
Computational design and experimental verification of pseudoknotted ribozymes
- 1INRS - Institut Armand-Frappier, Laval, QC H7V 1B7, Canada
- 2Software Engineering and Computer Science Department, Concordia University, Montreal, Quebec H3G 1M8, Canada
- 3Electrical and Computer Engineering Department, Concordia University, Montreal, Quebec H3G 1M8, Canada
- Corresponding authors: jonathan.perreault{at}iaf.inrs.ca, nawwaf.kharma{at}concordia.ca
-
↵4 These authors contributed equally to this work.
Abstract
The design of new RNA sequences that retain the function of a model RNA structure is a challenge in bioinformatics because of the structural complexity of these molecules. RNA can fold into its secondary and tertiary structures by forming stem–loops and pseudoknots. A pseudoknot is a set of base pairs between a region within a stem–loop and nucleotides outside of this stem–loop; this motif is very important for numerous functional structures. It is important for any computational design algorithm to take into account these interactions to give a reliable result for any structures that include pseudoknots. In our study, we experimentally validated synthetic ribozymes designed by Enzymer, which implements algorithms allowing for the design of pseudoknots. Enzymer is a program that uses an inverse folding approach to design pseudoknotted RNAs; we used it in this study to design two types of ribozymes. The ribozymes tested were the hammerhead and the glmS, which have a self-cleaving activity that allows them to liberate the new RNA genome copy during rolling-circle replication or to control the expression of the downstream genes, respectively. We demonstrated the efficiency of Enzymer by showing that the pseudoknotted hammerhead and glmS ribozymes sequences it designed were extensively modified compared to wild-type sequences and were still active.
Keywords
INTRODUCTION
Natural functions of noncoding RNAs (ncRNAs) are numerous, but they also have various applications in engineering biological systems (Auslander et al. 2010; Prommana et al. 2013; Cameron et al. 2014; Zalatan et al. 2015) and therapeutics (Ruder et al. 2011; Esposito et al. 2014; Lienert et al. 2014; Halloy et al. 2022), as well as in nanotechnology (Grabow and Jaeger 2014; Afonin et al. 2015; Shu et al. 2015). Some ncRNAs with complex structures, such as riboswitches that act as receptors binding specific metabolites, are involved in controlling gene expression (Serganov and Nudler 2013). Other intricate RNA structures can confer catalytic activities to RNA, like for the small self-cleaving ribozymes. The best-known example is the hammerhead ribozyme (HHRz), which is involved in producing single-copy genomes out of the multimeric RNA resulting from rolling-circle replication of viroids (Prody et al. 1986). There are also hundreds of examples of different hammerhead-type ribozymes for which RNA self-cleavage still has no obvious function (Hammann et al. 2012). The core of the so-called minimal version of the hammerhead ribozyme has been studied extensively over more than three decades; atomic resolution structures are available (Martick and Scott 2006), as well as extensive biochemical data strongly supporting the requirement of the core consensus sequence (Nelson and Uhlenbeck 2008). In the case of the hammerhead ribozyme, a tertiary interaction between stem I and II was found to have a critical impact on its activity (De la Pena et al. 2003; Khvorova et al. 2003). Different configurations of pseudoknots were later found to hold stem I and II to achieve the same impact and also make the ribozymes over 1000-fold more active than a version without this interaction (Jimenez et al. 2011, 2015; Perreault et al. 2011). Another example of a ribozyme, the glmS ribozyme, has a clear regulatory function. This ribozyme cleaves its 5′-UTR (UnTranslated Region) by using glucosamine-6-phosphate (GlcN6P) as a cofactor, leading to degradation of the mRNA and repression of genes involved in GlcN6P synthesis when the latter is in sufficient concentrations (Winkler et al. 2004; Collins et al. 2007). While not studied as extensively as the hammerhead ribozyme, the archetypal model for RNA structure/function studies, several atomic resolution structures of glmS ribozyme are also available (Klein and Ferre-D'Amare 2006; Cochrane et al. 2009), as well as a wealth of experimental data (Ramesh and Winkler 2014).
It is widely accepted that the function of ncRNAs is attributed to their structure (Leontis et al. 2006; Mortimer et al. 2014), which is determined by the nucleotide composition of the RNA polymer. An RNA strand folds to form an array of secondary structures which then form stable tertiary structures (Batey et al. 1999). The diverse range of functions of ncRNAs, as well as the relationship between their sequence-structure and function, highlight the importance of methods for analysis and design of ncRNAs with desired structural attributes.
Formation of the secondary structure is the first step in RNA folding. It starts by forming the stem–loops before getting to the formation of pseudoknots to have the final tertiary structure (Batey et al. 1999). For an RNA sequence, a secondary structure is defined by a set of base pairs (ϕi, ϕj), where 1 < i < j < n, such that positions i and j are paired. For two base pairs (ϕi, ϕj) and (ϕk, ϕl), a nonnested loop or a pseudoknot forms if either of the nesting rules i ≤ k ≤ j ≤ l or k ≤ i ≤ j ≤ l is violated. Pseudoknots are abundant in nature (Zandi et al. 2016), exist in specific types (Condon et al. 2004), and are known to play key roles in the functionality of active RNAs (Staple and Butcher 2005), including rRNAs (Powers and Noller 1991), riboswitches (Gilbert et al. 2008), and ribozymes (Harris et al. 2015). Experimental methods, such as nuclear magnetic resonance (NMR) spectroscopy (Varani and Tinoco 1991) or X-ray crystallography (Muchmore et al. 1996), which are used for determining RNA structure, are complex and time-consuming. Hence, computational approaches provide an attractive alternative to study and analyze RNA structures. In addition, computational methods can generate thousands of RNA sequences in short amounts of time, at relatively low cost, while providing the means to predict key sequence attributes useful for further analysis and experimentation.
The classical definition of computational RNA structure prediction or RNA folding (Nussinov and Jacobson 1980) is to find the set of base pairs which are predicted to exist in RNA's most stable structure called the minimum free energy (MFE) structure at thermodynamic equilibrium. The MFE structure can be predicted by computational methods using an energy model. Over the past few decades, several energy models (Freier et al. 1986; Serra and Turner 1995; Mathews et al. 1999; Dirks and Pierce 2003), as well as computational structure prediction methods such as RNAfold (Hofacker 2003), mfold (Zuker 2003), HotKnots (Ren et al. 2005), RNAstructure (Reuter and Mathews 2010), IPknot (Sato et al. 2011), pKiss (Theis et al. 2010), and NUPACK-analyze (Zadeh et al. 2011), have been developed. Conversely, for “inverse folding” rather than predicting the RNA folding, a structure of RNA can be used as a model template to generate new sequences that should fold in the same structure. Early RNA inverse folding programs, such as RNAinverse (Hofacker 2003), designed sequences to have an MFE structure that precisely matches a desired target structure, although they did not take pseudoknots into consideration at the time. Besides the classical MFE as the design criteria, other criteria such as Boltzmann probability or ensemble defect optimization criteria have also been used. In the context of RNA inverse folding, Boltzmann probability quantifies the probability of an RNA sequence folding into a given structure, where the ensemble defect quantifies the expected number of incorrectly paired nucleotides at thermodynamic equilibrium. The value of ensemble defect should be minimized as much as possible to avoid misfoldings in double and single stranded regions of structure (Dirks et al. 2004). It has been shown that the sequences designed to maximize the Boltzmann probability or minimize ensemble defect tend to be thermodynamically more stable than those designed to satisfy the MFE criteria (Zadeh et al. 2011; Zandi et al. 2016).
The RNA design problem is computationally complex (Schnall-Levin et al. 2008) and to find solutions, most existing algorithms resort to heuristics and a combination of local, global, and stochastic search methods. Generally, a random seed RNA sequence is first generated, then the seed is iteratively mutated until the predicted folding attributes of the design candidate converge to the desired values. In our recent work, Enzymer (Zandi et al. 2016) utilizes an adaptive, weighted sampling strategy to design RNA secondary structure with low ensemble defect. The in silico simulations showed that Enzymer generates RNAs that are thermodynamically more stable, have higher Boltzmann probability of folding into the desired target, and have lower ensemble defect than those generated by other state-of-the-art pseudoknot designer methods such as MODENA (Taneda 2011) and antaRNA (Kleinkauf et al. 2015).
The wealth of existing computational RNA secondary structure design methods, where each method utilizes a different design criterium and sequence optimization strategy, makes it a difficult task for the experimental biologists to choose the right method for their particular RNA design purpose. For instance, some methods such as NanoTiler (Bindewald et al. 2008) or NUPACK-design are more suited for the design of nonfunctional RNA nanostructures, while other methods such as Frnakenstein are specialized in designing RNA switches (Lyngso et al. 2012). Another decisive factor in the applicability of an RNA designer method is its ability to realize all the structural elements necessary for a particular design objective. For instance, only NanoTiler (Bindewald et al. 2008), MODENA, antaRNA, and Enzymer can handle the pseudoknots. Another important consideration is related to the applicability of the underlying energy model used by each design method. Still another important consideration is the availability of experimental evidence in support of the applicability of a design method for a specific design purpose. Ultimately, it is wet-lab experimental data that provide the most reliable measure of the usefulness of a design method. To the best of our knowledge, as summarized by Churkin et al. (2017), there seems to be no comprehensive report in the literature that provides experimental evidence on the applicability of all inverse RNA folding methods to the design of pseudoknotted ncRNAs.
In this study, we demonstrate that Enzymer can be used as a reliable method for the design of pseudoknotted ribozymes. We used Enzymer to reengineer three naturally occurring ribozymes: a self-cleaving HHRz from a mouse gut metagenome, a self-cleaving HHRz of Yarrowia lipolytica (Perreault et al. 2011), as well as a self-cleaving glmS ribozyme (Klein and Ferre-D'Amare 2006). For each ribozyme, we obtained the minimally required catalytic core extracted from multiple sequence alignment data and used the catalytic core as a design template for Enzymer.
RESULTS
Synthetic hammerhead ribozymes with a pseudoknot for the stem I–II interaction
The activity of an RNA depends on its folding in defined secondary and tertiary structures (Bhartiya and Scaria 2016). To have a functional synthetic RNA, the structure of the original RNA used as template should be conserved. In our study, we used Enzymer to design sequences that fit the functional structures of given templates. To validate the efficiency of Enzymer's designed sequences, we experimentally tested two types of self-cleaving ribozymes: HHRz and glmS ribozymes.
Numerous HHRzs were previously designed “by hand” by many researchers, where for typical minimal HHRzs, all three stems had four or more base pairs, with few exceptions (Kuwabara et al. 1999). It was noted that cleavage rate measurement was performed with 25 mM Mg2+. On the other hand, we have chosen, on purpose, more challenging target models, that is, with a stem II of only two canonical base pairs. Two HHRz structures from different genomes and context were used as templates: one from the mouse gut metagenome and the other from Yarrowia lipolytica (Perreault et al. 2011). Both ribozymes are type I hammerhead with a pseudoknot occurring between stem I and the loop of stem II. The generated ribozymes should fold in the same secondary structure as the chosen ribozymes by respecting the position of canonical base pairs and single stranded regions. Enzymer does not change the size of the sequences generated as they should be exactly the same as the template sequence. The two base-pair stem II of the mouse gut metagenome HHRz (Fig. 1A,B; Supplemental Fig. S1 [for the WT version]) makes it a good model to test Enzymer since misfolding could easily prevent stem II from folding correctly following a faulty design. Using the secondary structure of this HHRz (Fig. 1A,B), we selected the 14 sequences with the lowest ensemble defects. At least two such sequences were selected among ten designed per set of differing parameters, namely different optimal temperatures for folding, which we thought of as a way to have structures with a gradient of stability. In this way, HHRz 1 to 6 were set for optimal folding at 37°C, HHRz 13 and 14 at 17°C, 11 and 12 at 27°C, 7 and 8 at 47°C, and 9 and 10 at 57°C. For self-cleavage tests to compare their activity to the wild type, all ribozymes were assayed during transcription at 37°C.
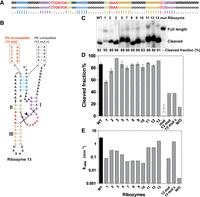
Ribozymes designed according to Mouse gut metagenome ribozyme structure. (A) Design template and secondary structure of the “mouse gut metagenome HHRz” structure used as the input for Enzymer. All of the original sequences were modified except the red nucleotides forming the catalytic core. (B) Secondary structure with a new sequence generated by Enzymer, with a color scheme corresponding to that of the design template in A, with mutations compatible or incompatible with the pseudoknot illustrated. Cleavage site is shown by an arrowhead. (C) Cleavage % of the ribozymes during transcription (for 2.5 h) for the self-cleaving ribozymes compared to the WT. (D) Cleavage extent of the cis-acting ribozymes (WT, 1 to 13 and mut). For Rzs 13 mut, 13 mut c, and N/O (non-optimized), the cleavage of trans-acting ribozymes was used. Average of three experiments with standard deviation. Comparison of cleavage extent for cis- and trans-acting ribozymes is available in Supplemental Material (Supplemental Fig. S4). (E) Cleavage rate (kobs) of ribozymes.
The HHRz 4, 6, and 14 sequences could not be transcribed or assayed properly for the trans version (some sequences are harder than others to transcribe and after a few attempts, we decided to focus on the other ribozymes), but the other 11 ribozymes were active compared to the wild type, which cleaved at 86% (Fig. 1C). Ribozymes 3 had a slightly higher fraction cleaved than the wild type with a cleavage of 96%. The rest of them have variable end-point cleavage efficiency similar or slightly lower than the wild type, except ribozyme 1 that clearly cleaved less (56%) (Fig. 1C,D). For more precise measurements of the activity of the ribozymes, we followed the kinetics of each one of them during 64 min. For these assays, trans-acting ribozymes were used to facilitate measurements of rate constants independently of transcription. The ribozyme sequences were divided into two parts: the template and the ribozyme part (Supplemental Table S1; Supplemental Fig. S1). The results show that all the ribozymes were able to cleave the substrate, but with different rates and efficiencies. The kobs (rate constant) of the wild-type ribozyme was the highest (3 min−1) followed by HHRz 13 (1.6 min−1), HHRz 11 (0.54 min−1), and HHRz 12 (0.54 min−1). Most of the ribozymes had cleavage rates 10- to 100-fold slower (ranging roughly from 0.03 to 0.3 min−1) with no apparent correlation with the end-point cleavage (full curves in Supplemental Fig. S2).
Interestingly, WT HHRz was the fastest cleaving ribozyme; although HHRz 13 has a very similar activity, other ribozymes cleave much more slowly, even hundreds of folds more slowly. This contrasts with the computed ensemble defects (Table 1) which are optimized for the designed ribozymes, and thus very low, as opposed to the WT for which the calculated ensemble defect was higher than most designed ribozymes.
RNA sequence data generated by Enzymer for all ribozymes
In addition to these results and to prove that ribozymes are able to fold in the correct tertiary structure, HHRz 13 activity was compared to a mutated version with a disrupted pseudoknot and another mutated version compatible with the pseudoknot (with all other positions of HHRz 13 kept identical). As we expected, the mutated version with the disrupted pseudoknot lost the cleavage activity by ∼20-folds (kobs = 0.07 min−1); and when the mutations were compatible with the pseudoknot the activity is recovered (kobs = 1.5 min−1). However, cleavage efficiency or maximum fraction of substrate cleaved is not entirely restored, likely due to the fact that contrary to the original sequence designed by Enzymer, this one is not optimized. For another test of a ribozyme generated by Enzymer, but without the iterations that lead to optimized ensemble defect and thus named “non-optimized” (N/O), there was less cleavage detected than most other designs and it had the smallest kobs, 2 × 10−3 min−1, which is 100-fold less than the average of the other 11 designs for the same model (Fig. 1E; Supplemental Table S2; Supplemental Fig. S2). This indicates that, with this model-ribozyme, ensemble defect optimization was important to obtain efficient ribozymes that have high kobs and high fraction cleaved, as without this optimization, even if all base pairing rules were followed, the cleavage rate was a few orders of magnitude lower. Moreover, the much slower HHRz 13 with the disrupted pseudoknot, compared to a mutant compatible with pseudoknot formation, which cleaves almost as fast as the “WT HHRz 13,” supports the claim that Enzymer was able to design active hammerhead ribozymes using the inverse folding approach by conserving the pseudoknotted structure.
Pseudoknotted hammerhead ribozymes overlapping coding sequence
Next, we wanted to highlight how Enzymer could be used to include additional sequence constraints to obtain efficient ribozymes. As illustrated by the N/O control in Figure 1, the “potential capability” to fold in the active structure is not enough to ensure it will do it efficiently. We thus designed ribozymes flanking the beginning of the coding sequence for the red fluorescent protein (RFP). As a template, we used a type I hammerhead ribozyme found in the Yarrowia lipolytica genome (Fig. 2A,B; Perreault et al. 2011). Sequence constraints include the conserved catalytic core, the Shine–Dalgarno (SD) sequence and the first 10 codons of RFP (Fig. 2A). Identity of the rest of the nucleotides could vary in order to accommodate these constraints and permit proper folding of the HHRz, while ensuring that we did no modifications to the RFP protein. The aim is to have a nondefective protein and to be sure that if there is a decrease in the protein's expression it is caused by the ribozyme's activity and not a mutation in the protein's sequence, not even a synonymous substitution. In this design, an active ribozyme is expected to cause a cleavage inside the coding sequence, which results in a decrease in the protein expression level. It should be noted that while stem II was designed to form two base pairs (as in the WT), the conserved G-C base pair of stem II was changed to U-A because “A” was a constraint from the SD sequence at that position. Such transversions are expected to reduce rate by >>100-fold (Ruffner et al. 1990); this is thus in principle a very detrimental change. Nevertheless, this study was in the minimal HHRz model and in this pseudoknotted version, the extent of cleavage was similar to that of WT and the rate was reduced by “only” 30-folds for HHRz YL1 compared to WT according to our results.
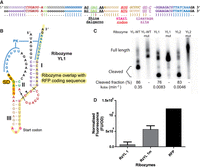
Yarrowia lipolytica ribozymes overlapping RFP coding sequence. (A) Design template and secondary structure of the “Yarrowia lipolytica HHRz” structure used as the input for Enzymer. The original sequence of the ribozyme was modified, except the red nucleotides forming the catalytic core. Nucleotides corresponding to sequence constraints for fusion with coding sequence and Shine–Dalgarno are indicated. (B) Secondary structure with a new sequence generated by Enzymer, with a color scheme corresponding to that of the design template in A. Portions corresponding to sequence of the RFP gene are underlined in yellow for the coding sequence and in orange for the Shine–Dalgarno (SD). Cleavage site is shown by an arrowhead. (C) Cleavage activity of the ribozymes during transcription (for 2.5 h) compared to the WT. Cleaved products of YL1 and YL2 are different from wild type because we reduced stem III by four base pairs and one nucleotide from the loop as compared to WT to permit proper spacing between SD and start codon. (D) The RFP repression assay in which the red fluorescence intensity is measured after 3 h induction with IPTG and compared between the active ribozymes (RzYL 1) and the mutated inactive version (RzYL 1 m), with one control of the unmodified RFP expression (RFP). The bands on the gel, appearing in the uncleaved control lanes (mut), are likely due to a nonspecific degradation of RNA.
To have uncleaved size markers, we mutated both HHRz YL ribozymes in the catalytic core without affecting the coding sequence. The ribozymes were tested for their cleavage efficiency during in vitro transcription and their activity was compared to the wild type. Both ribozymes were active with fractions cleaved of 76% for the HHRz YL1 and 83% for the YL2, which are close to the cleavage efficiency of the wild type (86%) (Fig. 2C). The ribozymes were divided into two parts in the same way as for the mouse gut metagenome ribozymes (Supplemental Table S1 for DNA sequences). The ribozyme WT was able to cleave 54% of the substrate after 8 min compared to the two ribozymes designed by Enzymer (YL1 and YL2) that attained only 28% and 25% of the cleavage rate after 64 min. While end-point cleavage was lower, it was still within a few folds of the WT; on the other hand, kobs was orders of magnitude lower.
In spite of an activity much lower than for the ribozymes from Figure 2C, expression of RFP was repressed in the presence of the active version of the ribozyme (RzYL 1) as compared to the inactive mutant version (RzYL 1m), as illustrated by the lower RFP fluorescence (Fig. 2D). The background, represented by E. coli without any RFP construct, was relatively high; raw fluorescence results are therefore presented in Figure 2D to avoid misinterpretation. Otherwise, subtracting the background from both the active RzYL 1 and its inactive version suggest an eightfold decrease in expression due to ribozyme cleavage. An example of RFP expression without any potentially hindering secondary structures in its 5′-UTR (such as HHRz stems and pseudoknot) is also provided for indicative purposes.
Synthetic glmS ribozymes activated by glucosamine-6-phosphate
To be able to see if Enzymer could successfully design structures more complex than the HHRz, we used the glmS ribozyme as another template. This ribozyme has a multistem structure, three pseudoknots and many highly conserved nucleotides (Fig. 3A,B). To design such a ribozyme, we used the glmS ribozyme from Thermoanaerobacter tengcongensis (Klein and Ferre-D'Amare 2006).
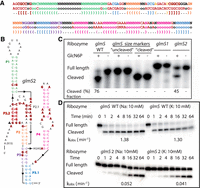
glmS ribozymes. (A) Design template and secondary structure of the glmS ribozyme used as the input for Enzymer. All of the original sequence was modified except the red nucleotides forming the catalytic core. (B) Secondary structure with a new sequence generated by Enzymer, with a color scheme corresponding to that of the design template in A. Cleavage site is shown by an arrowhead. Red nucleotides in bold are those conserved from the WT sequence. (C) Cleavage end point of the ribozymes during transcription (for 2.5 h) compared to the WT with and without glucosamine-6-P (GlcN6P). Inactivating mutations were made to the WT sequence to get the glmS uncleaved size marker, and glmS cleaved is a shorter RNA corresponding to the expected size of the cleaved product. (D) Kinetics of the ribozymes WT and glmS2 (for 64 min) compared to the WT with glucosamine-6-P (GlcN6P) and K+ or Na+.
We used two glmS sequences generated by Enzymer to compare their activity to the wild-type glmS ribozyme. These ribozymes were transcribed in the presence or absence of the GlcN6P, where activity could be observed only in the former case. The wild type has a cleavage end point of 76% and glmS2 designed by Enzymer has a 45% cleavage efficiency, while no activity was detected for glmS1 in these conditions (Fig. 3C). We also assayed glmS ribozymes after purification to evaluate rate constants (Fig. 3D). While cotranscriptional cleavage was not observed for glmS1, we could detect a small amount of cleavage for glmS1 in these conditions, but both glmS1 and glmS2 cleaved much more slowly than the WT (1.7 min−1 vs. 5.2 × 10−7 and 0.052 min−1) (Supplemental Fig. S2). Still, as for the wild type, self-cleavage glmS2 is completely GlcN6P dependent since we could not detect cleavage in its absence, showing that in addition to self-cleaving capabilities, the designed glmS2 also have a competent binding pocket for GlcN6P.
To better understand the low cleavage rate of glmS2 compared to the WT ribozyme, we wanted to test if the several GGGs, present in multiple positions in the glmS2 sequence, form a G-quadruplex. If this is the case, a G-quadruplex might affect the formation of the correct active structure of the ribozyme. Our results have shown almost no difference in activity and cleavage rate of glmS2 in the presence of either KCl or NaCl (0.052 and 0.041 min−1), which was an indication that no G-quadruplex is formed, at least in these conditions.
For further confirmation of Enzymer's ability to design RNAs folding in the same structure as the WT, we used the in-line probing technique to compare the degradation profile of each one of the glmS ribozymes (WT, 1 and 2). The in-line probing shows that the three sequences share a similar structure in the 5′ part of the ribozyme (from G17 to G73, Fig. 4C). glmS2 ribozyme has more similarity in the structure with the WT in the part closer to the 3′ (after G73) compared to glmS1. These results may explain the cleavage activity of this sequence compared to glmS1. This difference may be the reason for the instability of glmS1's structure causing the cleavage reaction's absence or the inability of the ribozyme to bind the cofactor correctly.
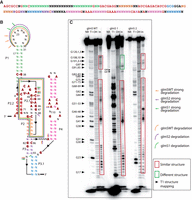
In-line probing of glmS ribozymes. Designed glmS ribozyme structures were compared to the WT glmS ribozyme by in-line probing. (A) Design template with consensus sequence. (B) Secondary structure of glmS WT. (C) glmS2 shows more similarity to the WT in the degradation profile (red boxes).
DISCUSSION
Even if any inverse folding strategy will ultimately be limited by the folding algorithm it uses, an optimization can only be as good as the structure prediction tools used. In this work, we proved that Enzymer can successfully design functional pseudoknotted RNAs. We experimentally tested a total of 18 ribozymes designed by this new inverse folding algorithm. Despite the fact that many ribozymes cleave more slowly, results show that Enzymer could design pseudoknotted functional ribozymes. Even the very significantly reduced rate of cleavage in trans for RzYL 1 appears to be sufficient for biologically significant activity according to RFP reduced expression with the high cis cleavage activity. We also demonstrate for the first time that the Dirks energy model (Dirks and Pierce 2003), which was developed for characterization of pseudoknotted RNA shapes, can be effectively used to model and design functional ribozymes. The fact that most HHRz designs have much lower kobs, compared to WT, suggests that the ensemble defect is better for optimizing the end point cleavage percentage, which means the cis-acting versions of ribozymes, for which most HHRz designs were comparable to WT, rather than the cleavage rate, for which only HHRz 13 was comparable to WT.
Previous attempts reengineered the glmS ribozyme (Lau and Ferre-D'Amare 2013, 2016; Lau et al. 2017), where sequences differed from the WT by as few as three mutations, with many other examples having ∼10 mutations and as many as 26 mutations, although the majority of these were not assayed individually, or ∼3 to ∼5 mutations on average in work from Link et al. (2006). The glmS ribozyme sequences we designed differed from their model natural template by 92 and 79 bases (out of 152) for glmS1 and glmS2, respectively, and they have the least sequence similarity with the WT ribozyme sequence for any designed glmS ribozyme, as well as the least overall sequence similarity with natural sequences (Supplemental Table S3). Interestingly, both glmS1 and glmS2 diverge slightly from the more recent alignment consensus found in McCown et al. (2011), with five positions for glmS1 and seven for glmS2, that differ from positions with at least 90% of conservation. Interestingly, one of the P2 base pairs from our model is not conserved in the McCown et al. (2011) consensus (see Supplemental Fig. S3). The fact that our designs have a G-C bp rather than a G-U (differences indicated in Fig. 4 with a star next to the base pair), like the WT template used from Klein and Ferre-D'Amare (2006) at that position would stabilize a bp that could potentially be only transient in the folding pathway leading to catalysis. Together, this suggests that rather than being a faulty Enzymer design, glmS1 could be cotranscriptionally inactive because the provided model lacked important information. Indeed, glmS1 was generated in our early attempts to design a sequence for this complex ribozyme and as such it lacked some constraints (colored in blue in Table 1). For instance, the different potential to form purine stacks in an internal loop of P4, which is described as the “floor” of the GlcN6P binding site by Klein and Ferre-D'Amare (2006), would likely impact ribozyme activity if it affects binding GlcN6P or its positioning relative to the cleavage site. As opposed to HHRz, for which the catalytic core has been studied extensively and is very well defined, we may have overlooked important constraints in the glmS ribozyme, explaining that the two tested designs had a considerably decreased activity compared to WT. It is the case especially for glmS1 ribozyme that had very low to absent cleavage activity compared to the glmS2 that had a good cleavage efficiency and structure similar to the WT. Such constraints could be related to the target structure itself, but could also include additional structures corresponding to intermediates during catalysis: prebound, bound precleaved, cleaved states. Previous studies (Klein and Ferre-D'Amare 2006; Cochrane et al. 2009) indicate global rigidity of this RNA and no significant conformational change between these states. The fact that the cleaved state remains unchanged, in spite of losing a portion of sequence, may be important as the designs and corresponding ensemble defects were computed for the full sequence only. A more recent study shows that the ribozyme folding in the structure allowing the binding of the ligand is unstable and even the cofactor binding to its binding pocket is unable to stabilize this conformation. These findings suggest that the structure is too dynamic and that there should be an equilibrium between the structure's stability and the ligand binding for the cleavage to occur efficiently. This might help explain the low activity of the two glmS ribozymes designed by Enzymer (Savinov and Block 2018). Alternatively, it could also highlight challenges that still need to be overcome for inverse folding. For instance, the WT sequence likely has a favorable folding landscape to achieve the “target structure” while the best way for Enzymer to simulate this is to have a minimal ensemble defect. Multiple instances of kinetic traps or misfolded dead-ends exist in the literature, which have been shown to be reduced by specific folding intermediates (Brown et al. 2004; Cao and Chen 2007). If the strategy of using ensemble defect to reduce such misfolded RNAs seems to work, at least to some extent, it may also have limitations. Taking into account multiple substructures may represent one of the future challenges of inverse folding but represents a challenge for experimentalists as well.
Interestingly, when comparing the rate constants with the design temperatures, we observe an inverse correlation between the ribozyme design temperature and their activity, as opposed to end-point cleavage assays. The most active designed ribozymes were HHRz 13 followed by HHRz 12 and HHRz 11, respectively designed for 17°C, 27°C, and 27°C (details in Supplemental Table S4, Supplemental Fig. S1). This may illustrate an advantage of having less stable (less rigid) structures, which could concurrently, also potentially reduce likelihood of misfolded dead-ends. The use of trans-cleaving ribozymes facilitates the measurement of time series to obtain rate constants but may not always be representative of the cis-cleaving version, which is normally expected to cleave more efficiently.
To our knowledge, only two groups actually tested ribozymes designed through inverse folding. The first group (Dotu et al. 2014) used a type III HHRz from a portion of the plus polarity strand of peach latent mosaic viroid (PLMVd). To make their design, they used RNAiFold and tested the generated ribozymes experimentally. These ribozymes were active, but RNAiFold does not take into account pseudoknots. On the other hand, others have designed pseudoknotted ribozymes, like the HDV ribozyme, but without experimental validation (Taneda 2011). More recently, Yamagami et al. (2019) have designed active ribozymes (∼50-fold slower than WT), which do have pseudoknots, but this work, to our knowledge, is the only other experimental validation of pseudoknotted ribozymes designed by inverse folding. They only focused on HDV ribozymes, which have a different topology than glmS and the two HHRz types that we have used as templates for designs. In that article, Yamagami and coworkers had fixed base identity for several positions, reducing the sequence space to search from 53 to 43 Ns (64, 38, and 101 Ns for HHrz from mouse gut, HHRz from Yarrowia and glmS, respectively, in this study). Among the fixed base identity, five “As” were assigned to the loop 4 of HDV, interestingly, beyond our sequence constraints, multiple contiguous “As” were often noticed in regions designed to be single stranded. Perhaps this occurred as minimization of ensemble defect that might preferentially assign A instead of U or G to form single stranded regions. This is because Us or Gs have more base pairing capability due to wobble G-U base pairs in addition to G-C. Also, Cs are probably avoided because they can form stronger base pairs with G than As with U.
The ability of Enzymer to design sequences for complex active structures with many sequence constraints, such as for the Yarrowia lipolytica HHRz example, paves the way for combinations of sequence elements. Indeed, we were able to design functional ribozyme structures with unprecedented nonribozyme sequence constraints (where >40% of the ribozyme sequence was fixed to fit the RFP sequence). Interestingly, we noted that glmS2 had several GGGs, including four which were close and that would theoretically have the potential to form a G-quadruplex according to G4Hunter (Brazda et al. 2019). Even if our comparison of activity in NaCl versus KCl suggests that G-quadruplexes do not actually form, it nevertheless highlights the potential uses of being able to push the boundaries in terms of divergence from the natural sequences. With proper design, many useful arrangements can be made to engineer new regulatory elements by overlapping important sequences, such as coding sequence or splicing sites, with structures, such as ribozymes or riboswitches, to gain new functions. Another example would be a riboswitch controlled by two or more aptamers with an expression platform than can be a ribozyme or one similar to the platforms of natural riboswitches.
MATERIALS AND METHODS
We generated a population of candidate sequences for each ribozyme. For each ribozyme, we sorted the generated sequences by their predicted normalized ensemble defect value, chose a small set for in vitro studies and measured their catalytic activities. We also designed a non-HHRz ribozyme: the GlcN6P-dependent glmS ribozyme.
Designing ribozymes
We obtained the secondary structures as well as the minimum catalytic core (for review, see Nelson and Uhlenbeck 2008) for the HHRzs from Perreault et al. (2011). For the glmS ribozyme, we used the secondary structure as well as the minimum catalytic core from the model presented by Klein and Ferre-D'Amare (2006), but in its natural cis-acting form. Given a secondary structure and a set of nucleotides representing the minimal catalytic core, the step to initialize a design template is to generate a seed sequence using the catalytic core and then using the letter “o” for any position which is not part of the catalytic core, or highly conserved nucleotides, of the ribozymes. In other words, positions considered to be involved only in secondary structure (standard base pairs, including pseudoknots), as well as single stranded “nonessential positions,” are open for change of nucleotide identity. To account for the three-dimensional structure essential for ribozyme activity, all conserved positions considered important for tertiary contacts are fixed in the provided model.
We ran Enzymer using the design templates as input and set the maximum number of iterations to 600. We generated 14, two, and two sequences for the mouse gut metagenome hammerhead, Yarrowia lipolytica hammerhead, and the glmS ribozymes, respectively, in independent trials. For most trials, we chose the Mathews parameters (Mathews et al. 1999), but for optimal folding at temperatures other than 37°C we used the Serra and Turner energy parameters (Serra and Turner 1995). For all cases, we used the additional parameters for pseudoknots from the Dirks and Pierce model (Dirks and Pierce 2003). More details on how to use Enzymer are available in Najeh et al. (2021) and on the algorithm in Zandi et al. (2016). In short, users need to provide a secondary structure model with sequence constraints, as well as parameters as described above. The sequence constraints will typically correspond to the conserved catalytic core of the ribozyme (or of the binding pocket of riboswitches for instance), as well as any other nucleotides which participate in noncanonical base pairs that are important to properly fold in the active tertiary conformation. For the HHRz, these bases are very well defined, but for glmS these constraints were determined from a combination of sequence conservation (McCown et al. 2011) and information from the structure (Klein and Ferre-D'Amare 2006). The sequences we generated are presented in Table 1 together with their design templates (which are also included in each relevant figure); additional details on sequence generation are available (Supplemental Text).
Transcription
The synthesis of the cis-acting ribozymes by in vitro transcription was made in the presence of radioactive Uridine Triphosphate (alpha-32P-labeled UTP) to be able to visualize and quantify the self-cleavage activity during the transcription. The reaction of 50 µL contained the DNA produced by PCR, 2 mM rNTPS (2 mM each of rATP, rGTP, rCTP) and 0.8 mM UTP, 1× transcription buffer (80 mM HEPES-KOH pH 7.5, 24 mM MgCl2, 40 mM DTT, 2 mM spermidine), 1 U/µL of inorganic pyrophosphatase (Sigma Aldrich), 1 U/µL RNase inhibitor (Thermo Fisher), 1 U/µL T7 RNA polymerase (produced by a laboratory at University of Sherbrooke) and milliQ water for 2 h at 37°C. Then 1 U/µL of DNase I (RNase free) (NEB) was added to each reaction and they were incubated for 30 min at 37°C. For the glmS ribozymes, for each ribozyme we had two reactions with or without 1 mM of GlcN6P.
The RNAs are then precipitated and a sample is taken and run on a denaturing polyacrylamide gel to visualize the cleavage reaction and measure the fraction cleaved of each ribozyme during transcription.
For the trans-acting ribozymes (divided into two parts: the ribozyme and the substrate) and the glmS ribozymes used for the in-line probing, transcription was made in the absence of radioactivity. RNAs were extracted by phenol/chloroform extraction followed by ethanol precipitation. The RNAs were later separated on 10% denaturating polyacrylamide gel, the corresponding bands were cut and RNAs were eluted overnight at 4°C.
RNA 5′ labeling
After elution, the RNAs were precipitated by ethanol and the substrates were dephosphorylated using 1U antarctic phosphatase (NEB) in the presence of 1× buffer (50 mM bis tris propane-HCl, pH 6, 1 mM MgCl2, 0.1 mM ZnCl2) during 20 min at 37°C. The enzyme was inactivated by heating at 65°C during 5 min. The RNA labeling was done using 1U of T4 polynucleotide kinase at 37°C during 1 h in the presence of gamma-32P-labeled-ATP and 1× of the enzyme buffer (70 mM tris-HCl pH 7.6, 10 mM MgCl2, 5 mM DTT). The labeled substrates were purified on a 10% denaturating polyacrylamide gel and precipitated with ethanol. For the glmS ribozymes, the same conditions were used except for the buffer where the tris-HCl was replaced by the HEPES solution in order to inhibit the self-cleavage of the ribozyme in the presence of the tris.
Self-cleavage analysis
An aliquot of 1 µL of each ribozyme transcription was taken and diluted in 9 µL of milliQ water. To each aliquot an equal volume of the 2× dye formamide buffer (95% formamide, 10 mM EDTA, 0.025% bromophenol blue, and 0.025% xylene cyanol blue) was added. The full length and the cleaved ribozymes were separated by electrophoresis on a 10% polyacrylamide gel in 1× TBE buffer (89 mM Tris, 89 mM boric acid, 0.02 M EDTA). The gel was exposed to a storage phosphor screen for 30 min to 1 h, scanned with a Typhoon FLA9500 (GE Health Care) and quantified with ImageQuant.
Kinetics measurements
Hammerhead ribozymes
Each trans-acting ribozyme (5 pmol/µL) was preincubated in the presence of the cleavage buffer (50 mM Tris pH 7.5, 100 mM NaCl, and 25 mM KCl) at 85°C for 1 min then snap cooled on ice. Trace amounts of radiolabeled substrate and 4 pmol/µL of nonradiolabeled substrate (to ensure similar concentrations for all ribozymes) were added to the reaction that was initiated by adding 1 mM MgCl2 and incubated at 37°C. The timer starts at the moment the MgCl2 is added, then the reaction is followed during 64 min. Aliquots of the reaction were taken at different times (20 sec, 40 sec, 1 min, 2 min, 8 min, 16 min, 32 min, and 64 min) and the reaction is stopped by putting the aliquot in the stop dye (2× formamide buffer). The RNAs were fractioned on a 10% denaturating PAGE and the results visualized and quantified as described. The cleavage rates (kobs) were calculated for each ribozyme using GraphPad by using the equation of “one phase decay.” It should be noted that we only have a slight excess of ribozyme over substrate, contrary to typical single turnover kinetics, which can affect the actual values of constants determined, but still allow us to compare results from different ribozymes.
glmS ribozymes
The kinetic measurements for the glmS ribozymes were done using the cis-acting versions of the ribozymes. The denaturation-renaturation of these ribozymes was tried in two ways. The ribozymes were denaturated by incubating them at 85°C for 1 min then either directly put on ice, or, in the second assay, after denaturation the RNAs were left to cool at room temperature and this gave a better result for the ribozymes cleavage rate.
To start the reactions, 1 mM of GlcN6P was added and reactions were incubated at 37°C for 1 d. For these ribozymes, in addition to time points mentioned above for the hammerhead ribozymes, aliquots were also taken after 2, 4, and 24 h.
In-line probing of glmS ribozymes
The radiolabeled ribozymes were incubated for 16 h at 37°C in the in-line probing buffer (2×) (100 mM Tris-HCl, pH 8.3, 200 mM KCl, and 40 mM MgCl2) and in the absence of GlcN6P. The stop dye was added to stop the reaction. Two controls were used to identify the positions of the nucleotides on the gel:
The T1 reaction:RNA is incubated in the presence of 0.25 M sodium citrate, pH 5.0, and formamide for 2 min at 56°C. Then 1.5 µL of diluted RNase T1 (1/100) is added and the reaction is incubated at 56°C during 5 min.
OH reaction:RNA was put in the presence of 2 µL of Na2CO3 buffer (10×) (0.5 M Na2CO3, pH 9.0, and 10 mM EDTA).
After the two controls were prepared, RNA fractions were separated on an 8% denaturating polyacrylamide gel.
RFP repression
The ribozyme overlapping the RFP sequence (Rz YL1) and its inactive version (Rz YL1m) were cloned in pUC57 with the inducible Lac promoter, upstream of the rest of the RFP sequence (ordered from Biomatic). The plasmids were transformed in E. coli BL21DE3. The plasmid pSEVA3b67Rb (ordered from Addgene in E. coli DH10B), including a version of RFP without ribozymes, was used as positive control to ensure proper fluorescence detection. As a negative control, we used E. coli BL21DE3 without any plasmid, and therefore lacking RFP, to evaluate background fluorescence, which was subtracted from other values.
The bacteria were precultured in Lysogeny Broth (LB) overnight with ampicillin (100 µg/mL) for pUC57 and chloramphenicol (6 µg/mL) for pSEVA3b67Rb. Then each culture was diluted in M9 minimal mineral media to an OD600 of 0.2. When the OD600 was around 0.5–0.6, expression was induced using 1 mM IPTG. After 3 h induction, the fluorescence intensity was measured using a Typhoon FLA9500 and was normalized with the OD600.
SUPPLEMENTAL MATERIAL
Supplemental material is available for this article.
ACKNOWLEDGMENTS
This work was supported by the Natural Sciences and Engineering Council of Canada (NSERC) (418240 to J.P.). J.P. is a Junior 2 FRQS Research Scholar. S.N. received a fellowship from the Armand-Frappier Foundation and FRQNT. The authors also wish to thank Elvira Rwasamanzi for technical help.
Footnotes
-
Article is online at http://www.rnajournal.org/cgi/doi/10.1261/rna.079148.122.
- Received March 2, 2022.
- Accepted May 27, 2022.
This article is distributed exclusively by the RNA Society for the first 12 months after the full-issue publication date (see http://rnajournal.cshlp.org/site/misc/terms.xhtml). After 12 months, it is available under a Creative Commons License (Attribution-NonCommercial 4.0 International), as described at http://creativecommons.org/licenses/by-nc/4.0/.








