
“Crowd-control” by RNA: a pervasive theme in biology
- Centre for Cancer Biology, an Alliance of SA Pathology and University of South Australia, Adelaide, SA 5000, Australia
- Department of Medicine, University of Adelaide, Adelaide, SA 5005, Australia
- Corresponding author: cameron.bracken{at}unisa.edu.au
Abstract
As we continue to find new regulatory roles for RNAs, a theme is emerging in which regulation may not be mediated through the actions of a specific RNA, as one typically thinks of a regulator and target, but rather through the collective nature of many RNAs, each contributing a small degree of the regulatory load. This mechanism has been termed “crowd-control” and may apply broadly to miRNAs and to RNAs that bind and regulate protein activity. This provides an alternative way of thinking about how RNAs can act as biological regulators and has repercussions, both for the understanding of biological systems, and for the interpretation of results in which individual members of the “crowd” can replicate the effects of the crowd when overexpressed, but are not individually significant biological regulators.
Keywords
HISTORICAL PERSPECTIVE
Our understanding of noncoding RNAs has advanced rapidly in recent decades, in step with improving technologies that allow transcriptomic sequencing at ever-increasing depth. Historically, noncoding RNAs are understood as facilitators of protein production, dating back to the 1940s when ribosomal RNAs were found to be abundant at sites from which proteins were produced (ribosomes). The other key pillars of the “central dogma” were in place by the early 1960s, by which time transfer RNAs (tRNAs) were known to act as adaptors for the incorporation of amino acids and messenger RNAs (mRNAs) identified as the intermediate between genes and proteins. The 1980s brought further understanding of small nuclear RNAs (snRNAs) as critical elements for mRNA splicing, and small nucleolar RNAs (snoRNAs) as facilitators of rRNA and tRNA modification. However, despite the expanding noncoding repertoire, in each case the function of the RNA was still, directly or indirectly, facilitating the production of protein that was viewed as the functional output of gene expression.
This narrow view of the role of RNA began to expand in the 1980s, when the use of antisense RNAs became widespread, not as a means to enable the production of protein, but instead as a means to regulate it. Reports of regulatory roles then exploded over the subsequent decade, with microRNAs (miRNAs) found to control gene expression, not just in C. elegans in which they were initially found, but across all multicellular organisms. This led to a “gold-rush” of discovery, including of the mechanisms through which short-interfering RNAs (siRNAs) and miRNAs regulate gene expression for which the 2006 Nobel Prize was awarded. In a sense, the “gold-rush” has continued to this day, with many tens of thousands of publications building to an understanding that noncoding RNAs play critical roles in the regulation of biological processes; a function facilitated by not only miRNAs, but potentially also by tens of thousands of long noncoding RNAs (lncRNAs) and circular RNAs (circRNAs) whose importance is rapidly emerging.
“CROWD-CONTROL”: A KEY TO UNDERSTANDING THE REGULATORY ROLE OF MANY RNAs
Fueled by both an expectation of how regulatory systems “should” be controlled, and the necessity to explain mechanisms behind observations, there are an abundance of reports in which a single regulatory RNA:target interaction is put forward as a mechanism by which a phenotypic effect is perpetrated. There are many instances where this is accurate, but it is worth considering what is really being claimed in many such reports. In the case of a miRNA for example, hundreds of miRNAs are each capable of directly regulating, at least weakly, hundreds if not thousands of transcripts. In the case of competitive endogenous RNAs, where RNA interactions act as competitive sponges to sequester regulatory molecules, any one specific transcript in all but the most extreme of scenarios represents only a tiny fraction of possible interactors within the cellular milieu. These observations draw into question how often a single regulatory RNA is biologically meaningful.
A recent study that sheds light on this concept came from the laboratory of Matthias Hentze, who found that the binding of RNA regulates the activity of glycolytic enzymes. They reported that a single enzyme is bound by several thousand distinct transcripts, with seemingly any one of these capable of exerting a regulatory effect if expressed at supraphysiological levels (Huppertz et al. 2022). This was termed “crowd-control”; a form of biologically meaningful regulation that is exerted by the collective actions of many RNAs (“the crowd”) and characterized by extreme redundancy of the individual regulators. The depletion of individual RNAs in loss-of-function experiments therefore has negligible effects; however, significant regulation would still be expected in gain-of-function experiments where an individual RNA is overexpressed. Regulation should therefore not be attributed to any one specific RNA in isolation, though if a researcher was to select a candidate RNA for gain-of-function experiments, results could easily be interpreted to indicate that the selected RNA is more significant than it actually is. This commentary discusses that beyond enolase-1, crowd-control may be an accurate framework through which to view the activities of many RNAs more broadly. This is important for two reasons. Firstly, to gain an accurate understanding of how genes are regulated, and secondly so that gain-of-function data is correctly interpreted.
DIVERSE RNA REGULATORS OF PROTEIN FUNCTION
It has become increasingly clear that RNAs can regulate protein activity through multiple mechanisms, both indirect and direct. Examples of indirect regulation include the organization of chromatin or membrane-less compartments such as paraspeckles and Cajal bodies by long noncoding RNAs (Mao et al. 2011; Quinodoz et al. 2021). Examples of RNAs directly regulating protein activity include the activation of innate immunity through the binding of foreign RNA to nucleic acid sensors (Jensen and Thomsen 2012), or the inhibition of autophagy by the binding of vault RNAs to p62 (Horos et al. 2019). However, there are over a thousand RNA-binding proteins (RBPs) (Castello et al. 2016), so the opportunities for RNA-mediated control are extensive. It is also noteworthy that of the RNA binding sites that have been identified, nearly half exist in intrinsically disordered regions and are often coincident with hotspots for post-translational modification such as lysine acetylation and tyrosine modification.
One recent elegant example of a novel mechanism of RNA-mediated protein regulation is that of enolase-1, a key enzyme within the glycolytic pathway that was found to associate with approximately 2000 different RNA transcripts in an acetylation-dependent manner (Huppertz et al. 2022). During differentiation where the rate of glycolysis decreases, acetylation promotes the binding of a diverse array of RNAs that inhibit enzymatic activity. Importantly, RNA-mediated inhibition of enolase-1 can be exerted by a number of these RNAs when expressed at high (100 nM) levels, even if the RNAs selected are unremarkable examples from the 2000 bound transcripts identified. The “crowd-control” aspect is therefore that one can recapitulate the mechanism of inhibition using any of these RNAs if present at a sufficiently high level, but in endogenous conditions it is via their cumulative actions that the regulation of glycolysis emerges.
There are several implications that can be drawn from this work. One of course is the increasingly varied roles that RNAs play in biology, but another implication is methodological. If one is unaware of the crowd-control hypothesis, then one could easily select a specific RNA from among those bound to enolase-1, perhaps on the basis of its particular enrichment or the known roles of that transcript, then manipulate its expression to demonstrate a specific role for that regulator. However, this would be a disservice to the true story of collective binding that emerges from a pool of RNA regulators.
CROWD-CONTROL AS APPLIED TO miRNAs
One could speculate the failure to appreciate the breadth of “crowd-control” may also be applied to other areas of RNA biology, perhaps most especially miRNAs. This is because every miRNA has potentially thousands of binding partners, and every transcript is potentially bound by dozens of different miRNAs. Thus, although the molecular mechanisms are completely different between how miRNAs work and how enolase-1 is regulated, the conceptual framework of crowd-control applies to both because it is the sum of interactions through which regulation is exerted. There are also parallels between the study of miRNAs and RBPs such as enolase-1, as the failure to appreciate the potential for crowd-control can lead to overstating the importance of individual RNAs in gain-of-function experiments. The miRNA field is rife with such examples, making the concept of crowd-control an important lens through which to evaluate new data.
MiRNAs serve as the guidance component of a larger ribonucleoprotein complex, RISC (RNA-induced silencing complex), providing binding selectivity for members of this complex whose role it is to silence expression of the transcripts to which they are bound. One of the challenges for the field is created by the short interface through which miRNAs interact with their targets. As the primary interface is only 6–8 nt long, many 3′UTRs are predicted to possess dozens of candidate miRNA response elements (MREs) (Fig. 1A). Similarly, each miRNA can bind between 5% and 30% of the protein-coding transcriptome, meaning most genes are bound by multiple miRNAs (Hafner et al. 2010). As such, one can easily form hypotheses that link almost any gene to miRNAs and disease.
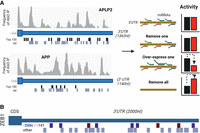
(A) MicroRNA-guided AGO interaction sites identified by AGO-HITS-CLIP (high-throughput sequencing, cross-linked immunoprecipitation) indicate multiple sites of miRNA binding. Black bars indicate predicted binding sites for miRNAs that are in the top 100 most expressed miRNAs in MDA-MB-231 cells (in which the experiment was performed). Gray bars indicate all other candidate binding sites. Canonical binding sites were predicted by TargetScan. The crowd-control hypothesis states that each of the interacting miRNAs are likely to have minimal effects by themselves, but collectively can have a meaningful impact on gene expression. This can be recapitulated by any individual miRNA when expressed at supraphysiological levels. (B) Predicted miRNA binding sites within the ZEB1 3′UTR indicate unusually strong targeting by the miR-200 family. Mir-200 encompasses five family members with two different targeting specificities. miR-200c/-200b/-429 (blue bars) have six predicted sites. miR-141/-200a (red bars) have three predicted sites. Light gray bars indicate potential binding sites for an additional 19 miRNAs (from among the 86 miRNAs that are expressed at a level >0.1% of all miRNAs in HMLE cells—an epithelial cell line in which ZEB1 is actively repressed). A single predicted binding site is present in the ZEB1 3′UTR for 16 of these miRNAs. Two predicted binding sites are present for each of the remaining three miRNAs.
Tens of thousands of publications now exist in which a miRNA:target relationship is claimed to be functionally significant, though the sheer volume of reports strains credulity that there could be so many critical switches involving a single miRNA and only one (or a few) specific targets. This is especially the case given that the vast majority of miRNA interactions exert only weak regulatory activity, as measured at either the RNA (Hunter et al. 2013; Ma et al. 2018) or protein (Selbach et al. 2008) level.
The issue of the vast number of publications that claim major effects for individual interactions was addressed in an influential review published earlier this year, in which the authors attribute the breadth of these reports to a “lack of minimum controls and experimentation necessary to make convincing conclusions” (Kilikevicius et al. 2022). Whilst this is undoubtedly true in many instances, it is also consistent with a crowd-control interpretation for miRNA function, where for any given 3′UTR, multiple (perhaps dozens of) miRNAs that are capable of binding, each exert a modest repressive activity. One can imagine scenarios where this might occur; for example, dampening the expression of proliferation genes in a nonproliferative state, or preventing spurious up-regulation from transient signals or genetic noise. In such cases, and in parallel to the report regarding enolase-1, care must therefore be taken not to mistake the effect of supraphysiological expression as an indicator of endogenous activity.
Such a model could also explain why large numbers of miRNA regulators are often ascribed to well-studied genes. This follows the logic that miRNAs will be capable of binding essentially any transcript and are thus theoretically capable of suppressing almost any gene. Thus, for genes that are more heavily studied, many more instances of miRNA-mediated repression will have been reported. To illustrate, we recently conducted a survey across the literature and found that no less than 61 separate miRNAs were reported to directly bind and negatively regulate the 3′UTR of the transcript that encodes ZEB1, a transcription factor that enforces a mesenchymal phenotype (Migault et al. 2022). There are some specific miRNAs, such as members of the miR-200 family which promote an opposing epithelial phenotype, that genuinely seem to be important regulators of ZEB1. Evidence to support this includes an unusually high number of confidently predicted binding sites for miR-200 within the ZEB1 3′UTR (Fig. 1B), and multiple studies, including those using loss-of-function experiments, that show strong suppression of ZEB1 looking at both reporter genes and endogenous mRNA and protein expression (Gregory et al. 2008; Park et al. 2008). However, in contrast, only single or unremarkable binding sites are predicted for most of the other miRNAs that are claimed to regulate ZEB1, and studies that experimentally demonstrate suppression of ZEB1 are often heavily reliant upon overexpression. The collective weight of such studies may therefore serve to obfuscate the functions of miRNAs more than they clarify them. However, even if their individual roles are often overstated, the crowd-control that is exerted by their collective actions might still be important. In Figure 1B, for example, all of the potential miRNA binding sites in the ZEB1 3′UTR are for miRNAs that are expressed in epithelial cells, so it would not be surprising they share collective actions in repressing the expression of a mesenchymal-promoting gene. In the specific case of miRNAs, collective actions may also be enhanced by TNRC6, a family of three paralogous proteins capable of simultaneously binding two or three miRNA:AGO complexes, depending upon the TNRC6 isoform (Briskin et al. 2020). Proximity of miRNA sites is known to increase target repression beyond the independent activities of the sites (Grimson et al. 2007; Saetrom et al. 2007). The cooperative binding that is facilitated by TNRC6 provides a mechanism through which miRNA sites in close proximity are more effective, and thus might enhance the efficiency of crowd-control.
Large, highly connected networks are intrinsically unstable (May 1972). Broad and weak repression cumulatively enhances the stability of gene regulatory networks more effectively than a lower number of stronger interactions (McCann et al. 1998; Chen et al. 2019). Modest repression by a miRNA should therefore not simply be dismissed as ineffective if the nature of the control is crowd-mediated, and if the miRNA is a component of that crowd which regulates individual genes and provides greater stability to the overall gene expression network. The same is true of other RNAs that bind and regulate RBPs; a field that can be expected to grow over the forthcoming years.
ACKNOWLEDGMENTS
The author would like to acknowledge funding from the Australian Research Council (FT190100544) and The Hospital Research Foundation (2022-S-DTFA-007).
Footnotes
-
Article is online at http://www.rnajournal.org/cgi/doi/10.1261/rna.079644.123.
-
Freely available online through the RNA Open Access option.
This article, published in RNA, is available under a Creative Commons License (Attribution-NonCommercial 4.0 International), as described at http://creativecommons.org/licenses/by-nc/4.0/.








