39 modification (Lecointe et al. 1998).
Lack of the TruA/Pus3 pseudouridylases results in several defined phenotypes (Fig. 4). E. coli and S. typhimurium truA (hisT) mutants de-repress the histidine operon, and have a modest-to-severe reduction in growth rate that depends on growth supplements (Chang et al. 1971; Tsui et al. 1991), and S. cerevisiae pus3Δ mutants grow slowly and are temperature sensitive (Carbone et al. 1991; Lecointe et al. 2002; Han et al. 2015). In contrast, in humans PUS3 mutations have been linked to intellectual disability, associated with loss of Ψ in a representative tRNA with Ψ39 (Shaheen et al. 2016; Abdelrahman et al. 2018).
The roles of Pus3 pseudouridylation in tRNA function are not yet clear. As Pus3 targets both U38 in the ACL and U39 in the closing base pair of the anticodon stem, lack of Pus modifications could in principle have different roles in each capacity. In S. cerevisiae, loss of Ψ38 impaired function of one tRNA examined, loss of Ψ39 impaired function of one of three tRNAs examined, and the growth defect of pus3Δ trm10Δ double mutants, lacking Ψ38, Ψ39, and m1G9, was primarily due to reduced function of a single tRNA with Ψ39 and m1G9, although three other tRNAs have Ψ39 and m1G9, and four others have Ψ38 and m1G9 (Han et al. 2015). Curiously, in addition, a higher frequency of −1 frameshifting in test sequences in S. cerevisiae was correlated with tRNAs with Ψ39 and found to be reduced in pus3Δ mutants (Bekaert and Rousset 2005). As biochemical and structural analysis has shown that Ψ stabilizes both duplex and single-stranded RNA due to coordination of a water molecule and to enhanced stacking of its favored 3′ endo conformation (Arnez and Steitz 1994; Davis 1995; Durant and Davis 1999; Charette and Gray 2000), it seems plausible that the selective Ψ39 effects will be explained by similar physical or structural effects. The effects of Ψ38 may be due to related stabilization effects or may also be due in part to alteration of the noncanonical 32–38 interactions found in many tRNAs (Auffinger and Westhof 1999).
m5C34,40,48,49,50 and Trm4/NSUN2
Understanding the biology of m5C and the corresponding Trm4/NSUN2 methyltransferase family is complicated by its presence at different locations in tRNA, as well as in other RNAs. In S. cerevisiae and in mouse, Trm4/NSUN2 catalyzes m5C formation in tRNA substrates at C34 in the ACL, at C40 in the anticodon stem, at C48 in the variable loop, and at C49 and C50 in the T-stem (Motorin and Grosjean 1999; Brzezicha et al. 2006; Tuorto et al. 2012). In contrast, S. pombe has two Trm4 paralogs, with Trm4a methylating C34 and C48 and Trm4b methylating C49 and C50 modification (Muller et al. 2019b). Furthermore, multiple m5C-modified sites have been found within mRNAs and other RNAs in human cells by bisulfite sequencing methods (Squires et al. 2012; Amort et al. 2017; David et al. 2017). As m5C modifications can contribute differently to tRNA function in the tRNA ACL, the tRNA body, and in other RNA substrates, it has been difficult to sort these out to ascribe known biological phenotypes to specific m5C modification sites.
Lack of m5C modification in fungi results in relatively mild phenotypes. S cerevisiae trm4Δ mutants have little growth defect in a variety of normal media conditions, but have increased sensitivity to paromomycin (Wu et al. 1998) and are sensitive to oxidative stress (Chan et al. 2010), which has been linked in WT cells to a 70% increase in m5C34 in tRNALeu(CAA), improved tRNALeu(CAA) decoding, and increased translation of proteins rich in UUG codons, including a specific ribosome subunit that is important for survival of peroxide stress (Chan et al. 2012). Curiously also in S. cerevisiae, m5C levels in tRNAHis (but not in two other tRNAs), are increased after amino acid starvation, rapamycin treatment, growth to stationary phase, and growth of temperature sensitive strains at nonpermissive temperature, although it is not clear why this occurs (Preston et al. 2013). In S. pombe, lack of either or both of Trm4a and Trm4b results in little or no growth defect under a variety of conditions, including oxidative stress conditions and paromomycin sensitivity, although trm4aΔ mutants are mildly resistant to CaCl2, suggesting some type of mitochondrial function (Muller et al. 2019b).
Lack of NSUN2 and m5C modifications has been linked to numerous phenotypes in mammals. In humans, NSUN2 mutations are linked to autosomal recessive intellectual disability (Fig. 4; Abbasi-Moheb et al. 2012; Khan et al. 2012; Martinez et al. 2012), and mouse Nsun2−/− mutants have aberrant stem cell differentiation in hair follicle stem cells (Blanco et al. 2011), blocked meiotic progression into pachytene in testis germ cells (Hussain et al. 2013), reduced survival of neurons, and reduced spatial working memory (Blanco et al. 2014). Furthermore, lack of NSUN2 in mouse and human skin cells results in increased sensitivity to UVB radiation and oxidative stress, resulting in accumulation of tRNA-derived fragments and downstream consequences, as discussed further below (Blanco et al. 2014; Gkatza et al. 2019). However, mRNA levels could also contribute to phenotypes of NSUN2 mutants, as m5C-modified mRNAs are recognized by ALYREF/THOC4 in mammals to promote mRNA export (Yang et al. 2017), and in zebrafish m5C-modified maternal mRNAs have higher stability during the maternal-to-zygotic transition, mediated by interaction with the m5C-RNA binding protein Ybx1 (Yang et al. 2019).
There is also a report that lack of NSUN2/TRM4 might result in reduced tRNA stability. A. thaliana trm4b mutants have shorter primary roots and are sensitive to different oxidative stresses (David et al. 2017), and have reduced levels of tRNAAsp(GTC), a representative tRNA substrate with m5C at C48, C49, and C50 (David et al. 2017). Although mouse Nsun2−/− mutants are not known to result in reduced levels of tRNAs in mouse (Blanco et al. 2014), it is known that Nsun2−/− Dnmt2−/− mouse mutants, lacking both sources of m5C in tRNA, are mostly inviable, and survivors have reduced levels of tRNAs with both modifications (Tuorto et al. 2012).
MODIFICATIONS IN THE tRNA BODY
m1A9 and m1G9 and the catalytically versatile Trm10 family
The m1A9 and m1G9 (collectively m1R9) modifications are frequently found in tRNAs from eukaryotes and archaea. Seminal prior work provided compelling evidence that the m1A9 modification prevents misfolding of mitochondrial tRNALys(UUU), as the unmodified tRNA could adopt an alternative structure with an elongated acceptor stem, which was prevented by m1A9 (Helm et al. 1998, 1999; Helm and Attardi 2004). Subsequently, it was shown that S. cerevisiae Trm10 is responsible for m1G9 modification of all nine known substrate tRNAs in vivo, and that the Trm10 family is widespread in eukaryotes and archaea, with two or three paralogs in different metazoans (Jackman et al. 2003). Although S. cerevisiae trm10Δ mutants do not have any obvious growth phenotype in rich or minimal media over a range of temperatures (Jackman et al. 2003), the mutants are unusually sensitive to the anticancer drug 5-fluorouracil (Fig. 4; Gustavsson and Ronne 2008).
Further investigation has revealed several surprises in the biology of the Trm10 family and m1R9 modifications. Because guanosine and adenosine are chemically very different, it was a distinct surprise to discover that the Crenarchaean Trm10 ortholog from Sulfolobus acidocaldarius catalyzes formation of m1A9, rather than m1G9, on a tRNAiMet transcript, and that Trm10 from the Euryarchaean Thermococcus kodakaraensis catalyzes both m1A9 and m1G9 formation on different tRNAs (Kempenaers et al. 2010). Remarkably also, the human TRMT10C ortholog and the short chain dehydrogenase/reductase SDR5C1 comprise two of the three different subunits of the human protein-only RNase P trimer, and moonlight as an m1R9 methyltransferase subcomplex that catalyzes formation of both m1A9 and m1G9 on mitochondrial substrate tRNAs (Vilardo et al. 2012), all of which bear m1R9 in bovine mitochondrial tRNAs with a purine at this residue and canonical cloverleaf structure (Suzuki and Suzuki 2014). Moreover, it is now known that whereas human TRMT10A catalyzes m1G9 modification on all tested cytoplasmic tRNAs known to have the modification, the human TRMT10B ortholog is specific for m1A9 modification of tRNAAsp(GUC) (Howell et al. 2019), the sole human cytoplasmic tRNA with m1A9 (Clark et al. 2016), and knockout cell lines prove the mutually exclusive specificity of TRMT10A for m1G9 and TRMT10B for m1A9 in vivo (Vilardo et al. 2020).
Structural and biochemical analyses have clarified aspects of the mechanism of the Trm10 protein family (for review, see Krishnamohan and Jackman 2019). The structure of Trm10 from S. cerevisiae and S. pombe shows the typical fold of the SPOUT family of methyltransferases, but acting as a monomer (Shao et al. 2014), and biochemical analysis has revealed a noncanonical methyltransferase mechanism. Thus, the mechanism of S. cerevisiae Trm10 involves a collaborative catalytic role for two highly conserved aspartate residues near the active site of S. cerevisiae Trm10, with added contributions from a nearby glutamate residue (Krishnamohan and Jackman 2017). Consistent with these findings, catalysis by the dual specificity T. kodakareinsis Trm10 is also synergistically inhibited in the corresponding double carboxylate variants (Singh et al. 2018).
Nonetheless, it remains to be determined exactly how specificity for m1A9 and/or m1G9 modification are established within Trm10 family members. Whatever the mechanism, the catalytic difference between TRMT10A acting on m1G9 and TRMT10B on m1A9 is not reflected in binding affinity, as each protein bound substrate and nonsubstrate tRNAs with comparable affinity (Howell et al. 2019). It also remains to be determined why S. cerevisiae Trm10 modifies certain tRNAs with equal efficiency in vitro but is more selective in vivo (Swinehart et al. 2013).
Although human cell lines lacking TRMT10A, TRMT10B, or both have no obvious growth defect accompanying the lack of m1G9 and/or m1A9 in their cytoplasmic tRNAs (Vilardo et al. 2020), mutations in TRMT10A result in human disorders (Fig. 4). Thus, early onset diabetes and microcephaly was linked to a TRMT10A-R127* nonsense mutation (Igoillo-Esteve et al. 2013), and a homozygous TRMT10A–G206R mutation was associated with defective glucose metabolism, microcephaly, and intellectual disability (Gillis et al. 2014), and in each case the corresponding TRMT10A variant was catalytically inactive (Gillis et al. 2014) or the corresponding patient cell lines had no detectable m1G9 in tRNAs examined (Cosentino et al. 2018). Initial study showed that TRMT10A knockdown is associated with apoptosis in rat and human β-cells, as well as in a rat pancreatic β-cell line, in this case after treatment with fatty acids, high concentration of glucose, or any of several ER-stressors (Igoillo-Esteve et al. 2013) and, as described further below, these have been linked to tRNA-derived fragments (Cosentino et al. 2018).
Three other phenomena related to TRM10 biology have appeared, each of which adds new dimensions to its roles. First, TRMT10A appears to functionally interact with the mRNA m6A demethylase FTO to regulate m6A levels in mRNA, as endogenous TRMT10A and FTO reciprocally coimmunopreciptate, TRMT10A knockdown or knockout results in increased m6A levels in poly(A) mRNAs, TRMT10A stimulates FTO m6A-demethylation activity in vitro, and there is significant overlap among mRNAs with increased m6A-modification in TRMT10A knockdowns and mRNAs identified by CLIP-seq of TRMT10A and FTO (Ontiveros et al. 2020). The consequences of this potential coregulation of tRNA m1G9 modification and mRNA m6A modification are not yet known. Second, natural S. cerevisiae TRM10 variants have been reported to affect UGA stop codon readthrough efficiency, and SUP45 (encoding the polypeptide release factor eRF1) and TRM10 variants are found in distinct linkage disequilibrium, suggesting evolutionary pressure to moderate termination readthrough efficiency (Torabi and Kruglyak 2011). Third, the biology of S. cerevisiae TRM10 is even more intricate than expected due to the 18-mer ncRNA derived from TRM10 that down-regulates translation (Pircher et al. 2014).
m2,2G26 and Trm1/TRMT1
The m2,2G26 modification occurs frequently in tRNAs from eukaryotes and archaea. Among a set of characterized tRNAs with an encoded G26, the m2,2G26 modification is found in 127 of 160 eukaryotic cytosolic tRNAs (including 21 of 22 in S. cerevisiae and 8 of 10 in humans) and 14 of 32 mitochondrial tRNAs (three of nine in S. cerevisiae and one in humans) (Juhling et al. 2009). In S. cerevisiae and humans, the methyltransferase Trm1/TRMT1 modifies both cytoplasmic and mitochondrial tRNAs to m2,2G26 (Phillips and Kjellin-Straby 1967; Hopper et al. 1982; Ellis et al. 1986; Liu and Straby 2000; Dewe et al. 2017), but it should be noted that the m2G26 modification often found at G26 is also likely catalyzed by Trm1 family members (Edqvist et al. 1994; Urbonavicius et al. 2006). S. cerevisiae Trm1 is localized to the inner nuclear rim (Li et al. 1989) and trm1Δ mutants are temperature sensitive due (Fig. 4) to decay of substrate tRNAs by the RTD pathway (Dewe et al. 2012; discussed further below). In humans, TRMT1 mutations have been linked to intellectual disability (Fig. 4; Najmabadi et al. 2011; Davarniya et al. 2015; Blaesius et al. 2018; Zhang et al. 2020), associated with reduced m2,2G26 in patient-derived lymphoblastoid cell lines (Zhang et al. 2020) and with reduced m2,2G26, reduced proliferation, and sensitivity to oxidative stress in HEK 293T TRMT1 knockout cells (Dewe et al. 2017).
4-acetylcytidine at C12, ac4C12, and the Tan1/THUMPD1:Kre33/NAT10 complex
The ac4C modification is typically found at C12 in eukaryotic tRNAs with a long variable arm, including all but one of the 26 characterized cytoplasmic tRNALeu species, 15 of 19 cytoplasmic tRNASer species, and five mitochondrial tRNALeu species with these properties (Juhling et al. 2009). The ac4C modification is typically found at the middle cytidine of a CCG motif, and remarkably, in archaea, ac4C is found in numerous tRNAs at numerous positions, as well as in other RNAs (except for mRNA), and its levels are dramatically increased at high temperature (Sas-Chen et al. 2020).
Biochemical and genetic analysis has shown that the ac4C12 modification of tRNA requires a complex of Tan1/THUMPD1 and Kre33/NAT10. A prior genetic screen revealed that S. cerevisiae Tan1 was required for ac4C12 formation (Johansson and Bystrom 2004). Subsequent work showed that ac4C12 formation is catalyzed in yeast and humans by Kre33/NAT10 in complex with Tan1/THUMPD1, and surprisingly, that Kre33/NAT10 also acts independently of Tan1 to catalyze acetylation of two cytidine residues in 18S rRNAs in S. cerevisiae and humans (Sharma et al. 2015). This requirement for Tan1 to direct Kre33/NAT10 to tRNA for acetylation is consistent with the tRNA binding activity of Tan1 (Johansson and Bystrom 2004). Kre33/THUMPD1 has a helicase domain that is important for function (Sharma et al. 2015), like that of the related E. coli TmcA protein that acetylates C34 of elongator tRNAMet (Ikeuchi et al. 2008; Chimnaronk et al. 2009), but its role is not yet known.
Tan1 is biologically important in both S. cerevisiae and humans (Fig. 4). In S. cerevisiae, tan1Δ mutants are temperature sensitive due to degradation of substrate tRNAs by the RTD pathway (Chernyakov et al. 2008; Kotelawala et al. 2008; Dewe et al. 2012, discussed further below). In humans, mutations in THUMPD1 are associated with developmental delay, intellectual disability, and behavioral abnormalities, associated with loss of ac4C12 modification of tRNA (Broly et al. 2022).
m7G46 and the Trm8/METTL1:Trm82/WDR4 complex
The m7G46 modification is widely found in tRNAs from prokaryotes and eukaryotes, as well as in some mitochondrial and plastid tRNAs, when G46 is the middle residue of a tRNA with a 5 nt variable loop (Okamoto et al. 2004; Matsumoto et al. 2007; Juhling et al. 2009), comprising the RAGGU motif in yeast and humans (Lin et al. 2018). The corresponding tRNA m7G46 methyltransferase is a two subunit complex in S. cerevisiae and humans, encoded by TRM8/METTL1 and TRM82/WDR4, both components of which are highly conserved in eukaryotes (Alexandrov et al. 2002), although they can be functionally replaced in S. cerevisiae by single subunit bacterial Trm8 homologs from E. coli or Thermotoga maritima (Alexandrov et al. 2005; for review see Tomikawa 2018). Structural analysis has revealed a substantial interaction surface between yeast Trm8 and Trm82 including two salt bridges whose residues are highly conserved (Leulliot et al. 2008). A slightly different interaction surface is found in the human METTL1:WDR4 complex (Li et al. 2023; Ruiz-Arroyo et al. 2023), stabilized by two different salt bridges as well as three hydrogen bonding interactions, and biochemical experiments show that each of three mutations that disrupt these intersubunit interactions eliminates activity (Li et al. 2023). Intriguingly, comparison of crystal and cryo-EM structures with and without tRNA and cofactor (S-adenosylmethionine or S-adenosylhomocysteine) show that tRNA and cofactor binding is accompanied by substantial changes in METTL1 and bending of the tRNA, resulting in local unwinding of the variable loop and base flipping of G46 into the active site, with a prominent role for the previously disordered METTL1 amino terminus (Li et al. 2023; Ruiz-Arroyo et al. 2023).
Trm8 and Trm82 are biologically important in eukaryotes (Fig. 4). Lack of TRM8 and/or TRM82 is associated with temperature sensitivity in S. cerevisiae and S. pombe, linked to decay of substrate tRNAs by the RTD pathway (Alexandrov et al. 2005; Chernyakov et al. 2008; Dewe et al. 2012; De Zoysa and Phizicky 2020, discussed further below). In humans, mutations in WDR4/TRM82 are linked to microcephaly and primordial dwarfism (Fig. 4), and in each of two unrelated families, the same conserved R170 residue is mutated to leucine or glutamine (Shaheen et al. 2015; Trimouille et al. 2018). Consistent with this clinical manifestation, the WDR4-R170L mutation results in reduced tRNA m7G modification activity in vitro and reduced or eliminated activity in vivo (Shaheen et al. 2015; Li et al. 2023), and the WDR4-R170Q mutation results in the elimination of activity in vitro and in vivo (Li et al. 2023). In addition, human METTL1 is subject to phosphorylation at S27 by Akt (protein kinase B) and RSK in vitro and in vivo, which resulted in loss of m7G modification activity in vitro (Cartlidge et al. 2005), consistent with the lack of function of the corresponding yeast and human phosphomimetic mutants (Cartlidge et al. 2005; Li et al. 2023; Ruiz-Arroyo et al. 2023). The consequences of this regulation are not yet known.
Recent results in mammals have revealed profound biological effects associated with lack of m7G46, linked to reduced levels of tRNAs. Mouse embryonic stem cells (mESCs) lacking METTL1 have substantially reduced levels of six of the 22 tRNA species normally bearing m7G46, associated with increased ribosome pausing at the corresponding codons, reduced translation efficiency of mRNAs rich in these codons, and defective self-renewal and neural differentiation (Lin et al. 2018).
Remarkably, recent experiments show that m7G modification of one specific tRNA by METTL1/WDR4 drives oncogenic transformation. In support of the causal link between METTL1/WDR4 expression and oncogenic transformation, METTL1 and WDR4 expression and m7G46 modification are up-regulated in certain cancers, associated with poor prognosis (Dai et al. 2021; Orellana et al. 2021); knockout of METTL1 or WDR4 results in a reduction in cell proliferation, oncogenicity, and tumor growth, which is associated with reduced m7G46 modification and reduced levels of several m7G46-modified tRNAs; and overexpression of METTL1/WDR4 (but not catalytic dead variants) results in increased oncogenicity and increased translation of a subset of genes associated with cell cycle regulation (Dai et al. 2021; Orellana et al. 2021). Furthermore, tRNAArg(TCT) accounts for most of this biology, as translation is most affected for genes particularly enriched in AGA codons, which is decoded by tRNAArg(TCT); tRNAArg(TCT) expression correlates with METL1/WDR4 expression in tumors and is associated with poor survival; and overexpression of tRNAArg(TCT) is oncogenic and phenocopies many of the properties of METTL1/WDR4 overexpression (Orellana et al. 2021).
m1A58, Trm6:Trm61, and tRNAiMet
The m1A58 modification is ubiquitous in tRNA from eukaryotes, occurring in 33 of the 55 tRNA isodecoders of cytosolic tRNA from S. cerevisiae and in the majority of those from human cytosolic tRNAs (Saikia et al. 2010; Cozen et al. 2015; Boccaletto et al. 2022), as well as in a limited number of tRNAs in prokaryotes, archaea, and mitochondria of animals and plants (Juhling et al. 2009). Formation of m1A58 is catalyzed by the essential Trm6:Trm61 (Gcd10:Gcd14) complex in S. cerevisiae (Anderson et al. 1998, 2000), which is widely conserved in eukaryotes (Bujnicki 2001; Ozanick et al. 2005). This complex is comprised of a dimer of Trm6:Trm61 heterodimers in which the noncatalytic Trm6 subunit positions the tRNA for Trm61 to modify at A58, which is exposed by separation of the T-loop and D-loop (Finer-Moore et al. 2015).
The m1A58 modification is crucial for structure and function of initiator tRNA, tRNAiMet(CAU) (for review, see Kolitz and Lorsch 2010). Thus, although m1A58 is found on numerous S. cerevisiae tRNAs, the lethality of a trm6Δ mutation is suppressed by overexpression of tRNAiMet(CAU), suggesting that this is the only biologically important substrate (Anderson et al. 1998). This finding is consistent with the unique structure of initiator tRNAiMet, which features a tRNA substructure involving hydrogen bonding interactions between A58 and residues A54 and A60 in the T-loop, and between A20 of the D-loop and G57, A59, and A60 of the T-loop (Basavappa and Sigler 1991). It seems highly likely that this substructure is uniquely common to all eukaryotic initiator tRNA species, as the residues A20, A54, and A60 (and the lack of N17) are normally found in initiator tRNA, but are only rarely found among elongator tRNAs (particularly not in combination), and m1A58 is found in all but one characterized eukaryotic tRNAiMet (Marck and Grosjean 2002; Kolitz and Lorsch 2010; Boccaletto et al. 2022).
The lack of m1A58 in S. cerevisiae leads to reduced levels of tRNAiMet(CAU), due to decay (Anderson et al. 1998), by both the nuclear surveillance pathway (Kadaba et al. 2004) and the RTD pathway (Tasak and Phizicky 2022), as discussed further below. The m1A58 modification is also likely important for cell health and tRNAiMet stability in other eukaryotes. In mammalian cells, knockdown of either TRM6 or TRM61 in a rat glioma cell line was reported to result in a slow growth phenotype and reduced levels of tRNAiMet, which could be partially rescued by overexpression of tRNAiMet, and overexpression of TRM6–TRM61 resulted in increased levels of tRNAiMet, as well as of tRNAeMet (Macari et al. 2016). In Arabidopsis, lack of either the TRM6 or the TRM61 ortholog leads to embryo arrest and seed abortion and reduced TRM61 expression is associated with reduced levels of tRNAiMet (Tang et al. 2020). It remains to be determined how the levels of tRNAiMet(CAU) are reduced in these and other multicellular organisms.
Lack of m1A58 can also affect the function of at least one other tRNA. Thus, cells from patients with MERFF (myoclonus epilepsy, ragged-red fibers) due to an A54G mutation in mitochondrial tRNALys lack m1A58 as well as the taurine modification normally associated with this disease, and the lack of m1A58 was directly linked to reduced translation by mitochondrial tRNALys (Richter et al. 2018).
It is now known that m1A58 levels in tRNAs are subject to regulation by members of the AlkB family of dioxygenases with tRNA m1A demethylase activity. AlkBH1 was documented to have tRNA m1A demethylase activity based on CLIP-Seq experiments showing binding to mature tRNAs, in vitro assays that documented tRNA m1A demethylation activity, accompanied by ALKBH1 knockdown experiments that resulted in increased m1A levels in specific tRNAs, and ALKBH1 overexpression experiments that resulted in reduced levels of m1A in these tRNAs (Liu et al. 2016). Strikingly, it was also shown that transient ALKBH1 knockdown led to a threefold increase in levels of tRNAiMet, associated with increased cellular proliferation, and increased translation, and that glucose starvation led to increased ALKBH1 expression, reduced m1A levels in tRNA targets, and reduced tRNAiMet levels and reduced translation (Liu et al. 2016). In addition, m1A levels in tRNAs may also be regulated by two other members of the AlkB protein family. Thus, ALKBH3 has tRNA m1A demethylation activity in vitro and is highly expressed in tumor cells, and its knockdown in tumor cells reduces proliferation (Ueda et al. 2017), and FTO catalyzes m1A demethylase activity in vitro in addition to its known m6Am and m6A demethylation activity, and FTO knockdown in cell lines and in Fto−/− MEFs resulted in increased m1A levels in specific tRNAs (Wei et al. 2018).
In addition, as dicussed further below, it is now known that increased TRMT6:TRMT61-dependent m1A levels in some tRNA-derived fragments leads to their reduced gene silencing activity (Su et al. 2022).
tRNA TURNOVER PATHWAYS
The half-life of typical tRNAs is extraordinarily long, estimated to be 44 and 50 h in Euglena gracilis and chicken muscle, respectively, similar to that of rRNAs (Nwagwu and Nana 1980; Karnahl and Wasternack 1992) and ∼9 h in S. cerevisiae (Gudipati et al. 2012). However, although tRNAs are stable, lack of any of several tRNA body modifications in S. cerevisiae and S. pombe leads to decay of a specific subset of the hypomodified tRNA species by either of two decay pathways. The nuclear surveillance pathway targets pre-tRNAs for 3′–5′ exonucleolytic decay shortly after transcription, and the RTD pathway targets mature tRNAs for 5′–3′ exonucleolytic decay after maturation (Figs. 1, 7; Supplemental Table S1). These pathways are described in more detail below.
Two different tRNA decay pathways in S. cerevisiae. (Left) A pre-tRNAiMet molecule is depicted in the typical secondary structure shortly after transcription, with uncolored circles representing tRNA residues, pale red circles representing the 5′ leader nucleotides, pale blue circles representing the 3′ trailer nucleotides, and a bright red circle indicating the site for m1A58 modification. A pre-tRNAiMet lacking m1A58 is targeted for decay by the nucelar surveillance pathway in S. cerevisiae, involving oligoadenylation of the pre-tRNA by Trf4 of the TRAMP complex, and then 3′–5′ exonucleolytic degradation of the pre-tRNA by Rrp6 and Rrp44 of the nuclear exosome. Spliced leader-containing pre-tRNAs are also targeted for decay by the nuclear surveillance pathway (Kramer and Hopper 2013; Chatterjee et al. 2022). (Right) A mature tRNA with a CCA end is depicted in its typical secondary structure, with residues that are normally modified to form ac4C12 in yellow, m2,2G26 in blue, m7G46 in green, and m1A58 in red. Specific mature tRNAs lacking one of these modifications are targeted for decay by the rapid tRNA decay pathway in S. cerevisiae, involving 5′–3′ exonucleolytic decay of the tRNA by Rat1 and Xrn1 in the nucleus and cytoplasm, respectively, both of which are inhibited by pAp, which accumulates in met22Δ mutants.
The nuclear surveillance pathway
Earlier groundbreaking work in S. cerevisiae defined the nuclear surveillance pathway by identification and characterization of spontaneous suppressors of the temperature sensitivity of trm6-504 mutants (Kadaba et al. 2004), which was known to be due to reduced levels of tRNAiMet (Anderson et al. 1998). Thus, the isolation of suppressing mutants in TRF4 and in RRP44, encoding, respectively, a protein with poly(A) polymerase activity and a 3′–5′ exonuclease in the exosome, led to the definition of a pathway (Fig. 7) in which pre-tRNAiMet lacking m1A58 was targeted for 3′–5′ exonucleolytic decay by Rrp6 and the nuclear exosome after polyadenylation by Trf4 (Kadaba et al. 2004). Biochemical analysis showed that Trf4 was part of the TRAMP complex, along with Air1 or Air2 and the RNA helicase Mtr4 (LaCava et al. 2005; Vanacova et al. 2005), and that the TRAMP complex and nuclear exosome could degrade a mature tRNAiMet transcript but not native tRNAiMet, and a tRNAAla(GGC) transcript with a destabilizing D-stem mutation, but not the corresponding WT tRNAAla(GGC) transcript (Vanacova et al. 2005). Further, in vitro analysis showed that the TRAMP complex and recombinant Rrp44, the sole nuclease of the core exosome (Dziembowski et al. 2007), specifically acted on mature tRNAiMet lacking m1A58, but not on any of several other tRNAs examined, in a preparation of bulk RNA (Schneider et al. 2007). This in vitro specificity for tRNAiMet lacking m1A58 recapitulated the specificity observed in vivo and was consistent with the known unique involvement of residue A58 of initiator tRNAiMet in tertiary interactions with A54 and with A60 as part of the unique substructure of tRNAiMet (Basavappa and Sigler 1991). Although there is a similar TRAMP5 complex containing the highly related Trf4 homolog Trf5 (Houseley and Tollervey 2006), its role in tRNA decay is less clear.
Further in vivo analysis in S. cerevisiae provided evidence that the nuclear surveillance pathway also targets a large portion of newly transcribed pre-tRNAs in WT cells (Gudipati et al. 2012). Thus, tiling arrays revealed a global increase in steady state tRNA levels in mutants conditionally lacking Dis3/Rrp44 or lacking Rrp6, and a synergistic increase in tRNA levels in rrp6Δ dis3− double mutants. This data, combined with pulse chase experiments, showed that more than 50% of the global population of transcribed tRNAs is degraded by the nuclear surveillance pathway as pre-tRNAs, while also revealing that the half-life of mature tRNAs in S. cerevisiae is ∼9 h, and is independent of Dis3. The cause of this strikingly high level of pre-tRNA decay is unknown, but was speculated to be due to some combination of pre-tRNA misfolding after transcription, pre-tRNA instability, stochastic mutations arising during transcription, and competition between the maturation machinery and the nuclear surveillance pathway for normally folded pre-tRNAs (Gudipati et al. 2012).
The nuclear surveillance pathway in S. cerevisiae is known to compete with early steps of tRNA processing and 3′ end formation. Failure of proper 3′ trailer removal by Trz1, Rex1, and Rrp6 can lead to polyadenylation and pre-tRNA decay by the nuclear surveillance pathway, as documented for two pre-tRNAs with longer structured 3′ trailers that are targeted by Trz1 (Skowronek et al. 2014), and for three pre-tRNAs with longer 3′ ends, including two pre-tRNAiMet species lacking m1A58 that are normally processed by Rex1 (Ozanick et al. 2009). Consistent with direct competition between the nuclear surveillance pathway and the tRNA 3′ end formation machinery, overexpression of the La protein (Lhp1) prevents decay of pre-tRNAiMet lacking m1A58 by the nuclear surveillance pathway (Anderson et al. 1998).
It also appears that the nuclear surveillance pathway can target pre-tRNAs at different points in the biogenesis pathway in S. cerevisiae. An early analysis showed that the species of pre-tRNAiMet lacking m1A58 that was targeted by the nuclear surveillance pathway had complete 5′ leaders and a portion of their 3′ trailers, implying targeting shortly after initial transcription (Kadaba et al. 2004, 2006; Ozanick et al. 2009). However, the nuclear surveillance pathway also appears to target end-matured unspliced pre-tRNAs that are 3′ trimmed by Rex1, and competition also occurs at this stage, as overexpression of La prevents access of Rex1 to the 3′ ends and the ensuing decay (Copela et al. 2008). Given the very different stages in biogenesis of these pre-tRNA targets, it seems likely that the nuclear surveillance pathway targets pre-tRNAs at all nuclear steps of biogenesis, in competition with components of the maturation machinery.
It seems likely that the nuclear surveillance pathway will target pre-tRNAs for decay widely throughout eukaryotes. The nuclear exosome is widely conserved across eukaryotes (Houseley and Tollervey 2009; Januszyk and Lima 2014), as are the components of the TRAMP complex (Win et al. 2006; Schmidt and Butler 2013), and it is known in S. pombe that pre-tRNAs lacking La protein are targeted by Rrp6 of the nuclear exosome (Huang et al. 2006). Although there is both a nucleolar and a nuclear TRAMP complex in S. cerevisiae (Wolin et al. 2012), and an analogous nucleolar TRAMP complex in S. pombe to recruit RNAs (Win et al. 2006), in humans there is both a nucleolar TRAMP complex and a nuclear exosome targeting (NEXT) complex that recruits RNA substrates (Lubas et al. 2011, 2015; Schmidt and Butler 2013). Although tRNA transcription and early tRNA processing events are nucleolar in yeast (Thompson et al. 2003), and other tRNA processing steps take place in the nucleoplasm or at the inner nuclear membrane (Rose et al. 1995; Murthi and Hopper 2005), it is not known where in the nucleus tRNA biogenesis takes place in other organisms. Thus, although the nuclear exosome has a wide swath of other RNA substrates (Houseley et al. 2006; Wolin et al. 2012), it is plausible that in other organisms the TRAMP and/or the NEXT complexes have roles in targeting pre-tRNAs for decay by the nuclear exosome. Global analysis of the effects of the nuclear exosome on tRNA levels or stability has not been examined in organisms other than S. cerevisiae.
The rapid tRNA decay pathway
In S. cerevisiae, lack of m7G46, ac4C12, or m2,2G26, alone or in combination with other modifications, is associated with temperature sensitivity due to 5′–3′ exonucleolytic decay of a subset of the mature hypomodified tRNAs by Xrn1 and/or Rat1 of the RTD pathway (Fig. 7). Thus, the temperature sensitivity of trm8Δ trm4Δ mutants, lacking m7G46 and m5C49, is due to decay of tRNAVal(AAC), as levels of tRNAVal(AAC) but not other hypomodified tRNAs were reduced at high temperatures, the temperature sensitivity was suppressed by overexpression of tRNAVal(AAC), and both decay and temperature sensitivity were suppressed by mutation of RAT1, XRN1, or MET22 (Alexandrov et al. 2006; Chernyakov et al. 2008). Mutation of MET22 suppresses decay by the RTD pathway (Fig. 7) because it leads to increased levels of the metabolite pAp, an inhibitor of Rat1 and Xrn1 (Murguia et al. 1996; Dichtl et al. 1997; Yun et al. 2018). A similar set of experiments showed that the temperature sensitivity of tan1Δ trm44Δ mutants (lacking ac4C12 and Um44) and of trm1Δ trm4Δ mutants (lacking m2,2G26 and m5C) was due to RTD of tRNASer(CGA) and tRNASer(UGA), and not other tRNAs with the corresponding modifications (Chernyakov et al. 2008; Whipple et al. 2011; Dewe et al. 2012), and showed that this decay occurred at the level of mature tRNAs, rather than pre-tRNAs (Alexandrov et al. 2006; Chernyakov et al. 2008). Additional experiments expanded the scope of the RTD pathway to single mutants lacking m7G46, m2,2G26, or ac4C12, as the temperature sensitivity of each single mutant was suppressed by a met22Δ mutation and associated with tRNA decay at the restrictive temperature (Dewe et al. 2012).
Mechanistic studies suggest that the RTD pathway targets tRNAs that expose their 5′ end due to reduced stability, which can arise from lack of stabilizing body modifications or from destabilizing mutations in the tRNA body, and which is amplified by growth at higher temperatures. Temperature is a prominent feature of the RTD pathway as each of the hypomodified strains implicated in RTD is temperature sensitive due to the RTD pathway, although decay is still evident at lower temperatures (Chernyakov et al. 2008; Whipple et al. 2011; Dewe et al. 2012). To analyze the importance of destabilizing mutations and lack of body modifications in triggering RTD, the growth properties of strains expressing variants of the essential tRNASer(CGA) gene SUP61 were compared in WT and tan1Δ trm44Δ backgrounds (with and without a met22Δ mutation to inhibit RTD), and then compared to the predicted stabilities of the variants in the combined acceptor and T-stem, which are normally stacked in mature tRNA (Whipple et al. 2011). This analysis revealed that lack of ac4C12 and Um44 in tan1Δ trm44Δ strains acts as if it destabilizes the tRNA by 1.0–1.5 kcal/mol, and that met22Δ strains (in which RTD is inhibited) could tolerate ∼1.5–2 kcal/mol more destabilization than WT strains in the acceptor and T-stems. In addition, biochemical analysis of 5′ end accessibility of purified tRNAs showed that the 5′ end was more sensitive to purified Xrn1 or calf intestinal phosphatase in tRNAs lacking modifications that triggered RTD than in WT tRNAs, and in variants with destabilizing mutations in the acceptor and T-stem than in WT tRNAs (Whipple et al. 2011).
As ac4C12 and Um44 are both located in residues known to participate in tertiary interactions (Fig. 2; Kim et al. 1974a,b; Giege et al. 2012), this data supports a model in which lack of these modifications destabilize the tertiary structure of the tRNA, which is known to be the initial step in the overall melting of tRNAs (Shelton et al. 2001; Wilkinson et al. 2005), making it more likely for the subsequent helix unwinding to expose the 5′ end to exonucleases. In support of this argument, the other modification mutants implicated in the RTD pathway lack m7G46 or m2,2G26, and now also m1A58 (see below), and all of these residues are known to participate in tertiary interactions in some tRNAs (Kim et al. 1974a,b; Basavappa and Sigler 1991; Giege et al. 2012). This tertiary structure destabilization model also explains why a number of variants of fully modified SUP4oc (tRNATyr(GUA) with a UUA anticodon) that trigger RTD at low temperature (28°C) have mutations in the D-stem–loop, the V-loop or the T-loop, as well as in the acceptor and T-stems (Guy et al. 2014).
Additional studies emphasize the importance of both reduced overall tRNA stability and of higher temperature in increased susceptibility to RTD. Thus, high-throughput analysis of S. cerevisiae SUP4oc variants reveals a correlation between RTD at 28°C and a reduction in the predicted ΔΔG° of variants (Guy et al. 2014), and shows that the pervasive temperature sensitivity of SUP4oc variants observed between growth at 28°C and 37°C is highly correlated with susceptibility to RTD, suggesting that temperature sensitivity is frequently due to RTD (Payea et al. 2018).
Recent results show that the RTD pathway is conserved in the phylogenetically distant fission yeast S. pombe, and extend the use of the RTD pathway to another body modification mutant in both S. pombe and S. cerevisiae. Thus, S. pombe trm8Δ mutants are now known to be temperature sensitive due to decay of tRNATyr and to some extent tRNAPro(AGG) by the RTD pathway, as levels of these tRNAs were reduced at high temperature, overexpression of these tRNAs suppressed the temperature sensitivity, and each of four spontaneous suppressors of the temperature sensitivity had mutations in the Rat1 ortholog DHP1 and prevented decay of these tRNAs (De Zoysa and Phizicky 2020).
Similarly, S. pombe trm6Δ mutants are now known to be temperature sensitive due to decay of tRNAiMet by the RTD pathway, as tRNAiMet levels were reduced at high temperature, overexpression of tRNAiMet suppressed the temperature sensitivity, each of three spontaneous suppressors of the temperature sensitivity and tRNA decay had mutations in DHP1 or the MET22 ortholog TOL1, and each of nine suppressors of the exacerbated growth defect of trm6Δ imt06Δ mutants (also lacking one of four copies of the tRNAiMet gene) had mutations in DHP1 or TOL1 (Tasak and Phizicky 2022). Furthermore, the TRAMP complex had little role in quality control of tRNAiMet in S. pombe trm6Δ mutants, as deletion of the TRF4 ortholog CID14 in trm6Δ mutants had little effect on growth or in preventing tRNAiMet decay (Tasak and Phizicky 2022). Moreover, reexamination of S. cerevisiae trm6 mutants showed a prominent role of the RTD pathway in preventing tRNAiMet decay, in addition to the known role of the nuclear surveillance pathway. Thus, both the temperature sensitivity and the tRNAiMet decay observed in S. cerevisiae trm6-504 mutants were suppressed by mutation of any of the components of the RTD pathway (RAT1, XRN1, and MET22), and the lethality of S. cerevisiae trm6Δ mutants could be suppressed by mutation of both the nuclear surveillance pathway (trf4Δ) and the RTD pathway (met22Δ) but not by either alone (Tasak and Phizicky 2022).
As S. pombe and S. cerevisiae diverged ∼600 Mya (Parfrey et al. 2011), these results fuel speculation that the RTD pathway will target decay of specific hypomodified tRNAs throughout eukaryotes in these and other body modification mutants. Thus, it seems plausible that the RTD pathway is responsible for the reduced levels of specific hypomodified tRNAs in mammals lacking m7G46 (Lin et al. 2018; Dai et al. 2021), and in mouse strains lacking m5C in their tRNAs (Tuorto et al. 2012; Hussain et al. 2013). Indeed, there is evidence that the RTD pathway acts in humans, as heat stress at 43°C in HeLa cells resulted in loss of tRNAiMet levels which was prevented by knockdown of XRN1 and the human RAT1 ortholog XRN2 (Watanabe et al. 2013).
Consistent with its targeting of mature tRNAs, the RTD pathway competes with elements of the translation pathway. Thus, RTD is prevented by overexpression of elongation factor 1A (EF-1A), which normally binds charged tRNA to escort the tRNA to the ribosome A-site, and is enhanced by reduced expression of EF-1A (Dewe et al. 2012; Turowski et al. 2012). Similarly, overexpression of ValRS suppresses RTD of tRNAVal(AAC) in trm8Δ trm4Δ mutants (Turowski et al. 2012). Competition might also explain the apparent paradox of why reduced Pol III transcription, resulting from overexpression of the negative regulator Maf1 or from Pol III mutants, protects against RTD, as the reduced numbers of tRNAs would be more easily protected by the available EF-1A (Turowski et al. 2012).
The RTD pathway may also compete with the cellular retrograde tRNA transport pathway in which tRNAs are imported from the cytoplasm to the nucleus (Shaheen and Hopper 2005; Takano et al. 2005), as discussed further below. Thus, tRNATyr(GUA) and tRNALys(UUU) lacking m2,2G26 accumulate in the cytoplasm if retrograde transport is inhibited genetically by either of two mechanisms, as well as in cells lacking the RTD exonuclease Xrn1 (Kramer and Hopper 2013). The parsimonious explanation of these results is that the RTD pathway and the retrograde transport pathway are in competition for the same substrate tRNAs lacking m2,2G26, to degrade them or give them a second chance to be modified (Kramer and Hopper 2013).
It also seems likely that 5′ end capping of pre-tRNA transcripts competes with 5′–3′ decay by the RTD pathway, as the preferential accumulation of 5′ capped pre-tRNA (relative to uncapped pre-tRNA) that occurs when RNase P is inhibited is exacerbated in a met22Δ derivative strain, and in some cases in an xrn1Δ-derivative strain, indicating decay of uncapped pre-tRNA by an RTD-like mechanism (Ohira and Suzuki 2016).
In addition, recent experiments establish that the onset of the RTD pathway in both S. pombe and S. cerevisiae triggers activation of the GAAC pathway (the integrated stress response pathway in humans), which reprograms transcription and translation after stress treatments leading to uncharged tRNA and/or ribosome collisions (Hinnebusch 2005; Udagawa et al. 2008; Castilho et al. 2014; Duncan et al. 2018; Wu et al. 2020; Yan and Zaher 2021; Kim and Zaher 2022). Thus, in S. pombe, mutations in any of four genes of the GAAC pathway fully suppressed the temperature sensitivity of trm8Δ mutants and partially restored tRNATyr(GUA) levels, and temperature shift experiments showed that the growth defect, tRNATyr(GUA) decay, and GAAC induction start at exactly the same temperature, and are due to the tRNA decay and not the temperature shift itself (De Zoysa and Phizicky 2020). Furthermore, S. cerevisiae modification mutants subject to RTD also activate the GAAC pathway, but with the opposite effect on growth (De Zoysa and Phizicky 2020). Thus, for the well-studied S. cerevisiae trm8Δ trm4Δ mutant, GAAC activation occurs at the lowest temperature at which the growth defect and decay of tRNAVal(AAC) is observed, and deletion of any of several genes in the GAAC pathway exacerbates the temperature sensitivity and the loss of tRNAVal(AAC). As a gcn2Δ mutation exacerbates the temperature sensitivity of each of four other S. cerevisiae modification mutants that undergo RTD, it seems likely that activation of the GAAC pathway is a general consequence of onset of the RTD pathway in S. cerevisiae (De Zoysa and Phizicky 2020).
Intriguingly, the TOR pathway is also possibly linked to RTD in HeLa cells, as the XRN1 and XRN2/RAT1-mediated decay of tRNAiMet that occurs under heat stress is inhibited by rapamycin, concomitant with increased nucleolar and reduced nucleoplasmic localization of XRN2/RAT1 (Watanabe et al. 2014).
The Met22-dependent pre-tRNA decay pathway in S. cerevisiae
Recent results document the existence of an additional tRNA decay pathway in S. cerevisiae that is related to RTD by its Met22-dependence, but different due to the nature of its tRNA substrates. Whereas the RTD pathway typically targets mature tRNAs (Alexandrov et al. 2006; Chernyakov et al. 2008), this MPD pathway acts specifically on end-matured intron-containing pre-tRNAs, which accumulate due to mutations in the anticodon stem–loop (ASL) or the introns that perturb ASL-intron structure (Payea et al. 2020). As described above, eukaryotic tRNA introns form a characteristic BHB or BHL structure with the ASL that is recognized by the endonuclease (Thompson and Daniels 1988, 1990; Xue et al. 2006; Yoshihisa 2014; Schmidt and Matera 2020), and tRNA variants that disrupt this structure trigger MPD (Payea et al. 2020). Moreover, MPD is quantitatively comparable to that observed for classical RTD substrates with acceptor stem mutations, and removal of the intron eliminates most of the observed decay (Payea et al. 2020). It remains to be determined which Met22-dependent exonucleases or endonucleases act in MPD, although presumably the nucleases are inhibited by the pAp that builds up in met22Δ mutants (Dichtl et al. 1997; Yun et al. 2018). It also remains to be determined if the MPD pathway extends to intron-containing tRNAs in other eukaryotes (Chan and Lowe 2016; Schmidt and Matera 2020).
tRNA NUCLEAR-CYTOPLASMIC SUBCELLULAR DYNAMICS
The tRNA retrograde process
tRNAs surprisingly travel bidirectionally between the nucleus and cytoplasm in both budding yeast (Shaheen and Hopper 2005; Takano et al. 2005) and vertebrate cells (Zaitseva et al. 2006; Shaheen et al. 2007). The distribution of tRNAs between the nucleus and the cytoplasm results from the balance between: (1) tRNA nuclear export to the cytoplasm after transcription (primary tRNA nuclear export); (2) retrograde import of cytoplasmic tRNA into the nucleus (tRNA retrograde nuclear import); and (3) return of tRNAs that have been imported into the nucleus to the cytoplasm (tRNA reexport) (Fig. 8).
Bidirectional tRNA trafficking between the nucleus and cytoplasm and generation of tRNAPhe yW37 in S. cerevisiae. Step 1. Upon 5′ and 3′ processing and addition of several nucleoside modifications to newly transcribed intron-containing tRNAs, Los1, Mex67–Mtr2, and Crm1 escort the end-processed, partially modified intron-containing tRNAs to the cytoplasm via the primary tRNA nuclear export step. The tRNAs are then spliced on the mitochondrial outer membrane. Numerous additional nucleoside modifications also occur in the cytoplasm after splicing. Cm32 and Gm34 (orange circles) modifications added in the cytoplasm are important for yW biogenesis. Step 2. Spliced, modified tRNAs are returned to the nucleus via the tRNA retrograde nuclear import step. Mtr10 functions indirectly in tRNA nuclear import both constitutively and upon amino acid deprivation (red symbol), whereas Ssa2 functions only upon amino acid deprivation. tRNAPhe imported into the nucleus is further modified at G37 (yellow circle) to m1G37 (empty colored circle). Step 3. Msn5, Los1, Mex67–Mtr2, and pehaps also Crm1, escort the imported tRNAs back to the cytoplasm via the tRNA reexport step. Once reexported to the cytoplasm, tRNAPhe m1G37 is further modified to yW (black circle). Red circles indicate anticodon nucleotides 34, 35, and 36.
Initial studies of tRNA nuclear/cytoplasmic distribution were conducted by using Xenopus oocyte injections (Arts et al. 1998a; Kutay et al. 1998; Lund and Dahlberg 1998), reconstituted nuclear import assays using vertebrate cells (Zaitseva et al. 2006), and tRNA fluorescence in situ hybridization, FISH (Hellmuth et al. 1998; Sarkar and Hopper 1998; Shaheen and Hopper 2005; Takano et al. 2005; Shaheen et al. 2007). More recently, organelle fractionation and RNA-seq have been used (Schwenzer et al. 2019). Furthermore, analysis of the kinetics of tRNA nuclear export vs. nuclear import has become possible by using microinjection of tagged tRNAs and confocal imaging of the tagged tRNA in single vertebrate cells (Dhakal et al. 2019).
For vertebrate cells, the ability to distinguish primary tRNA nuclear export from tRNA reexport generally relies upon employment of transcription inhibitors (Shaheen et al. 2007; Schwenzer et al. 2019). In contrast, in budding and fission yeast and Trypanosoma brucei, it is possible to distinguish primary tRNA nuclear export from the reexport process because splicing of pre-tRNAs occurs in the cytoplasm; thus, those tRNAs encoded by genes possessing introns leave the nucleus in the primary export process with their introns, whereas tRNAs in the nucleus that have been spliced have undergone retrograde nuclear import, and exit the nucleus by reexport as spliced tRNAs (Yoshihisa et al. 2003; Murthi et al. 2010; Lopes et al. 2016; Kessler et al. 2018; Wan and Hopper 2018).
The distribution of tRNAs between the nucleus and the cytoplasm responds to nutrient status and environmental stresses. In budding yeast, cytoplasmic tRNAs accumulate in the nucleus upon amino acid, phosphate, and glucose deprivation (Shaheen and Hopper 2005; Hurto et al. 2007; Whitney et al. 2007). In vertebrate cells, cytoplasmic tRNAs accumulate in nuclei upon amino acid and glucose deprivation and in response to H202-induced oxidative stress (Shaheen et al. 2007; Dhakal et al. 2019; Schwenzer et al. 2019). Moreover, in vertebrate cells cytoplasmic tRNAiMet accumulates in nuclear granules upon temperature stress (Miyagawa et al. 2012; Watanabe et al. 2013). Furthermore, the tRNA nucleus–cytoplasm trafficking is relatively fast. Upon various stress impositions, for both budding yeast and vertebrate cells, cytoplasmic tRNAs rapidly redistribute to the nucleus and, likewise, rapidly return to the cytoplasm upon stress relief (Shaheen and Hopper 2005; Shaheen et al. 2007; Whitney et al. 2007; Dhakal et al. 2019; Schwenzer et al. 2019).
tRNA nuclear exporters
The first tRNA nuclear exporter identified was the conserved GTPase, Ran binding β-importin member, Los1 (budding yeast)/Xpo-t (fission yeast)/Exportin-t (vertebrates)/PAUSED (plants). This member of the β-importin family was identified decades ago (Hopper et al. 1980; Arts et al. 1998a; Hellmuth et al. 1998; Kutay et al. 1998; Lund and Dahlberg 1998; Sarkar and Hopper 1998; Hunter et al. 2003; Li and Chen 2003), and its interaction with tRNA substrates and RanGTP to form nuclear export complexes was described in a 3.2 Å resolution structure for the S. pombe Xpot in complex with a partial tRNA and RanGTP (Cook et al. 2009). However, insects lack a Los1/Exportin-t homolog (Lippai et al. 2000) and Los1 appears not to function in tRNA nuclear export in the kinetoplastid, T. brucei (Hegedusova et al. 2019). Furthermore, Los1 and its orthologs are unessential in every organism from which it has been deleted, including budding yeast, fission yeast, the plant A. thaliana, and haploid human cell lines (Hurt et al. 1987; Hunter et al. 2003; Li and Chen 2003; Cherkasova et al. 2012; Blomen et al. 2015; Hart et al. 2015; Wang et al. 2015; Azizi et al. 2020). Therefore, since tRNA nuclear export is essential for translation, eukaryotes must possess tRNA nuclear exporters that are independent of Los1 homologs.
For budding yeast there are at least three additional tRNA nuclear exporters that function in parallel to Los1: (1) the β-importin family member Msn5 which, for intron-containing tRNAs, appears to function solely in the tRNA nuclear reexport step (Murthi et al. 2010; Huang and Hopper 2015); (2) the mRNA nuclear exporter Mex67–Mtr2 heterodimer, which functions in both the tRNA primary and nuclear reexport steps (Wu et al. 2015; Chatterjee et al. 2017); and (3) the β-importin protein nuclear exporter, Crm1, which also functions in primary nuclear export (Fig. 8; Wu et al. 2015; Chatterjee et al. 2022). The RNA helicase, Dbp5, also functions in tRNA nuclear export (Lari et al. 2019), but it likely serves as an adapter/scaffold for Mex67–Mtr2 and/or Crm1.
Prior studies in vertebrate cells showed that, although unessential, Exportin-t appeared to be the dominant tRNA nuclear exporter (Arts et al. 1998a; Kutay et al. 1998), and that the Msn5 homolog Exportin-5, had a minor role in tRNA nuclear export (Bohnsack et al. 2002; Calado et al. 2002). However, a recent study documented that the human Mex67 homolog, NXF1, is a tRNA nuclear exporter (Chen et al. 2021), and the Mtr2 homolog, NXT1, has been reported to bind tRNA (Ossareh-Nazari et al. 2000). Further, in Arabidopsis deletions of both PAUSED and the exportin-5 homolog HASTY have a more severe phenotype than either individual deletion (Hunter et al. 2003), indicating that HASTY may function in tRNA nuclear export. Finally, in T. brucei, the Los1 and Msn5 homologs apparently have no important role in tRNA nuclear export; instead, Mex67 and Mtr2 function in tRNA nuclear export. Interestingly, as determined by RNA FISH studies, it appears that T. brucei Mex67 exports different families of tRNAs than does Mtr2 (Hegedusova et al. 2019). Unlike for budding yeast, the human Crm1 homolog, Xpo-1, appears not to play a role in tRNA nuclear export (Lund et al. 2004; Chen et al. 2021).
tRNA family preferences of the various tRNA nuclear exporters appears to be a theme, based on results from several approaches. In budding yeast, accumulation of end-processed, intron-containing tRNAs as assessed by northern analyses has served as a proxy for tRNA nuclear export due to the cytoplasmic location of SEN; thus, tRNAs retained in the nucleus are unspliced. More recently, in vivo copurification of nuclear exporters in complex with pre-tRNA cargo has served to assess tRNA nuclear export complexes (Huang and Hopper 2015). Both of these methodologies have documented that yeast Los1, Mex67–Mtr2, and Crm1 possess different preferences for each of the 10 different tRNA families encoded by intron-containing genes (Chatterjee et al. 2017, 2022). Moreover, vertebrate Exportin-t also has been reported to bind various tRNAs with different affinities (Li and Sprinzl 2006). tRNA family preferences for Los1/Exportin-t are surprising since this exporter is dedicated to tRNA nucleus–cytoplasm traffic and it interacts with the tertiary structure of mature tRNAs that is shared by all tRNAs (Arts et al. 1998b; Lipowsky et al. 1999; Cook et al. 2009).
The Los1/Exportin-t-independent tRNA nuclear exporters Mex67–Mtr2/NXF1–NXT1 and Crm1, which bind numerous RNAs and protein adapters (for reviews, see Kelly and Corbett 2009; Chatterjee et al. 2018), would not necessarily have been expected to recognize all tRNA families equally well. As predicted, Mex67–Mtr2 and Crm1 have been documented to possess tRNA family preferences and these preferences differ from each other and from Los1 (Chatterjee et al. 2022).
How tRNA family-specific tRNA nuclear export is achieved is not understood. Mex67–Mtr2 and NXF1–NXT1 function in mRNA nuclear export generally by binding RNA substrates via various protein adapters (for reviews, see Kelly and Corbett 2009; Chatterjee et al. 2018). Likewise, Crm1/Exportin-1 functions to export proteins from the nucleus to the cytoplasm via its interaction with proteins harboring leucine-rich nuclear export sequences (NES) (Fischer et al. 1995; Wen et al. 1995; Fornerod et al. 1997). Therefore, such proteins could preferentially bind subsets of nuclear tRNAs to function in preferential family-specific tRNA nuclear export. Other possible means for tRNA recognition include tRNA modifications and/or subnuclear locations of the nuclear exporters or the tRNAs.
In sum, there are multiple tRNA nuclear exporters in nature and their employment for efficient tRNA nuclear export differs among eukaryotic organisms. The implications of the individual nuclear exporters having tRNA family preferences could be far-reaching, as translation of the proteome could be affected by alteration of the cellular balance/activities of individual tRNA exporters. To fully understand tRNA family preferences for tRNA nuclear export, it will be important to understand how the various tRNA exporters interact with specific tRNA cargoes and to identify the protein adapters and possibly the RNA competitors.
tRNA nuclear importers
The gene products involved in retrograde tRNA nuclear import are not well defined. To date, two budding yeast proteins, the β-importin family member Mtr10 (Shaheen and Hopper 2005; Murthi et al. 2010) and the Ssa2 member of the chaperonin family (Takano et al. 2015) were shown to affect the levels of cytoplasmic tRNAs that accumulate in nuclei upon nutrient deprivation. Recently an assay has been developed that allows analysis of both constitutive and stress-induced tRNA nuclear import and reexport in budding yeast. This assay assesses modification of G37 of tRNAPhe to wybutosine (yW) (Fig. 6). yW modification of tRNAPhe requires all three steps of the tRNA retrograde pathway (Ohira and Suzuki 2011). The first step of the yW biogenesis is acquisition of m1G37 catalyzed by the nucleus-localized tRNA methyltransferase, Trm5. Trm5 can only modify spliced tRNAs; so, intron-containing pre-tRNAPhe is first exported via the primary tRNA nuclear export step to the cytoplasm where it is spliced and further modified by Trm7. Upon nuclear import of the spliced tRNA, Cm32 and Gm34 modified tRNAPhe G37 becomes a Trm5 substrate and G37 is thus modified to m1G37. Then, the m1G37 bearing tRNAPhe returns to the cytoplasm where m1G37 is further converted to yW via catalysis by the four cytoplasmic enzymes, Tyw1, Tyw2, Tyw3, and Tyw4. Thus, completion of yW modification of tRNAPhe requires primary nuclear export, tRNA nuclear retrograde import, and tRNA reexport (Ohira and Suzuki 2011). tRNAPhe possessing yW37 can be cleaved at position 37 upon treatment with HCl followed by aniline treatment (Thiebe and Zachau 1968; Ladner and Schweizer 1974) to generate tRNA halves, which are easily detected by northern analysis (Nostramo and Hopper 2020). Using this HCl-aniline assay, Mtr10 was shown to function in tRNA retrograde nuclear import both constitutively and upon amino acid deprivation, whereas Ssa2 functions in amino acid deprivation-induced tRNA nuclear import but not detectably in constitutive tRNA nuclear import (Nostramo and Hopper 2020). Ssa2, binds tRNAs and the nuclear pore protein, Nup116, and therefore, Ssa2 likely functions directly in tRNA retrograde nuclear import (Takano et al. 2015). In contrast, in vivo pull-down studies failed to document physical interactions between Mtr10 and tRNA (Huang and Hopper 2015) and therefore, it is not clear whether Mtr10 directly functions in tRNA nuclear import (Fig. 8). Whether there are additional budding yeast tRNA nuclear importers remains unknown but seems likely. The putative human Mtr10 homolog, Transportin 3, does not appear to affect tRNA nuclear import (Zhou et al. 2011) and, to date, no vertebrate tRNA nuclear importers have been reported.
Environmental stresses and tRNA nuclear/cytoplasmic dynamics
How environmental stresses result in redistribution of tRNAs between the nucleus and the cytoplasm is not well understood. As documented in budding yeast (Shaheen and Hopper 2005; Whitney et al. 2007; Takano et al. 2015; Lari et al. 2019; for reviews, see Huang and Hopper 2016; Chatterjee et al. 2018) as well as in vertebrate cells (Shaheen et al. 2007 PMCID: PMC1183567; Barhoom et al. 2011; Miyagawa et al. 2012; Watanabe et al. 2013; Dhakal et al. 2019; Schwenzer et al. 2019) environmental stresses affect both tRNA nuclear export and retrograde nuclear import steps. Recent studies using injected fluorescently tagged functional tRNAs measured the kinetics of tRNA subcellular movements and demonstrated that in response to nutrient deprivation, RNA nuclear import is down-regulated but tRNA nuclear export is nearly completely blocked, resulting in a net retrograde nuclear accumulation of tRNA (Dhakal et al. 2019).
There are different mechanisms that could cause altered nuclear vs. cytoplasmic pools of tRNAs upon stress (Huang and Hopper 2014). First, part of the pool of a given tRNA isoacceptor could be altered in response to stress. For example, budding yeast Ssa2, which is important for nuclear import of cytoplasmic tRNAs upon amino acid deprivation, preferentially binds tRNAs with destabilized aminoacyl acceptor stems (Takano et al. 2015). Likewise, using reconstituted nuclear import assays, the Fassati group reported that tRNAs deleted for 3′ nt, sometimes extending into the tRNA body, were preferentially imported into nuclei (Zaitseva et al. 2006). More recent tRNA sequencing studies of the tRNAs imported upon oxidative stress documented preferential nuclear accumulation of tRNA with 3′ deletions/truncations (Schwenzer et al. 2019). One possibility is that aberrant tRNAs might have preferential access to the tRNA nuclear importers because the truncated tRNAs are unable to interact with the translation machinery and thus are not otherwise engaged. Another possibility is that there are proteins that are able to recognize particular tRNAs and monitor integrity of the tRNA 3′ ends.
Alternatively, as documented by RNA sequencing of the tRNA cytoplasmic vs. nuclear pools upon stress in HeLa cells, there is tRNA family-specific tRNA nuclear accumulation upon stress (Schwenzer et al. 2019), possibly giving rise to changes of the proteome upon stress. tRNA family-specific nuclear accumulation could result from alteration of the levels of individual tRNA exporters and importers and/or the putative adaptors that may change in response to stresses, or the subcellular distributions of the exporters and importers themselves may be altered upon various stresses. Regarding the latter possibility, upon glucose deprivation and oxidative stress the steady state nuclear vs. cytoplasmic localization of the budding yeast tRNA nuclear exporters, Los1, Msn5, and Crm1, are inverted; normally, the steady state distribution of these proteins is nucleoplasmic or nuclear rim located, but under stress conditions the proteins appear to be predominately cytoplasmic (Quan et al. 2007; Huang and Hopper 2014). Likewise, glucose deprivation results in the inversion of the steady state nuclear/cytoplasmic distribution from primarily cytoplasmic to nucleoplasmic for the putative tRNA nuclear importer, Mtr10 (Huang and Hopper 2014). However, upon amino acid deprivation in budding yeast, Los1, Msn5, and Crm1 did not display altered nuclear vs. cytoplasmic distributions (Huang and Hopper 2014), whereas the level of the nucleus-located subpool of Ssa2 was reported to increase (Takano et al. 2015). To date, there have been no reports of similar studies of the subcellular locations of the budding yeast Mex67–Mtr2 tRNA nuclear exporter under conditions that alter tRNA subcellular dynamics; nor is it known whether there are altered levels or subcellular distributions of the various exporters in response to stresses in vertebrate cells.
The tRNA retrograde pathway and constitutive tRNA biogenesis/quality control
The tRNA retrograde pathway also serves important constitutive functions. One such function concerns tRNA modifications. As detailed above, yW modification of tRNAPhe requires tRNA nuclear import from the cytoplasm followed by reexport back to the cytoplasm. Similarly, queuosine (Q) modification of G34 of tRNATyr in T. brucei requires that pre-tRNATyr first be exported to the cytoplasm where tRNA splicing occurs (Lopes et al. 2016; Kessler et al. 2018). Upon tRNA retrograde nuclear import, the nucleus-localized tRNA-guanine transglycosylase, which has specificity for spliced tRNATyr as substrate, converts G34 to Q34. Then, the Q-modified tRNA exits the nucleus via Mex67–Mtr2 mediated tRNA reexport for appropriate function in translation in the cytoplasm (Kessler et al. 2018; Hegedusova et al. 2019). It is unknown whether other tRNA modifications in organisms that have cytoplasmic tRNA splicing also require tRNA retrograde nuclear import for tRNA modifications.
The tRNA retrograde pathway also serves an important role in tRNA quality control as demonstrated by studies in budding yeast (Kramer and Hopper 2013; Chatterjee et al. 2022). Errors sometimes occur such that aberrant tRNAs that are unprocessed at the 5′ and 3′ termini or that are hypomodified are prematurely exported to the cytoplasm. The levels of these inappropriate tRNAs increase upon deletion of the putative tRNA nuclear importer, Mtr10 (Kramer and Hopper 2013). Thus, tRNA retrograde nuclear import appears to remove aberrant tRNAs from the cytoplasm, returning them to the nucleus where they may be repaired and/or destroyed by the 5′ to 3′ nuclease Rat1 of the RTD pathway, or by the 3′ to 5′ nuclear surveillance pathway (Kramer and Hopper 2013; Chatterjee et al. 2022). As described above, the retrograde pathway is also in apparent competition with the cytoplasmic RTD quality control pathway that destroys hypomodified tRNAs or tRNAs with destabilizing mutations (Whipple et al. 2011; Kramer and Hopper 2013; Guy et al. 2014). Further, although Los1 and Crm1 have high fidelity in nuclear export of only those tRNAs with mature 5′ termini, Mex67 is able to export 5′ leader containing tRNAs to the cytoplasm, where they are spliced. Upon tRNA retrograde nuclear import these aberrant 5′ leader-containing spliced tRNAs can be destroyed by the 3′ to 5′ nuclear surveillance pathway, but not by the nuclear RTD process (Chatterjee et al. 2022). Protection from Rat1 turnover may be due to possession of the triphosphate of the initial tRNA transcripts or to caps at the 5′ temini (Ohira and Suzuki 2016; Chatterjee et al. 2022).
tRNA FRAGMENTS AND REGULATION OF GENE EXPRESSION
Although the canonical function of tRNAs as adaptors in gene expression, delivering amino acids to the translation machinery, has been appreciated since 1958 (Hoagland et al. 1958), numerous additional noncanonical functions for tRNAs have been described in the subsequent decades (for reviews, see Raina and Ibba 2014; Schimmel 2018). Most recently, this list of noncanonical functions has expanded due to the discoveries of tRNA fragments that serve numerous unanticipated roles in biology. The rapid pace of the discoveries and the important roles of the tRNA fragments in gene expression, development, and health have been summarized in numerous excellent review articles (Anderson and Ivanov 2014; Kumar et al. 2016; Oberbauer and Schaefer 2018; Guzzi and Bellodi 2020; Kim et al. 2020; Xie et al. 2020; Chu et al. 2022; George et al. 2022; Hou et al. 2022; Pekarsky et al. 2022). We direct the reader to these reviews regarding the roles of tRNA fragments in development, health and disease. Here, we highlight the various means by which the myriad of tRNA fragments are generated, the roles of nucleotide modifications in their production and functions, and the varying mechanisms by which tRNA fragments regulate gene expression in eukaryotic cells.
RNases involved in tRNA fragment production
The first discovered nucleases that cleave tRNAs were the E5 and D subsets of the bacterial colicins (Ogawa et al. 1999; Tomita et al. 2000; for review, see Ogawa 2016) and the fungal killer toxins from K. lactis and Pichia acaciae (Lu et al. 2005; Klassen et al. 2008). These secreted RNases cleave specific mature tRNAs in the ACL to generate half molecules, which reduces environmental competition by recipient cells via depletion of the recipient's active tRNA pools and therefore inhibition of their protein synthesis. Similarly, in bacterial and archaeal organisms the toxin–antitoxin systems that regulate cell growth upon various stresses can act via tRNA endonucleolytic cleavage. For example, the Type II MazF and VapB/C toxin–antitoxins cleave various tRNAs in the ACL to inhibit translation (Cintron et al. 2019; for review, see Walling and Butler 2019).
In contrast, the more recently discovered mechanisms that generate tRNA fragments in eukaryotic cells generally do not cause significant reduction of tRNA pools; rather, they generate novel noncoding RNAs that possess various activities able to regulate gene expression. These tRNA fragments have several different nomenclatures. Some RNases cleave specific mature tRNAs in or near the ACL and generate tRNA ∼half molecules (30–40 oligonucleotides) that are variously dubbed tRNA fragments (tRFs), tiRNAs (tRNA stress-induced RNAs), tsRNAs (tRNA-derived small RNAs), tdRs (tRNA-derived RNAs) or tRHs (tRNA halves); hereafter these tRNA fragments are referred to as tRHs (Fig. 9). RNases also cleave mature tRNAs in or near the D-loop or the T-loop to generate smaller, 13–26 nt fragments, named 5′- or 3′-tsRNAs or 5′- or 3′-tRFs (hereafter referred to as 5′- or 3′-tsRNAs). tRNA fragments can also be derived from pre-tRNAs; the 3′U tRFs are derived from the 3′ trailer of pre-tRNAs and the 5′ leader exon fragments are derived from initial tRNA transcripts containing the 5′ leader that have been cleaved in the ACL (Fig. 9; for reviews, see Anderson and Ivanov 2014; Raina and Ibba 2014). A single system for naming the myriad of tRNA fragments has been proposed (Holmes et al. 2023). Accordingly, the fragments will be referred to as tDRs (tRNA-derived RNAs) with numbers denoting the starting and ending positions of the mature tRNAs, according to conventional tRNA numbering (Sprinzl et al. 1998) (e.g., tDR-1:15); the specific tRNA from which the fragments are derived will also be designated (e.g., tDR-1:15-Val-AAC-1) and, finally the nomenclature will also contain information to link the particular tRNA fragments to tRNAs in the genomic database (http://gtrnadb.ucsc.edu) (see Fig. 9). This proposed nomenclature promises to eliminate future confusion; however, because the discoveries summarized here unfortunately generally do not have sufficient information to utilize the new systematic nomenclature, we will utilize the terms tRHs, 5′ or 3′-tsRNAs, and 3′U tRFs (Fig. 9).

Biogenesis of tRNA fragments. (A) tRNA fragments generated from pre-tRNAs prior to 5′ leader and 3′ trailer removal. Green font and bracket indicate tRNA fragments derived from 3′ trailers upon endonucleolytic cleavage by RNase Z. Blue bracket demarcates region of fragments resulting from cleavage of 5′ leader-containing pre-tRNAs in the ACL. (B) 5′ (left blue bracket) or 3′ (right blue bracket) ∼ half size tRNA fragments generated upon cleavage of mature tRNAs in the ACL. (C) 5′ (left puple bracket) or 3′ (right purple bracket) ∼ ¼ size tRNA framents resulting from endonucleolytic cleavage of mature tRNAs in the D- or T-loops, respectively. Black, blue, and purple fonts near brackets indicate the various names of the tRNA fragments; blue font nomenclature is used in this review. Red font refers to the proposed future systematic nomenclature for tRNA fragments. Arcs indicate the possible locations of loop cleavages. Names below each arc refer to the various endonucleases implicated in cleavages. Angiogenin (also refered to as ANG) is a vertebrate RNase A-like enzyme, and RNase L is an interferon induced 2′–5′ oligoadenylate synthetase-dependent RNase. Rny1 is a yeast T2-like endonuclease; Rnt2 and RNS1, RNS2, and RNS3 are plant T2-like endonucleases. Metazoan Dicer and plant Dicer-like DCLs are RNase III-like enzymes also functioning in pre-miRNA biogenesis.
Generation of tRNA halves
Numerous RNases function in the production of tRHs, 5′ or 3′-tsRNAs, and 3′U tRFs (Fig. 9), with some of the RNases functioning in tRNA cutting primarily under stress conditions. The discovery of amino acid starvation-induced cleavage of tRNAs in their ACLs in the protozoan Tetrahymena thermophila (Lee and Collins 2005) was followed by definition of other stress-induced tRNA cleavage events in other organisms, and definition of their mechanisms. Budding yeast Rny1 (vertebrate RNASET2) is an RNase T2-like endonuclease that, upon stress treatment, catalyzes cleavage of substrate tRNAs, rRNAs, and snRNAs. Rny1 cleaves some mature tRNAs in the ACL after exposure of cells to oxidative stress or high culture density/stationary phase (Thompson and Parker 2009). Rny1 is a resident of the yeast vacuole (lysosome in vertebrates); it is unknown whether tRNA cleavage results from release of Rny1 to the cytoplasm upon stress or, instead, whether tRNAs access the vacuole upon stress via autophagy (Luhtala and Parker 2012). Cleavages in the tRNA anticodon in Tetrahymena and the plant, Arabidopsis are catalyzed by combinations of multiple Rny1 orthologs, Rnt2 A, B, and C and RNS1, RNS2, and RNS3, respectively (Andersen and Collins 2012; Megel et al. 2019). In contrast, for vertebrate cells, generation of tRNA halves is generally catalyzed by ANG, an RNase A-like ribonuclease. Under normal environmental conditions, ANG is primarily localized in the nucleus; cytoplasmic pools exist in complex with an inhibitor, RNH1. Upon stress, nuclear ANG cleaves specific cytoplasmic tRNAs (for review, see Anderson and Ivanov 2014). Interestingly, the T. brucei protozoan genome does not encode either ANG-like or Rny1-like endonucleases (Fricker et al. 2019); so, it is unknown how the stress-induced tRNA half molecules are derived in this organism. Finally, a specialized mammalian endonuclease, RNase L, that is dependent upon 2′,5′ oligoadenylate for dimerization and activity, cleaves particular tRNAs in the ACL (Donovan et al. 2017 and references therein).
After cleavage in the ACL, the 5′ and 3′ halves may not be separated due to the base pairs that form the cloverleaf secondary structure. Indeed, it has recently been reported that tRNAs nicked in the ACL can be repaired (Chen and Wolin 2023; Costa et al. 2023). Identification of helicases that may aid separation of the nicked halves are a subject of current investigation (Drino et al. 2023).
Generation of 13–26 nt tRNA fragments
The 3′ U tRF species are comprised of the 3′ trailers of pre-tRNA transcripts, and result from the cleavage of these pre-tRNAs at the 3′ mature border by the endonuclease RNase Z (Trz1 in yeast) (Haussecker et al. 2010; Su et al. 2019; for reviews, see Anderson and Ivanov 2014; Keam and Hutvagner 2015; Xie et al. 2020). However, less is known regarding the enzymes required for generating the fragments from mature tRNAs that are smaller than tRNA halves (5′-tsRNAs and 3′-tsRNAs). The endonuclease, Dicer, which functions in the biogenesis of miRNAs, has been implicated in the generation of both the 5′-tsRNAs and 3′-tsRNAs in several biological systems (Cole et al. 2009; Haussecker et al. 2010; Durdevic et al. 2013b; Maute et al. 2013; Martinez et al. 2017; Luo et al. 2018). In contrast, other studies have shown that production of 5′- and 3′-tsRNAs can be independent of Dicer (Li et al. 2012; Kumar et al. 2014; for review, see Keam and Hutvagner 2015). For example, even though in Arabidopsis pollen cells a member of the Dicer family, DCL1, was reported to function in the generation of 5′-tsRNAs that target transposable element RNAs (Martinez et al. 2017), Arabidopsis missing all three of the unessential Dicer genes (DCL2, 3, and 4) and possessing a hypomorphic allele of the essential DCL1 gene (dcl1234) exhibited no differences in the tRNA cleavage products compared to the wild-type plants. Rather, deletion of the Rny1-like genes, RNS1, RNS2, and RNS3, affected the production of short tRNA fragments in a tissue-specific manner (Alves et al. 2017; Megel et al. 2019). Production of 5′- and 3′-tsRNAs also appears to be independent of ANG; for example, studies of stressed and unstressed human cells overexpressing ANG or possessing an ANG knockout reported comparable levels of 3′-tsRNAs (Su et al. 2019). A recent report documented that the tRNA substrate generating 3′-tsRNAs are mature aminoacylated tRNAs (Liu et al. 2021). A future challenge will be to detail the biogenesis pathways of the various small tRNA fragments. This may be complex given the exceedingly large number of tRNA fragments that can be generated from the various isoacceptor and isodecoder tRNAs encoded by eukaryote genomes and the numerous RNases with differing specificities.
tRNA modifications and tRNA fragments
RNA modifications play surprisingly important roles in tRNA fragment production and/or function (for review, see Lyons et al. 2018). Some tRNA modifications enhance tRNA cleavage. The fungal zymocin γ toxin subunit provides a eukaryotic example of this. K. lactis produces γ toxin that is toxic to S. cerevisiae, because when introduced into S. cerevisiae the γ toxin cleaves three different tRNAs, all of which possess the mcm5s2U34 modification; the modification is important for substrate cleavage, but additional surrounding nucleotides affect cleavage efficiency (Lu et al. 2005; Huang et al. 2008). There are examples of the requirements of tRNA modifications for the production of vertebrate tRNA fragments. For example, pseudouridylation functions in production of tsRNAs, as in human ECS cells pseudouridylation at U8 by Pus7 enhances production (or stability) and the activities of short 5′-tsRNAs that possess a terminal oligo guanosine (TOG) motif that are derived from tRNAAla(AGC/CGC/TGC), tRNACys(GCA), and tRNAVal(AAC) (Guzzi et al. 2018; for review, see Guzzi and Bellodi 2020).
Other modifications protect tRNAs from cleavage; there are many reports of such protection from several organisms/tissues involving several different modifications including Q34, m5C38, m5C48.49, C34m, m1G9. For example, in HEK293T and HeLa cells queuosine (Q34) modification of tRNAHis(GTC) and tRNAAsn(GTT/GTC) protected these tRNAs from cleavage by ANG in vitro and in vivo (Wang et al. 2018). Modification of m5C38 by Drosophila Dnmt2 protected several tRNAs from stress-induced cleavage; Dnmt2−/− mutants lacking m5C38, are sensitive to growth at high temperature and to oxidative stress (Schaefer et al. 2010). Similarily, the presence of m5C38 in mouse sperm inhibited fragmentation of tRNAGly into 5′ and 3′ tsRNAs (Zhang et al. 2018). m5C modifications catalyzed by Trm4/NSUN2 are also important for production of 5′ tRHs in mouse and human skin cells. Lack of m5C48,49 resulted in an ANG-dependent accumulation of 5′ tRHs from a subset of tRNA species (Blanco et al. 2014). Moreover, in human cell lines, C34 2′-O-methylation of tRNAeMet (C34m) is generated by small guide RNAs, SNOD97 and SCARNA97; this Cm34 modification protects tRNAeMet from stress-induced cleavage by ANG (Vitali and Kiss 2019). Further, m1G9 protects tRNAs from fragment production. Thus, lymphoblast cell lines derived from TRMT10A deficient patients accumulated tRNAGln 5′ tRHs as well as 5′ fragments of ∼22 nt. TRMT10A knockdown in a rat pancreatic β-cell line resulted in increased reactive oxidative species that led to apoptosis, and apoptosis was also caused by transfection of tRNAGln 5′ fragments into TRMT10A-competent EndoC-βH1 cells (Cosentino et al. 2018). Finally, in human cell lines, 5′ monophosphate methylation of tRNAHis(GTG) by BCDIN3D is reported to protect this mature tRNA from cleavage, resulting in reduced levels of 3′-tsRNAHisGTG (Reinsborough et al. 2019). As many of the discoveries of the roles of modifications in the biogenesis/function of tRNA fragments are recent, it is likely that future studies will uncover other such examples.
Diverse mechanisms of action of tRNA fragments
tRNA fragments can participate in some of the same noncanonical functions that mature tRNAs participate in. For example, both mature cytoplasmic tRNAs and tRHs are able to bind cytochrome C released from mitochondria and, in doing so, activate caspase and thereby inhibit apoptosis (Mei et al. 2010; Saikia et al. 2014). In another example, mature tRNAs prime retrotranscription by base pairing with the primer binding site (PBS) in retroviruses and endogenous LTR retroelements; likewise, the 5′ tRNAiMet derived tRH serves as the primer for Drosophila copia retroviral replication (Kikuchi et al. 1986), and 3′-tsRNAs that are complementary to human T-cell leukemia virus (HTLV) PBS serve as primers for reverse transcription in vitro (Ruggero et al. 2014). In contrast, in mouse 3′-tsRNAs with perfect complementarity to the PBS of retroelements compete with mature tRNA for PBS binding and thereby inhibit retrotranscription (Schorn et al. 2017; for review, see Schorn and Martienssen 2018).
tRNA fragments also serve unique functions that are unrelated to activities of full length tRNAs. These novel functions result from either binding of tRNA fragments to proteins or protein complexes or from complementary base pairing (often dependent upon Argonaute proteins) to target RNAs, thereby affecting the structure, stability, or activities of the target RNAs. Through these various mechanisms tRNA halves and tRNA small fragments can affect RNA transcription and epigenetic inheritance, RNA processing, RNA stability, RNA structure, or translation. Here, we provide a few examples of the various mechanisms of action by tRNA fragments.
tRNA fragments functioning via protein interaction
A well-described function of tRNA fragments is to inhibit protein synthesis initiation (for reviews, see Anderson and Ivanov 2014; Guzzi and Bellodi 2020). Ivanov et al. (2011) and Anderson and Ivanov (2014) reported that specific 5′ halves of tRNAAla and tRNACys, in combination with the YB-1 translational repressor, inhibit translation via displacement of initiation factor eIF4F from capped mRNAs, thereby globally inhibiting translation in response to stress. These tRHs contain the 4–5 5′ G nucleotides comprising the TOG motif that is important to generate the RNP complex. Similarly, in human ESC cells, TOG motif-containing small 5′-tsRNAs derived from tRNAAla(AGC/CGC/TGC), tRNACys(GCA), and tRNAVal(AAC), modified with Ψ8 (see above), bind the poly (A) binding protein, PABPC1, which is required for translation initiation (Guzzi et al. 2018). tRNA fragments have also been reported to affect translation by binding ribosomes. In the archaeon, Haloferax volcanii, stress-induced 5′-tsRNAs derived from tRNAVal bind to the small ribosome subunits and compete with mRNA binding, thereby inhibiting translation initiation (Gebetsberger et al. 2017). In contrast, in the protozoan, T. brucei, binding of a stress-induced tRNA 3′ tRH, derived from tRNAThr(AGU), to ribosomes or polysomes resulted in enhanced translation (Fricker et al. 2019). Interestingly, studies of differentiating mouse embryonic stem cells reported different modes of action of particular 5′ tRHs during differentiation: in stem and retinoic acid induced differentiating states, particular tRHs interact with ribosomes and ribosomal subunits, globally modulating translation; however, a set of tRHs also interact with and sequester the insulin growth factor-like mRNA binding protein, Igf2bp1, resulting in c-Myc mRNA instability (Krishna et al. 2019).
tRNA fragment interactions with proteins can also affect RNA processing. For example, 3′-tsRNAs from Tetrahymena affect pre-rRNA processing (Couvillion et al. 2012). These 3′-tsRNAs interact with a Piwi protein, Twi12, as well as other proteins to form an RNP complex that contains the 5′ to 3′ exonuclease Xrn2. The complex forms in the cytoplasm and it is required for Xrn2′s nuclear import/stability and its role in pre-rRNA processing.
tRNA fragments that function via RNA–RNA complementarity
There are numerous examples of tRNA fragments that cause down-regulation of specific mRNA targets via RNP complexes consisting of Argonaute proteins and tRNA fragments with limited complementarity to the target mRNAs (for review, see Kumar et al. 2014). This mechanism to regulate gene expression resembles the manner in which miRNAs and piRNAs affect gene expression. In fact, some small noncoding regulatory RNAs that were originally identified as miRNAs are actually 3′-tsRNA molecules derived from mature tRNAs (e.g., Haussecker et al. 2010; Maute et al 2013; Reinsborough et al. 2019) or 3′U tRFs derived from the 3′ trailers of pre-tRNAs (e.g., Haussecker et al. 2010; Pekarsky et al. 2016). However, the tRNA-derived fragments differ in important ways from miRNAs. First, they are transcribed by RNA polymerase III, rather than RNA polymerase II. Second, tRNA fragments generally have different biogenesis pathways than miRNAs (for review, see Ha and Kim 2014) or piRNAs (for review, see Han and Zamore 2014). Third, although complementary base pairing for some tRNA-derived fragments is similar to the mechanism by which miRNAs and piRNAs interact with target RNAs via short 7 nt 5′ seed sequences that base pair with the mRNA 3′ UTR (e.g., Kuscu et al. 2018), other tRNA fragments appear to interact with target RNAs differently. These tRNA fragments have been proposed to have seed sequences located in the 5′ ends, the middle, and/or 3′ ends of the tRNA fragments that are complementary with the 5′ UTR, the coding sequence, or the 3′ UTR of the target mRNAs (e.g., Luo et al. 2018). Finally, there are examples in which tRNA fragments affect target mRNAs via complementary base pairing, but independently of Argonaute proteins (Jehn et al. 2020).
Modifications also are implicated in tRNA fragment function via base pairing. For example, TRMT6/61A-dependent m1A modification in the seed region of particular 3′ts RNAs inhibits miRNA function. Inhibition is due to reduced base pairing with target mRNAs rather than to interaction with Argonaute. Over production of TRMT6/61A and fragment modification is correlated with bladder cancer (Su et al. 2022).
Although most known small RNAs that base pair with target mRNAs cause decreased gene expression, either due to increased turnover or to decreased translation, a tRNA fragment that enhances translation upon complementary base pairing with its target mRNA has been reported. Following up on the observations that a 3′-tsRNA derived from tRNALeu(CAG) in HeLa and HCT-116 cells is important for cell growth and efficiency of translation, the Kay group learned that this 3′-tsRNA possesses conserved complementarity with a region in the RPS28 coding sequence in mouse and human cells. It has been proposed that base pairing of the tRNA fragment with RPS28 mRNA alters mRNA structure, unfolding the mRNA to allow efficient translation at a step after initiation (Kim et al. 2017, 2019). There is no evidence for the interaction of this 3′-tsRNA with Argonaute proteins (Kim et al. 2017).
CONCLUDING REMARKS AND PERSPECTIVES FOR THE FUTURE
As documented above, the last several years have witnessed an explosion in our understanding of the biology of tRNA processing, tRNA modification, tRNA decay, and tRNA fragments. These advances set the stage for significant discoveries in the future, aided by ever more powerful new technologies. Four particularly interesting future topics are elucidated below.
First, there is great promise for breakthroughs in our understanding of the mechanisms of the numerous neurological, mitochondrial, and other disorders due to defects in tRNA processing. Multiple studies cited here and elsewhere (Suzuki 2021) have documented examples in which mutations leading to reduced function or to lack of different tRNA processing or modification components result in neurological or other disorders (Fig. 4). It seems highly likely that future studies will unravel why so many of these mutations selectively target the neurological system, why the mutations have different manifestations, and how they exert their effects at a mechanistic level. It also seems likely that some of the different manifestations will be due to tissue-specific differences in expression of isodecoders (Ishimura et al. 2014) or of different tRNA species.
Second, it seems likely that there will be significant new insights regarding the regulation of modifications. We described above a number of examples highlighting the variability of modifications in response to different stress or environmental conditions (Chan et al. 2010, 2012; Czech et al. 2013; Laxman et al. 2013; Preston et al. 2013; Alings et al. 2015; Damon et al. 2015; Han et al. 2015; Gupta et al. 2019; Cristodero et al. 2021; Huber et al. 2022), and in several cases there is significant understanding of the consequences of the altered modifications on translation (Chan et al. 2012; Czech et al. 2013), signaling pathway regulation (Damon et al. 2015), and metabolic regulation (Laxman et al. 2013; Gupta et al. 2019; Huber et al. 2022). Future analysis will undoubtedly reveal a more complete description of the pervasiveness of modification regulation, aided in part by the continued development of technology to facilitate collection of modification profiles of individual tRNAs (Liu et al. 2019; Furlan et al. 2021). The importance of modification regulation seems likely also to be extended by additional findings that tRNA modifications are removed in vivo in response to stress or other conditions, as shown for AlkBH1 demethylase (Liu et al. 2016), or findings that modification levels have tissue-specific differences due to variability in expression of the modification enzymes.
Third, it seems likely that there will be new surprises revealed about the interplay between tRNA biology and different regulatory or stress response pathways. Previous analysis has documented interactions between the Mod5 i6A modification enzyme and a central enzyme of sterol biosynthesis (Benko et al. 2000), reciprocal interactions between elongator function in xcm5U34 modification and the TORC1 and TORC2 signaling pathways (Candiracci et al. 2019), between xcm5U34 and the proteotoxic stress pathway (Nedialkova and Leidel 2015), several different interactions between the biology of different modifications and the GAAC pathway (Zinshteyn and Gilbert 2013; Chou et al. 2017; Han et al. 2018; De Zoysa and Phizicky 2020), and interactions between modifications such as s2U34 and queuine and metabolic pathways (Laxman et al. 2013; Gupta et al. 2019; Huber et al. 2022). It is likely that more such cross-pathway interactions will be discovered using the sophisticated modern arsenal of methodologies for analysis of transcription, translation, and the proteome.
Fourth, it is highly likely that there will be a huge increase in our knowledge of the biology of tRNA fragments. We cited above several well-studied examples in which tRNA fragments have been shown to inhibit apoptosis (Mei et al. 2010; Saikia et al. 2014), stimulate translation (Fricker et al. 2019), prime retroviral replication (Kikuchi et al. 1986), inhibit protein synthesis (Ivanov et al. 2011; Anderson and Ivanov 2014; Gebetsberger et al. 2017), impair production of siRNAs (Durdevic et al. 2013b), and affect pre-rRNA processing (Couvillion et al. 2012). Based on the large number of tRHs that continue to be found using modern sequencing methods, it is virtually certain that there will be additional insights into their different modes of regulation.
SUPPLEMENTAL MATERIAL
Supplemental material is available for this article.
ACKNOWLEDGMENTS
We thank Elizabeth Grayhack and members of the Phizicky and Hopper laboratories for valuable discussions during the course of this work. This research was supported by National Institutes of Health (NIH) grants GM052347 to E.M.P. and GM122884 to A.K.H.
Footnotes
-
Article is online at http://www.rnajournal.org/cgi/doi/10.1261/rna.079620.123.
-
Freely available online through the RNA Open Access option.
This article, published in RNA, is available under a Creative Commons License (Attribution-NonCommercial 4.0 International), as described at http://creativecommons.org/licenses/by-nc/4.0/.
REFERENCES
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵








