
Artifacts and biases of the reverse transcription reaction in RNA sequencing
- 1OncoRNALab, Cancer Research Institute Ghent, 9000 Ghent, Belgium
- 2Department of Biomolecular Medicine, Ghent University, 9000 Ghent, Belgium
- 3Center for Medical Genetics, Ghent University, 9000 Ghent, Belgium
- Corresponding author: jasper.verwilt{at}ugent.be
Abstract
RNA sequencing has spurred a significant number of research areas in recent years. Most protocols rely on synthesizing a more stable complementary DNA (cDNA) copy of the RNA molecule during the reverse transcription reaction. The resulting cDNA pool is often wrongfully assumed to be quantitatively and molecularly similar to the original RNA input. Sadly, biases and artifacts confound the resulting cDNA mixture. These issues are often overlooked or ignored in the literature by those that rely on the reverse transcription process. In this review, we confront the reader with intra- and intersample biases and artifacts caused by the reverse transcription reaction during RNA sequencing experiments. To fight the reader's despair, we also provide solutions to most issues and inform on good RNA sequencing practices. We hope the reader can use this review to their advantage, thereby contributing to scientifically sound RNA studies.
Keywords
INTRODUCTION
The study of RNA has propelled advances in understanding biochemical processes in health and disease and assisted in developing novel biomarkers and RNA-targeted therapies. RNA sequencing (RNA-seq) can characterize large numbers of RNA molecules in parallel and has quickly become the prevailing method to study the transcriptome of tissues and cells in various organisms. In its most widely adopted form, the quantification and characterization of RNA heavily rely on a reverse transcription (RT) step. The RNA is used as a template during the RT reaction to generate a complementary DNA (cDNA) strand. Each RT reaction uses at least four components: the template RNA, one or more oligonucleotide primers, a reverse transcriptase enzyme (RTase), and an RT buffer. This step has become so widely adopted that researchers do not hesitate to treat it like a comfortable black box: RNA in, DNA out. However, several studies challenged this mindset and revealed its flaws. Some researchers have gone against the current, showing how the RT reaction is more intricate than anticipated and depends on a subtle interplay of its components. As we will illustrate in this review, discrepancies can arise between the RNA and the resulting cDNA due to several factors. We group these inconsistencies into the generation of faulty molecules that differ in sequence from the template RNA (“RT artifacts”); and quantitative changes between nucleic acid fragments in the transcribed cDNA compared to the template RNA input (“RT bias”). The latter can be partitioned into intrasample bias (some [parts of] transcripts are more likely to be reverse transcribed than others) and intersample bias (due to inconsistencies between preanalytical variables or protocol choices). With this review, we hope to provide the community with a concise overview of the pitfalls and concerns inherent to the RT reaction, with a focus on RNA-seq experiments. We hope to engage researchers to include appropriate control measures and think twice about their novel or unexpected results: things are not always what they seem.
REVERSE TRANSCRIPTION BIASES
The RNA-seq community has thoroughly described the causes of quantification biases in RNA-seq experiments—such as GC content, transcript length, and sequence base composition—and developed numerous strategies to account for them (Oshlack and Wakefield 2009; Hansen et al. 2010; Li et al. 2010; Srivastava and Chen 2010; Roberts et al. 2011; Zheng et al. 2011). However, the RT reaction is hardly ever considered a source of these biases. In this section, we aim to uncover the biases originating from the RT reaction itself. All factors of RT can introduce individual biases, but they might also arise from interactions between multiple components. For clarity, we chose not to group the biases by cause but by whether they affect results in an individual sample (intra) or between samples (inter). Figure 1 provides an overview of the different intrasample biases. Table 1 summarizes these biases and provides recommendations to overcome them.
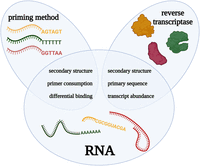
Overview of reverse transcription components and characteristics at the heart of intrasample quantification biases. Graphical overview of the intrasample biases akin to the reverse transcription reaction. The blue ellipses of “priming method” and “reverse transcriptase” overlap with the “RNA” circle as it is from the interaction of these factors that the biases originate. This figure was created using BioRender.
Overview of reverse transcription (RT) issues summarizing the cause, effect, and associated solution
INTRASAMPLE BIASES
Reverse transcriptase-RNA bias
To understand the reverse transcriptase-RNA bias, it is critical to understand that contemporary RTases are engineered versions of retroviral RTases, of which the wild-type versions have several characteristics helping with gene regulation and viral replication. First, retroviruses rely on an RNA secondary structure-dependence of RTases for their gene regulation and evolution (for review, see Smyth et al. 2018). Therefore, RT should be inherently dependent on RNA secondary structure. Second, retroviral RTases contain an RNase H moiety that is responsible for hydrolyzing the RNA of the formed cDNA:RNA duplex. This subunit is indispensable for retroviral replication as it frees the cDNA for the necessary “jumps” of the RTase within the genomic RNA. In vitro, however, RNase H activity can result in premature hydrolysis of the template RNA and interruption of the RT, introducing a negative bias toward longer transcripts (Kotewicz et al. 1988).
Commercially available RTases differ in sensitivity, specificity, reproducibility, and yield (Levesque-Sergerie et al. 2007; Sieber et al. 2010; Lindén et al. 2012; Zucha et al. 2020). These dissimilarities are inherent to the RTases due to their unequal capability of dealing with low template concentrations, specific RT conditions (such as temperature or buffer composition), and intensely structured RNAs. Therefore, before applying an RTase in the test tube, it is crucial to recognize its specific characteristics and tendencies. Most RTases have an RNase H subdomain responsible for hydrolyzing the RNA of the formed DNA:RNA duplex. Kotewicz et al. (1988) isolated an RTase lacking RNase H activity. Such recombinant enzymes synthesize cDNA from long RNA transcripts more efficiently (Kotewicz et al. 1988). However, discussions remain on the influence of the RNase H moiety of the RTase on the output of the RT reaction (Zucha et al. 2020). Enzymes with diminished or absent RNase H activity, such as Superscript IV (Thermo Fisher) and Maxima H Minus (Thermo Fisher), outperform others in terms of sensitivity, yield, and precision (Sieber et al. 2010; Zucha et al. 2020). RTases display a range of competencies in dealing with RNA folding and primary sequence, indicating an enzyme–structure and enzyme–sequence interaction (Brooks et al. 1995; Ståhlberg et al. 2004; Bustin et al. 2015; Minshall and Git 2020). More specifically, more than 100-fold cDNA yield differences can arise, purely derived from the RTase's handling of secondary structure (Ståhlberg et al. 2004). At higher RT temperatures, the RNA molecules dissociate, disrupting secondary structures. Therefore, enzymes have now been engineered to be more thermostable and which are able to operate at higher RT temperatures (Zucha et al. 2020). Consequently, we advise using thermostable RTases to mitigate RNA secondary structure biases.
Some researchers have demonstrated that the primary sequence of the RNA can introduce a bias during RT (Zheng et al. 2011). Several models exist that aim to remove this bias from the data. Li et al. (2010), for example, used the first several bases of the sequencing read (including the ones after the RT primer binding site) to predict up to 52% of the RNA abundance differences between transcripts. The random primer sequence only partially explains this bias: it is also observed in random primer-independent RNA-seq data, suggesting that the bases downstream from the priming site also play a role (Schwartz et al. 2011). However, because these studies rely on a PCR step as well, we cannot specifically pinpoint these biases to the RT reaction.
Primer-RNA bias
Depending on the RNA of interest, researchers can use different priming methods: oligo(dT) for reverse transcribing (mainly) mRNA (see below), targeted primers for transcripts of choice, or short random primers for any RNA. Still, these methods do not produce an unbiased cDNA representation of the RNA of interest. RNA molecules are highly structured and display numerous structural elements (Das 2021; Townshend et al. 2021). RNA secondary or tertiary structures can prevent priming. Researchers have exploited this observation by hybridizing short oligonucleotides to determine the structure of an RNA molecule of interest through the oligonucleotides’ binding capabilities (Lewis and Doty 1970; Wrede et al. 1978). From this observation, we can hypothesize that linear, lowly structured RNAs dominate the cDNA pool. Mir and Southern (1999) presented the secondary structure bias in microarrays, where less structured RNAs preferentially bind to the array. Upon RT, this bias induces seemingly differential abundance among transcripts with contrasting secondary structures within the same sample due to primer inaccessibility (Mir and Southern 1999; Ståhlberg et al. 2004; Bustin et al. 2015). Even within the same transcript, the choice of the targeted region can result in markedly differing abundance estimations (Kuo et al. 1997). Again, researchers can use thermostable RTases combined with a high RT temperature to partially overcome RNA folding-dependent biases (Malboeuf et al. 2018; Minshall and Git 2020). Still, there is always the chance that highly structured regions refold during primer annealing. Alternatively, researchers could use a thermostable group II intron reverse transcriptase (TGIRT)-based protocol. This approach uses a DNA:RNA hybrid RT primer with one overhanging random deoxynucleotide. Since the hybrid primer can only prime a single RNA nucleotide at the 3′-end (using the overhanging nucleotide), the priming is minimally dependent on the secondary structure of the target (Xu et al. 2019; Begik et al. 2023).
Random primers have other issues. First, Minshall and Git (2020) discuss that specific random primers are consumed by the abundant transcripts they associate with, thereby leaving less of these primers for the remaining transcripts and, thus, introducing a bias. Second, although random primers are random in their sequence, they are not random in their RNA-binding capacities and introduce a sizeable bias into transcript abundance (Hansen et al. 2012; Minshall and Git 2020). Hansen et al. (2012) were the first to describe this bias and propose a correcting model. This model uses a random primer-specific weight for each read and adjusts the abundance levels accordingly to improve quantification accuracy. These phenomena show that random priming of total RNA samples often unreliably quantifies the RNA transcripts of interest (Lekanne Deprez et al. 2002).
Similarly, gene-specific primers can have contrasting binding capabilities (due to targeted primary sequence and structure) and the relative abundance of multiple transcripts in such a study should be interpreted cautiously.
INTERSAMPLE BIASES
Intersample biases arise from the inconsistent use of reagents or discrepancies between RNA samples, such as RNA quantity, integrity, and purity (absence of proteins, DNA, enzymatic inhibitors, complexing agents, or nucleases; Fleige and Pfaffl 2006). Standardized execution and reporting (following MIQE-like guidelines; Bustin et al. 2009), accurate data normalization, use of equally qualitative samples, use of identical reagents and reagent quantities, and proper quality control checkpoints can mostly alleviate these biases. Because of the comparative nature of intersample analyses, such as differential expression analyses, intrasample biases are canceled out when taking the appropriate measures, ensuring unbiased conclusions on transcript abundance differences. We will not be expanding on these biases in this review.
REVERSE TRANSCRIPTION ARTIFACTS
Instead of presenting a divergent cDNA pool in terms of transcript abundances, RT can also generate inaccurate cDNA sequences, that is, cDNA molecules structurally different from their RNA templates. These are referred to as RT artifacts. In this section, we will discuss the primary artifacts (Fig. 2) and provide means to reduce them to a minimum (also summarized in Table 1).
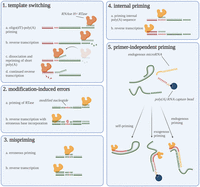
Overview of reverse transcription artifacts. Graphical overview of the five discussed priming artifacts. The reverse transcriptases are shown as yellow proteins (note the difference between yellow and orange reverse transcriptases). This figure was created using BioRender.
Template switching
Retroviruses are dependent on RTases for replication. The RTase scans the RNA sequence and synthesizes the complementary DNA (cDNA) sequence. However, the cDNA-RTase complex might bind to another RNA molecule with a sequence complementary to the cDNA's 3′-end mid-synthesis and proceed with synthesizing cDNA using the new RNA strand as a template. This phenomenon is called template switching (TS) and results in large deletions (intramolecular TS) or fused cDNA molecules (intermolecular TS) (Luo and Taylor 1990). Deletions resulting from intramolecular TS—which have recently been dubbed “falsitrons” (as in “false exitrons”; Schulz et al. 2021)—are typically characterized by the absence of a canonical splice site (Cocquet et al. 2006) and the presence of direct repeats flanking the deleted region (which often has a strong secondary structure bringing the repeats close together) (Pathak and Hu 1997). TS is certainly not a novel concept (Gilboa et al. 1979) and has been described in multiple retroviruses, such as HIV (DeStefano et al. 1992), but also in mitochondria (Sellem et al. 2000). This phenomenon is not confined to in vivo environments but also occurs when RTases are used in vitro. Unfortunately, these artifacts are hard to distinguish from real biological deletions, and care is advised when making claims about novel isoforms originating from a deletion. Such apparent deletions are caused by template switching, and there have been multiple efforts to quantify the extent of the problem in published isoforms (Geiszt et al. 2004; Cocquet et al. 2006; Schulz et al. 2021). In addition, when TS occurs on internal poly(A) stretches during oligo(dT)-based RT, artifacts similar to internal priming can be formed (Balázs et al. 2019). Lastly, TS events can resemble trans-splicing or back-splicing (on which circRNA formation relies) (McManus et al. 2010; Yu et al. 2014) or apparent fusion genes (Houseley and Tollervey 2010). There are, however, multiple options to reduce TS. First, the researcher can use a RTase lacking or having reduced RNase H activity. RNase H is essential to template switching as it hydrolyzes the RNA of the DNA:RNA duplex during RT, freeing up the cDNA and allowing it to find another RNA sequence to bind (Luo and Taylor 1990). Some discussion exists to what extent RNase H-negative RTases can still perform TS (Luo and Taylor 1990; Garces and Wittek 1991; Zaphiropoulos 2002; Zeng and Wang 2002; Zhu et al. 2002). Second, one can tweak some of the parameters of the RT reaction. Short reaction times, low RTase concentration, and low reaction temperature reduce the extent of TS (Ouhammouch and Brody 1992). TS is also modulated by the concentration of the acceptor RNA molecule (to which the RTase “jumps”), the number of nucleotides overlapping between the two RNA molecules (Luo and Taylor 1990), and the size of the “falsitron” (Delviks and Pathak 1999), but these are typically out of the researcher's hands. Last, suspicious results should always be validated by using a non-RT-dependent technique such as northern blot (Houseley and Tollervey 2010), direct RNA sequencing (Schulz et al. 2021), in situ hybridization, or RNA-templated DNA ligation.
Modification-induced errors
Even when the RTase correctly primes the transcripts of interest, it can still introduce errors. Actually, the errors generated by RTases contribute to the high mutational rate in retroviruses driving evolution in vivo (Mizutani and Temin 1976; Roberts et al. 1988; Svarovskaia et al. 2003). As RNA–cDNA discrepancies are usually not desired in RNA characterization, researchers have developed higher-fidelity RTases for in vitro applications (for review, see Svarovskaia et al. 2003). Several studies have quantified these errors for various RTases. When these errors happen randomly, they tend not to present significant problems. When these RTase errors always occur at the same position, the phenomenon becomes more problematic. Single-nucleotide polymorphism (SNP) calling from RNA-seq data, for example, requires a robust detection of a specifically altered nucleotide at a specific position. Without any accompanying data on the sequence of the native RNA and DNA, it is impossible to differentiate between RNA-editing events, genuine SNPs, and RT-induced sequence errors. Often, when these errors occur at specific positions, they result from the RTase misinterpreting modified RNA bases. Potapov et al. (2018) analyzed the mistakes made by three different RTases on modified and unmodified RNA. They noted that the presence of a pseudouridine (Ψ), 5-hydroxylmethyluridine (hm5U), or N6-Methyladenosine (m6A) nucleotides significantly increased the error rate of the RTase. To add to the problem, they discovered that specific RNA modifications differentially influence different RTases. ProtoScript II and AMV RTases, for example, significantly increase adenosine substitutions (Potapov et al. 2018). Researchers should apply various approaches to avoid making an incorrect assumption about a specific nucleotide and its potential modifications. Machine learning approaches have been developed to infer RNA modifications from RNA sequencing data, with varying success (Ryvkin et al. 2013; Werner et al. 2020; Tan et al. 2021).
Mispriming
Mispriming occurs when oligonucleotides prime noncomplementary RNA sequences, introducing mismatches in the resulting cDNA, typically in the first few nucleotides of the sequencing read (Zhang and Byrne 1999; Piskol et al. 2013; Van Gurp et al. 2013). Mispriming can arise when using oligo(dT) primers (as is the case with internal poly(A) stretches), when targeting a specific transcript of interest or sequencing adapters (leading to off-target effects) (Shivram and Iyer 2018), or when using random primers (Van Gurp et al. 2013). It happens nonrandomly with different nucleotides having differential sensitivity for mispriming (uracil residues are prone to mispriming, while cytosine residues prevent mispriming) (Van Gurp et al. 2013). This problem can often be alleviated by computationally trimming a set number of bases from the 5′ read end to remove the priming site from the final read.
Internal priming
mRNA quantification often relies on oligo(dT) RT primers that target the 3′-end of polyadenylated mRNAs. This type of priming is increasingly being used due to the rising popularity of cost-effective 3′-end sequencing at the tissue or single-cell level. However, nothing withholds the RTase and its primer from binding to an internal poly(A)-rich sequence (i.e., any poly(A) sequence not at the end of the transcript), thereby generating RT artifacts that can be falsely identified as mRNA molecules (i.e., when [contaminating] DNA is primed) or as alternative polyadenylation sites (Nam et al. 2002). However, a more recent article, challenged the hypothesis that these artifacts originate from internal priming by implying the internal poly(A) sequences are often too short for preferential binding; instead, they result from template switching after poly(A)-tail binding and dissociation (Balázs et al. 2019). Although this problem can (in part) be avoided using anchored oligo(dT) primers (Nam et al. 2002), these are not readily used and do not come standard in most RNA sequencing kits. The generated artifacts can also be filtered out post-sequencing during the bioinformatic analysis by inspecting the neighborhood of the transcript for genomic poly(A)-rich regions (Beaudoing et al. 2000; Tian et al. 2005; Zhang et al. 2005; Shepard et al. 2011; Graber et al. 2013; Wilkening et al. 2013). Recently, researchers developed specialized programs designed to remove internal priming artifacts and generated a list of transcripts prone to incorrect quantification due to internal priming (Svoboda et al. 2022). Of note, these artifacts are not always useless as RNA velocity analysis exploits them to determine the number of nonspliced transcripts (La Manno et al. 2018). To overcome internal priming during the library preparation, researchers can opt to use a TGIRT-based protocol (as explained above). In such setups, the RT primer exclusively binds to the 3′-end of the RNA transcript (Xu et al. 2019; Begik et al. 2023). Note this approach incorporates all transcripts instead of focusing on polyadenylated mRNA.
Primer-independent priming
Even without a purposely administered primer, RTases produce transcribed cDNA (Bernstein et al. 1983; Agranovsky 1992; Peyrefitte et al. 2003; Khraiwesh et al. 2010; Tuiskunen et al. 2010; Moison et al. 2011; Piché and Schernthaner 2018; Timofeeva and Skrypina 2018). These artifacts can have three origins: RNA self-priming (Bernstein et al. 1983; Tuiskunen et al. 2010; Timofeeva and Skrypina 2018), priming by endogenous nucleic acids (Khraiwesh et al. 2010; Timofeeva and Skrypina 2018), or priming by (contaminating) exogenous nucleic acids (such as transfer RNAs [tRNAs] or DNA present in RTase preparations) (Agranovsky 1992; Goldenberger and Altwegg 1995; Moison et al. 2011; Piché and Schernthaner 2018). Self-priming occurs when secondary structures are formed, creating double-stranded regions which the RTase recognizes as primed regions. The RT products generated from the double-stranded regions may be incorrectly classified as pseudogenes (Bernstein et al. 1983) or antisense products (Tuiskunen et al. 2010)—the latter is especially problematic during the strand-specific priming of viral RNA genomes, where strand-specific detection is critical. Finally, endogenous DNA fragments or RNA, such as microRNAs or tRNA (fragments), can initiate RT when they bind to other RNA transcripts (Agranovsky 1992; Khraiwesh et al. 2010). Exogenous nucleic acids from an upstream step or reagents can start the RT reaction. Piché and Schernthaner (2018) uncovered an intriguing example of this phenomenon: carry-over oligo(dT) oligonucleotides originating from mRNA extraction primed poly(A) tails during a primer-free RT reaction. Additionally, multiple available RTase solutions have repeatedly been shown to contain contaminating small RNA molecules that can initiate RT without an RT primer (Agranovsky 1992; Moison et al. 2011). These molecules are hypothesized to be highly associated with the RTase and are therefore difficult to remove (Moison et al. 2011). Generally, a “no-primer” control identifies said primer-independent RT products, after which they may be excluded from the analysis.
CONCLUSION
The simple linear and perfect conversion of RNA to cDNA is not a reality and should therefore be considered a black-box reaction to date. It relies on multiple factors and depends on close interactions between these different factors. This review highlights how the synthesized cDNA pool might differ from the original RNA repertoire. Where “RT biases” correspond to relatively different cDNA abundances of correctly transcribed molecules, the term “RT artifacts” refers to aberrant cDNA molecules. These can arise from incorrect primer binding or unexpected RTase behavior. We advise detailed transparent reporting and dedicated verification of (surprising) results using RT-independent techniques to reveal the mentioned biases and artifacts. We hope this review can incentivize researchers to be critical of blind trust in the RT reaction.
ACKNOWLEDGMENTS
The authors would like to thank their peers Marieke Vromman and Kathleen Schoofs for proofreading the final manuscript for correctness and clarity.
Footnotes
-
Article is online at http://www.rnajournal.org/cgi/doi/10.1261/rna.079623.123.
-
Freely available online through the RNA Open Access option.
This article, published in RNA, is available under a Creative Commons License (Attribution-NonCommercial 4.0 International), as described at http://creativecommons.org/licenses/by-nc/4.0/.








